Introduction
Perioperative acute hypoxic-ischemic cardiac
toxicity of anesthetics is one of the most serious complications
during surgical procedures, and occurs following the blockade of
sodium channels and negative inotropic effect (1–4). To
avoid cardiac toxicity, selection of a suitable drug for the
maintenance of anesthesia, especially during cardiac surgery, is
crucial (5,6). A combination of epidural and general
anesthetics have been commonly applied to reduce the use of general
anesthetics, such as combining ropivacaine, a local anesthetic, and
propofol, a general anesthetic (7,8).
Ropivacaine reportedly possesses lower cardiovascular and central
nervous toxicity in vivo, in comparison with bupivacaine
(9,10). However, its underlying mechanisms
remain unknown, and it is unclear whether the mechanism of action
is involved in the induction of perioperative ischemic and hypoxic
toxicity (11,12).
In contrast, due to its phenolic structure, propofol
can prevent ischemic/reperfusion injury (13,14).
Furthermore, propofol has an anti-oxidative effect in rat
cardiomyocytes and macrophages, which is associated with the
suppression of oxidative stress-related enzymes including inducible
nitric oxide synthase, superoxide dismutase (SOD) and neutrophil
cytosolic factor 1. In addition, propofol is associated with an
increase in intracellular nitric oxide release (15,16) and
the preservation of Bcl2 expression levels (17). Propofol was shown to protect
microglia from hypoxia-induced inflammation and apoptosis by
maintaining intracellular Ca2+ homeostasis and the
activation of the JNK1/Stat3 signaling pathway (18). Furthermore, propofol exerts
antiproliferative and anti-invasive effects on hepatocellular
carcinoma and rheumatoid arthritis fibroblast-like synoviocytes via
the Wnt/β catenin (19) and NF-κB
signaling pathways (20). Previous
studies demonstrated that propofol plays a variety of roles in
different cell types via various signaling pathways. However, the
effects of propofol, especially when combined with ropivacaine, on
human cardiomyocytes is not fully understood.
Therefore, the present study used human AC16 and HCM
cardiomyocytes treated with cobalt chloride (CoCl2) as
an in vitro model of cardiomyocyte ischemia. The present
study investigated the signaling pathways associated with propofol
and/or ropivacaine activity against oxidative stress injury in
cardiomyocytes.
Materials and methods
Cell culture
Human adult AC16 and HCM cardiomyocytes (21) (cat. nos. BNCC337712 and BNCC337719;
Suzhou BeNa Culture Collection Biotechnology Co., Ltd.) were
cultured in DMEM/F12 (Thermo Fisher Scientific, Inc.) supplemented
with penicillin 100 U/ml, streptomycin 0.1 mg/ml (Invitrogen;
Thermo Fisher Scientific, Inc.) and 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in a 5% CO2 incubator. To
establish hypoxic conditions, the cardiomyocytes were synchronized,
incubated in the complete DMEM/F12 with 500 µmol/l
CoCl2 (Sigma-Aldrich; Merck KGaA; CAS Number 7791-13-1),
propofol and/or ropivacaine at different concentrations for 12 h at
37°C in a 5% CO2 incubator.
Cell viability assay
To select the appropriate propofol and ropivacaine
concentrations, cell viability was assessed using the Cell Counting
Kit-8 (CCK-8; Beyotime Institute of Biotechnology) according to the
manufacturer's instruction. Cardiomyocytes were seeded at a density
of 3×103 cells in 96-well plates in sextuplicate and
incubated overnight in the complete DMEM/F12 with or without 500
µmol/l CoCl2 in an atmosphere with 5%
CO2 at 37°C. After removing the culture medium, cells
were treated with propofol (100, 50, 25, 12.5, 6.25 and 0
µg/ml) or ropivacaine at different concentrations (300, 150,
75, 37.5, 18.75 and 0 µg/ml in the absence of Cocl2
treatment experiments; 100, 50, 25, 12.5 and 0 µg/ml in the
500 µmol/l Cocl2-pretreatment experiments) in fresh medium,
repeated eight times, for 48 h at 37°C. In addition, to determine
the synergistic interactions between propofol and ropivacaine, AC16
and HCM cells were treated by 25 µg/ml of propofol and
different ropivacaine concentrations (200, 100, 50, 25, 12.5 and 0
µg/ml) for 48 h at 37°C. The supernatants were discarded and
the CCK-8 solution, (dilution, 1:10) was added to each well in
complete DMEM/F12, and the cells were incubated for 2 h at 37°C.
Absorbance at 450 nm was measured using a microplate reader (cat.
no. MQX200; BioTek Instruments, Inc.). All experiments were
repeated four times.
Flow cytometry analysis
Cell apoptosis and mitochondrial membrane potential
(Δψm) were evaluated using flow cytometry analysis. Cardiomyocytes
were incubated in complete DMEM/F12 with or without 50 µg/ml
propofol combined with 50 µM Juglanin (cat. no. 5041-67-8; HPLC
≥98%; Shanghai Zeye Biological Technology Co., Ltd.) for 2 h at
37°C, and treated with 500 µm CoCl2 in fresh medium for
12 h at room temperature. Following drug treatments, cells were
harvested, washed twice with PBS and stained with Annexin V-FITC/PI
Apoptosis Detection kit (cat. no. A211-01; Vazyme Biotech Co.,
Ltd.) in the binding buffer for 15 min in the dark at 25°C
according to the manufacturer's protocol. The fluorescence
intensity of Annexin V/PI-stained cells was analyzed using
FACSCanto II flow cytometry (Becton, Dickinson and Company) within
1 h. Early apoptotic Annexin V+/PI− cells
were counted using FlowJo software version 7.6.1 (Tree Star, Inc.)
and the quadrant percentage was considered to indicate the
apoptotic ratio.
To determine the Δψm, the remaining harvested cells
were incubated with JC-1 staining for 20 min at 37°C according to
the manufacturer's protocol of a JC-1 staining kit (cat. no.
KGA601; Nanjing Keygene Nanjing Kaiji Biotechnology Co., Ltd.)
After washing the samples twice with the incubation buffer in this
kit, cells were analyzed using the FACSCanto II flow cytometry
(Becton, Dickinson and Company). These experiments were repeated
three times.
Reactive oxygen species (ROS)
measurements
The effects of propofol on ROS production in
CoCl2-treated AC16 and HCM cells were determined using a
ROS-sensitive fluorescent probe (DCFH-DA) (cat. no. D6883;
Sigma-Aldrich; Merck KGaA). Then, cells were analyzed using the
FACSCanto II flow cytometry (Becton, Dickinson and Company). The
cells were seeded at a density of 2×105 cells/well into
6-well plates in triplicate in the following groups: i) Control
group, with the cells cultured in complete DMEM/F12; ii) chemical
hypoxia group, with the cells cultured in complete DMEM/F12 with
500 µmol/l CoCl2 for 12 h; and iii) propofol +
chemically-induced hypoxia group, with the cells pretreated with 50
µg/ml propofol for 2 h and then exposed to chemically-induced
hypoxia. After removing the culture medium, cells were washed with
PBS and incubated in 10 µm DCFH-DA in fresh serum-free medium for
30–40 min in a humidified incubator at 37°C with 5% CO2
in the dark. Labeled cells were analyzed using FlowJo software
version 7.6.1 (Tree Star, Inc.). All assays were performed in
duplicate.
SOD and malondialdehyde (MDA)
detection
Total intracellular expression levels of MDA and SOD
in AC16 and HCM cells were detected using commercially available
kits. Cells were divided into three groups, seeded at
1×106 cells/well and the following groups were analyzed:
i) Control group, chemically induced hypoxia group; and ii)
propofol + chemically induced hypoxia group. To detect MDA and SOD,
the supernatants were removed and cells were lysed in 1 ml of cold
cell lysis buffer, and then cells were crushed for 30 sec after 5
min. The lysis and further processing were repeated four times.
Lysed supernatants were collected by centrifugation at 12,000 × g
for 5 min at 4°C. Then, 0.25 ml of each supernatant, 1.5 ml reagent
C solution and 0.3 ml reagent D were mixed (Micro-MDA Assay Reagent
kit; cat. no. KGT003; Nanjing KeyGen Biotech Co., Ltd.) according
to the manufacturer's protocol, boiled at 95°C for 40 min and then
cooled in ice water for 10 min. After centrifugation at 3,000 × g
for 15 min at 4°C, absorbance values were detected at 532 nm using
a microplate reader. Cell lysis samples were mixed with the
reagents of the Superoxide dismutase (SOD) assay kit (cat. no.
KGT00150; Nanjing KeyGen Biotech Co., Ltd.) according to the
manufacturer's instructions. Then, samples were incubated at room
temperature for 10 min and the absorbance was detected at 550 nm
using a microplate reader. The relative activity levels of SOD
(U/ml) were calculated using the formula recommended by the
supplier of the kit, and expressed according to absorbance levels.
MDA and SOD concentrations were determined based on the constructed
standard curve and are expressed as nmol/(mg total protein). All
assays were performed in duplicate.
Western blot analysis
Western blotting was used to evaluate the activation
of the signaling pathways. Total proteins were extracted from cells
that underwent different treatments using ice-cold RIPA lysis
buffer (Beyotime Institute of Biotechnology) with a protease
inhibitor cocktail (Roche Applied Science), and total protein
concentration in every sample was determined using a BCA Protein
Assay kit (Beyotime Institute of Biotechnology). Then, 40 µg of
total protein solution for each sample was separated by 12%
SDS-PAGE and transferred onto PVDF membrane (Bio-Rad Laboratories,
Inc.). The membranes were incubated with primary antibodies against
JNK1 (1:1,000; cat. no. sc-137018; Santa Cruz Biotechnology, Inc.),
phosphorylated (p-)JNK (1:1,000; cat. no. sc-6254; Santa Cruz
Biotechnology, Inc.), p38 (1:1,000; cat. no. 8690; Cell Signaling
Technology, Inc.), p-p38 (1:1,000; cat. no. 4511; Cell Signaling
Technology, Inc.) and GAPDH (1:2,000; cat. no. 5174; Cell Signaling
Technology, Inc.) at 4°C overnight. Then, the membrane was
incubated with a horseradish peroxidase-conjugated secondary
antibody (goat anti-mouse IgG (H+L)-horseradish peroxidase (HRP)
conjugate; 1:5,000; A16072SAMPLE; Thermo Fisher Scientific, Inc.;
Goat anti-Rabbit IgG (H+L) Secondary Antibody-HRP; 1:5,000; Thermo
Fisher Scientific, Inc.) at room temperature for 1 h. Protein bands
were detected using the enhanced chemiluminescent (ECL) western
blotting detection kit (cat. no. E411-03, Vazyme Biotech Co., Ltd.)
and the intensity of each western blot band was quantified using
Quantity One 1-D Analysis software v4.5 (Bio-Rad Laboratories,
Inc.). All experiments were performed in triplicate.
Statistical analysis
Data are presented as mean ± SD and were analyzed
using one-way ANOVA followed by Tukey's test. All statistical
analyses were performed using GraphPad Prism 4.00 (GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Propofol increases cardiomyocyte
viability and inhibits CoCl2-induced cytotoxicity
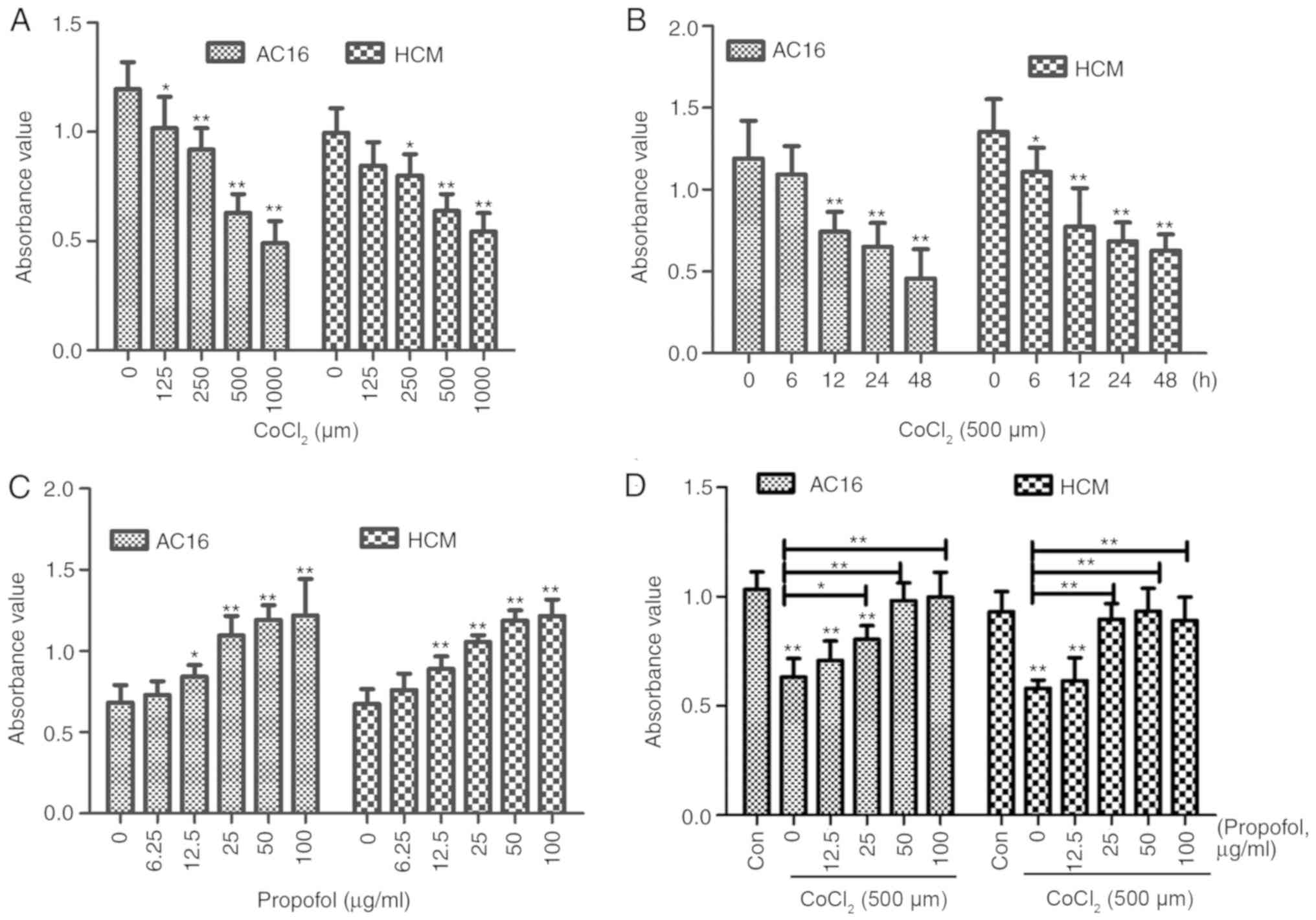
CCK-8 assay results suggested that cell viability
significantly decreased following a 12-h incubation with increasing
concentrations of CoCl2 (0, 125, 250, 500 or 1,000 µm;
Fig. 1A) and incubation with 500 µm
CoCl2 for different periods (0, 6, 12, 24 and 48 h;
Fig. 1B). AC16 and HCM cell
viability in the chemically induced hypoxia group was shown to
decrease to 62.30% (0.7419/1.1909) and 57.13% (0.7739/1.3546),
respectively, compared with the control groups following treatment
with 500 µm CoCl2 for 12 h (P<0.01). Therefore, these
conditions were selected for further experiments.
To determine the effects of propofol and ropivacaine
pretreatment on cardiomyocyte viability, the viability of AC16 and
HCM cells treated with propofol or ropivacaine at different
concentrations was determined using the CCK-8 assay. The present
results suggested that propofol significantly increased cell
viability under normal culture conditions in a dose-dependent
manner (Fig. 1C). The present
results indicated that propofol could reverse the decreased
viability of AC16 and HCM cells in a dose-dependent manner when
combined with CoCl2 treatment. Moreover, the protective
effects of propofol against CoCl2 hypoxiainduced injury
were identified to be the highest at 50 µg/ml propofol (Fig. 1D; P<0.01). Compared with the
viability observed following the application of propofol, combined
propofol and ropivacaine treatment did not increase the viability
of cells (Figs. S1 and S2). In addition, regardless of whether the
conditions were normoxic or hypoxic, ropivacaine did not affect the
viability of AC16 and HCM cells (Fig.
S3). Therefore, propofol was selected for further
investigation.
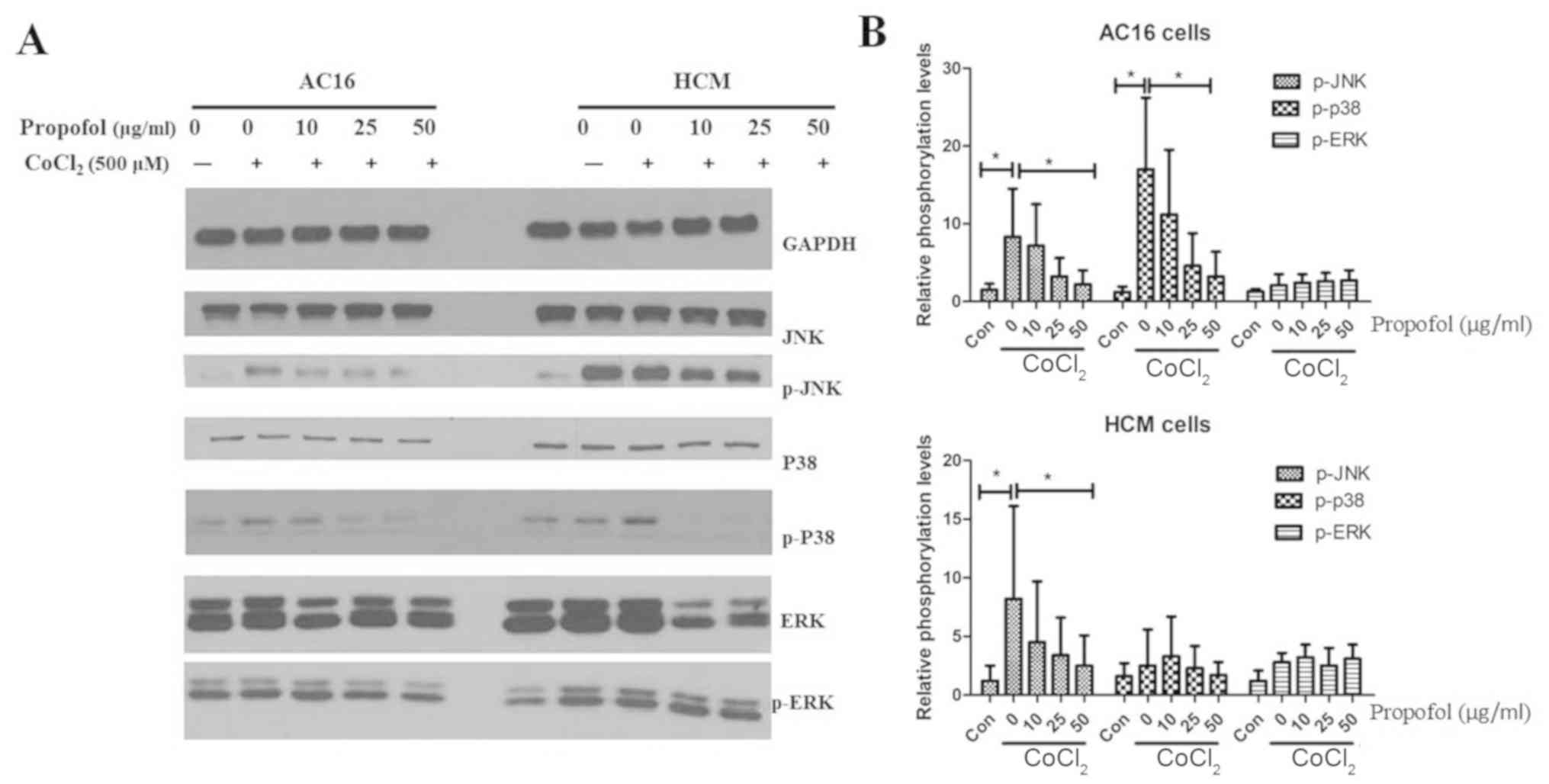
Propofol inhibits activation of the
JNK signaling pathways in AC16 and HCM cells exposed to
CoCl2
In order to identify the signaling pathways involved
in the anti-apoptotic functions of propofol, the present study
investigated the activation of the NF-κB and mitogen-activated
protein kinase (MAPK) signaling pathways. These pathways are
reported to be the key signal transduction pathways involved in
CoCl2-induced apoptosis in BV2 and HK2 cells (18). However, in the present study,
propofol did not affect the phosphorylation of p65 subunit, P38,
ERKs and JNK in either AC16 or HCM cells (Fig. S4). The present study also
investigated the signaling pathways that may underlie the
identified propofol effects. AC16 and HCM cells were pre-incubated
with propofol at different concentrations and treated with
CoCl2. CoCl2 was identified to promote the
phosphorylation of JNK and P38 in AC16 cells, and the
phosphorylation of JNK in HCM cells following 2 h treatment, but
these effects were significantly inhibited by propofol treatment
(Fig. 2). CoCl2 did not
increase the phosphorylation of P38 in HCM cells, which may be
caused by heterogeneity between cell types. The present results
indicated that JNK and p38, but not ERK signaling, represented the
main components of the apoptotic pathways contributing to
CoCl2-induced apoptosis of AC16 or HCM cells.
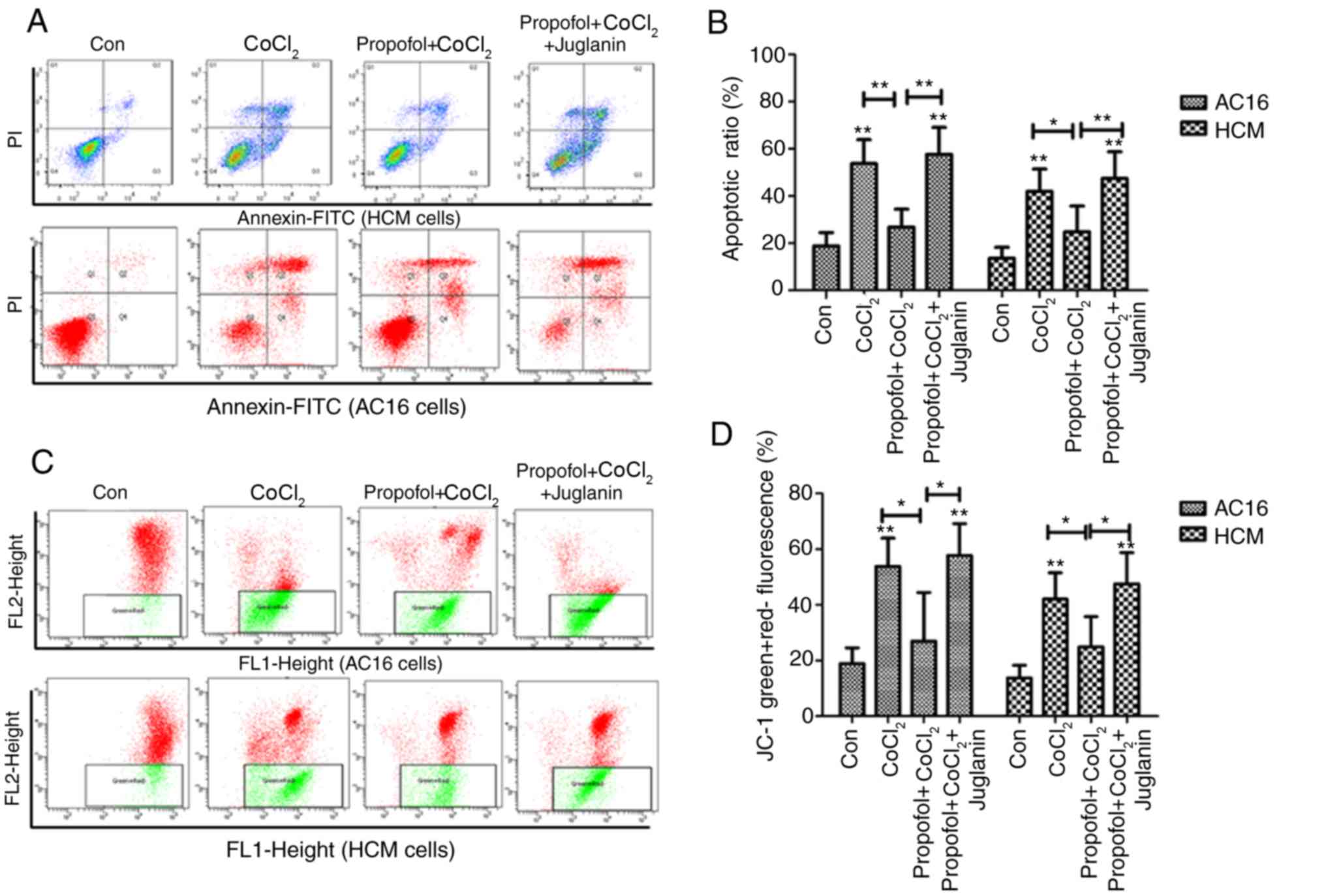
Propofol protects against
CoCl2-induced cardiac cell apoptosis
CoCl2 has been reported to induce
apoptosis of rat cardiac H9C2 cells (22). Therefore, the present study used
Annexin V/PI staining to investigate the apoptotic ratio of AC16
and HCM cells following different treatments (Fig. 3A). The ratio of cells in the 500 µm
CoCl2-induced hypoxia group was higher compared with the
control group (P<0.01). This ratio was significantly reduced
after 2 h pretreatment with 50 µg/ml propofol prior to the
induction of hypoxia. However, the JNK activator juglanin could
counteract this reduction and considerably suppressed the
anti-apoptotic effects of propofol pretreatment in
CoCl2-induced cardiac cells (Fig. 3B).
A hypoxiainduced injury may result in reduced
mitochondrial membrane potential (23). Therefore, the present study
investigated the effects of propofol on Δψm using JC-1 staining.
The present results suggested 50 µg/ml propofol significantly
inhibited CoCl2-induced decreases in the Δψm of AC16 and
HCM cells (Fig. 3C and D).
Additionally, the present results suggested that juglanin
significantly counteracted the recovery effect on Δψm induced by
pretreatment with propofol and promoted the reduction of Δψm
induced by CoCl2 (P<0.05).
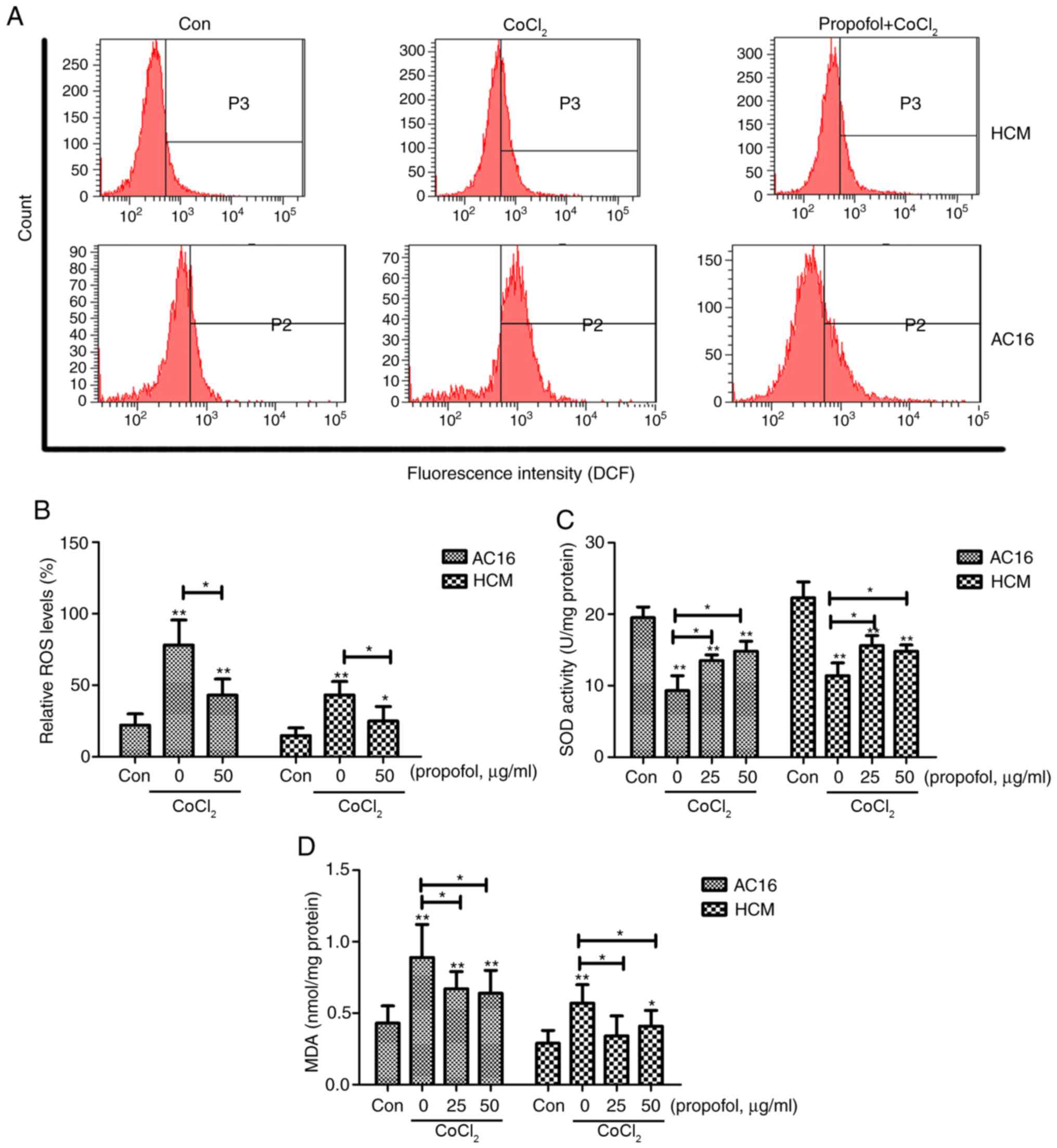
Propofol reduces
CoCl2-induced oxidative stress in AC16 and HCM
cells
Mitochondria are considered the main source of ROS
in cells (24). Therefore, the
present study investigated the mechanism underlying the
propofol-induced mitochondrial protection from oxidative stress.
The production of ROS was significantly increased in the
CoCl2 group compared with the control group (P<0.01),
and this was shown to be significantly decreased following propofol
pretreatment (Fig. 4A and B).
Intracellular MDA and supernatant SOD expression levels were also
investigated. The present results suggested that the production of
MDA was significantly increased in the CoCl2 group
compared with the control group, whereas the SOD levels
significantly decreased compared with the controls (P<0.01;
Fig. 4C and D). These effects of
CoCl2 significantly decreased following the application
of 25 and 50 µg/ml propofol for 2 h prior to CoCl2
treatment (P<0.05).
Discussion
Due to its rapid onset of action and rapid recovery
profile, propofol is widely used for the initiation and maintenance
of general anesthesia, sedation of mechanically ventilated adults,
procedural sedation and other similar purposes (25). However, side effects are inevitable
and include irregular heart rate, low blood pressure, burning
sensation at the site of injection and suppression of breathing
(26). Combined application of
epidural anesthesia, such as ropivacaine and lidocaine, may reduce
the propofol doses required for the induction and maintenance of
anesthesia, and further reduce its side effects in clinical
practice (27,28). The present study investigated the
effects of propofol and/or ropivacaine on cardiomyocytes in
vitro under normoxic or CoCl2-induced hypoxic
conditions.
In view of the higher detection sensitivity than
other tetrazolium salts such as an MTT assay, CCK-8 is widely used
for determination of cell viability in cell proliferation and
cytotoxicity assays (29). In the
present study, although absorbance values were different in control
groups of different cells, which may have resulted from different
incubation times, cell viability in all assays was accurately
assessed by CCK-8.
CoCl2 has been used for mimicking
pathophysiological hypoxia/ischemic conditions in vitro,
including ROS production, by activating the hypoxic signaling
pathway (23,30). The present results suggested that
CoCl2 decreased the viability of AC16 and HCM cells in a
dose and timedependent manner. To mimic a moderate hypoxic
environment, 500 µm CoCl2 treatment for 12 h was
selected for further experiments. The present results suggested
that this treatment induced cell apoptosis and ROS and MDA
production, decreased SOD production and disrupted the integrity of
the mitochondrial membrane leading to a reduction of Δψm. The
present results suggested that CoCl2 treatment may
induce the continuous flux of superoxide anions and hydrogen
peroxide, inducing oxidative stress in the cells, thus reducing the
activity of SOD. Therefore, CoCl2-induced cytotoxicity
was suggested to be ROS-dependent.
Propofol was previously reported to protect cells
against oxidative stress induced by hydrogen peroxide (31,32),
oxygen glucose deprivation (33) and
endotoxemia (34), and to inhibit
lipid peroxidation in various experimental cell models (35). The present results suggested that
propofol significantly increased cell viability under normal
culture conditions in a concentration-dependent manner, and the
protective effects of propofol pretreatment against
CoCl2 hypoxiainduced injury were greatest at a
concentration of 50 µg/ml. The present results indicated that
propofol pretreatment decreased cell apoptosis, prevented
impairment of mitochondrial membrane integrity, attenuated the
release of ROS and MDA and reversed the CoCl2-induced
SOD decrease. The present results suggested that propofol may exert
a strong protective effect against oxidative stress-induced injury
in cardiomyocytes.
The effects of propofol differ in various cell types
due to the activation or inhibition of different signaling pathways
(36). However, since ROS-dependent
intrinsic apoptosis is generally mediated by MAPK (37), the present study examined the
activation of the NF-κB and MAPK/p38/ERK/JNK signaling pathways,
which have been reported to be crucial for CoCl2-induced
apoptosis of BV2 (18) and HK2 cells
(38). Following activation of the
MAPK signaling cascade ERK plays an anti-apoptotic role, while JNK
and p38 exert pro-apoptotic effects during apoptosis. Moreover, the
activation of the JNK and p38 signaling pathways was significantly
inhibited following exposure to propofol. However, propofol was not
shown to affect the phosphorylation of p65, p38, ERK and JNK in
AC16 and HCM cells. The effect of CoCl2 and propofol
treatment was similar between the two cell lines but the magnitude
was different, which may be caused by cellular heterogeneity.
Therefore, further mechanistic studies are required to fully
elucidate the effects of propofol on human cardiomyocytes in order
to improve the efficacy and decrease the side effects of the
application of this anesthetic during cardiac surgeries.
In conclusion, the present results suggested that
pretreatment with propofol may protect human cardiac cells from
chemical hypoxiainduced injury via the regulation of the JNK
signaling pathways. The present results indicated that propofol may
be a promising cardioprotective against a variety of oxidative
stress injuries in the heart and may be a suitable drug for the
maintenance of anesthesia, especially during cardiac surgery.
However, it is not fully understood whether other pathways are
involved in these effects; therefore, larger cohort studies are
required.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Nanjing Science
and Technology Commission (grant no. 20150325).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH participated in the design of experiments,
carried out the molecular analysis of cells, interpretation and
analysis of data, and helped to draft the manuscript. QZ and YZ
were involved in drafting the manuscript and participated in all
experiments. YQ has been involved in all aspects of the study,
including experimental design, analysis and interpretation of data
and writing the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent to participate
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SOD
|
superoxide dismutase
|
|
ROS
|
reactive oxygen species
|
|
MDA
|
malondialdehyde
|
|
CCK-8
|
Cell Counting Kit-8
|
|
Δψm
|
mitochondrial membrane potential
|
|
PI
|
propidium iodide
|
|
MAPK
|
mitogen-activated protein kinase
|
References
|
1
|
Hoegberg LC, Bania TC, Lavergne V, Bailey
B, Turgeon AF, Thomas SH, Morris M, Miller-Nesbitt A, Mégarbane B,
Magder S, et al: Systematic review of the effect of intravenous
lipid emulsion therapy for local anesthetic toxicity. Clin Toxicol
(Phila). 54:167–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mather LE: The acute toxicity of local
anesthetics. Expert Opin Drug Metab Toxicol. 6:1313–1332. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Butterworth JF IV: Models and mechanisms
of local anesthetic cardiac toxicity: A review. Reg Anesth Pain
Med. 35:167–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weinberg G: Lipid rescue resuscitation
from local anaesthetic cardiac toxicity. Toxicol Rev. 25:139–145.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gray LD and Morris C: The principles and
conduct of anaesthesia for emergency surgery. Anaesthesia. 68
(Suppl):S14–S29. 2013. View Article : Google Scholar
|
|
6
|
Abubaih A and Weissman C: Anesthesia for
patients with concomitant sepsis and cardiac dysfunction.
Anesthesiol Clin. 34:761–774. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gottschalk A and Poepping DM: Epidural
analgesia in combination with general anesthesia. Anasthesiol
Intensivmed Notfallmed Schmerzther. 50:484–493. 2015.PubMed/NCBI
|
|
8
|
Hadimioglu N, Ulugol H, Akbas H,
Coskunfirat N, Ertug Z and Dinckan A: Combination of epidural
anesthesia and general anesthesia attenuates stress response to
renal transplantation surgery. Transplant Proc. 44:2949–2954. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Copeland SE, Ladd LA, Gu XQ and Mather LE:
The effects of general anesthesia on the central nervous and
cardiovascular system toxicity of local anesthetics. Anesth Analg.
106:1429–1439. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stewart J, Kellett N and Castro D: The
central nervous system and cardiovascular effects of
levobupivacaine and ropivacaine in healthy volunteers. Anesth
Analg. 97:412–416. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heavner JE, Dryden CF Jr, Sanghani V,
Huemer G, Bessire A and Badgwell JM: Severe hypoxia enhances
central nervous system and cardiovascular toxicity of bupivacaine
in lightly anesthetized pigs. Anesthesiology. 77:142–147. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rosen MA, Thigpen JW, Shnider SM, Foutz
SE, Levinson G and Koike M: Bupivacaine-induced cardiotoxicity in
hypoxic and acidotic sheep. Anesth Analg. 64:1089–1096. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Chen Z, Feng N, Tang, Zhao X, Liu
C, Xu H and Zhang M: Protective effect of propofol preconditioning
on ischemia-reperfusion injury in human hepatocyte. J Thorac.
9:702–710. 2017. View Article : Google Scholar
|
|
14
|
Li Y, Zhong D, Lei L, Jia Y, Zhou H and
Yang B: Propofol prevents renal ischemia-reperfusion injury via
inhibiting the oxidative stress pathways. Cell Physiol Biochem.
37:14–26. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsai YC, Huang CC, Chu LM and Liu YC:
Differential influence of propofol on different cell types in terms
of the expression of various oxidative stress-related enzymes in an
experimental endotoxemia model. Acta Anaesthesiol Taiwan.
50:159–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang B, Luo T, Chen D and Ansley DM:
Propofol reduces apoptosis and up-regulates endothelial nitric
oxide synthase protein expression in hydrogen peroxide-stimulated
human umbilical vein endothelial cells. Anesth Analg.
105:1027–1033. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang J, Xia Y, Xu Z and Deng X: Propofol
suppressed hypoxia/reoxygenation-induced apoptosis in hbvsmc by
regulation of the expression of bcl-2, bax, caspase3, kir6.1, and
p-JNK. Oxid Med Cell Longev. 2016:15187382016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu Y, Gu Y, Ding X, Wang J, Chen J and
Miao C: Intracellular Ca2+ homeostasis and JAK1/STAT3 pathway are
involved in the protective effect ofpropofol on BV2 microglia
against hypoxia-induced inflammation and apoptosis. PLoS One.
12:e01780982017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ou W, Lv J, Zou X, Yao Y, Wu J, Yang J,
Wang Z and Ma Y: Propofol inhibits hepatocellular carcinoma growth
and invasion through the HMGA2-mediated Wnt/β-catenin pathway. Exp
Ther Med. 13:2501–2506. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang S, Liang S, Zhao X, He Y and Qi Y:
Propofol inhibits cell proliferation and invasion in rheumatoid
arthritis fibroblast-like synoviocytes via the nuclear factor-κB
pathway. Am J Transl Res. 9:2429–2436. 2017.PubMed/NCBI
|
|
21
|
Li Q, Qi X and Jia W:
3,3′,5-triiodothyroxine inhibits apoptosis and oxidative stress by
the PKM2/PKM1 ratio during oxygen-glucose deprivation/reperfusion
AC16 and HCM-a cells: T3 inhibits apoptosis and oxidative stress by
PKM2/PKM1 ratio. Biochem Biophys Res Commun. 475:51–56. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mao SY, Meng XY, Xu ZW, Zhang WC, Jin XH,
Chen X, Zhou X, Li YM and Xu RC: The role of ZFP580, a novel zinc
finger protein, in TGF-mediated cytoprotection against chemical
hypoxia induced apoptosis in H9c2 cardiac myocytes. Mol Med Rep.
15:2154–2162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Solaini G, Baracca A, Lenaz G and Sgarbi
G: Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys
Acta. 1797:1171–1177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pascual-Ahuir A, Manzanares-Estreder S and
Proft M: Pro- and antioxidant functions of the
peroxisome-mitochondria connection and its impact on aging and
disease. Oxid Med Cell Longev. 2017:98608412017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feng AY, Kaye AD, Kaye RJ, Belani K and
Urman RD: Novel propofol derivatives and implications for
anesthesia practice. J Anaesthesiol Clin Pharmacol. 33:9–15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Keyl C, Schneider A, Dambacher M,
Wegenhorst U, Ingenlath M, Gruber M and Bernardi L: Dynamic
cardiocirculatory control during propofol anesthesia in
mechanically ventilatedpatients. Anesth Analg. 91:1188–1195. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiang Y and Li YH: Comparison of 1.5%
lidocaine and 0.5% ropivacaine epidural anesthesia combined with
propofolgeneral anesthesia guided by bispectral index. J Zhejiang
Univ Sci B. 8:428–434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Osaka Y, Inomata S, Tanaka E, Nakamura T,
Honda K, Miyabe M, Toyooka H and Tanaka M: Effect of propofol on
ropivacaine metabolism in human liver microsomes. J Anesth.
20:60–63. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ginouves M, Carme B, Couppie P and Prevot
G: Comparison of tetrazolium salt assays for evaluation of drug
activity against Leishmania spp. J Clin Microbiol.
52:2131–2138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jeon YJ, Kim HS, Song KS, Han HJ, Park SH,
Chang W and Lee MY: Protective effect of dieckol against chemical
hypoxia-induced cytotoxicity in primary cultured mouse hepatocytes.
Drug Chem Toxicol. 38:180–187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Romuk E, Szczurek W, Nowak P, Skowron M,
Prudel B, Hudziec E, Chwalińska E1 and Birkner E: Effects of
propofol on oxidative stress parameters in selected parts of the
brain in a rat model of parkinson disease. Postepy Hig Med Dosw
(Online). 70:1441–1450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen XH, Zhou X, Yang XY, Zhou ZB, Lu DH,
Tang Y, Ling ZM, Zhou LH and Feng X: Propofol protects against
H2O2-induced oxidative injury in differentiated pc12 cells via
inhibition of Ca(2+)-Dependent NADPH oxidase. Cell Mol Neurobiol.
36:541–551. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Z, Yang P and Qi Y: Role of
microRNA-134 in the neuroprotective effects of propofol against
oxygen-glucosedeprivation and related mechanisms. Int J Clin Exp
Med. 8:20617–206123. 2015.PubMed/NCBI
|
|
34
|
Gokcinar D, Ergin V, Cumaoglu A, Menevse A
and Aricioglu A: Effects of ketamine, propofol, and ketofol on
proinflammatory cytokines and markers of oxidative stress in a rat
model of endotoxemia-induced acute lung injury. Acta Biochim Pol.
60:451–456. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Eriksson O, Pollesello P and Saris NE:
Inhibition of lipid peroxidation in isolated rat liver mitochondria
by the general anaesthetic propofol. Biochem Pharmacol. 44:391–393.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang S, Liu Y, Huang L, Zhang F and Kang
R: Effects of propofol on cancer development and chemotherapy:
Potential mechanisms. Eur J Pharmacol. 831:46–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang Z, Song F, Li Y, Xue D, Zhao N,
Zhang J, Deng G, Li M, Liu X and Wang Y: Capsular polysaccharide of
mycoplasma ovipneumoniae induces sheep airway epithelial cell
apoptosis via Ros-dependent JNK/P38 MAPK pathways. Oxid Med Cell
Longev. 2017:61758412017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xiong T, Dong W, Fu H, Li Q, Deng C, Lei X
and Guo L: Involvement of the nuclear factor-κB pathway in the
adhesion of neutrophils to renal tubular cells after injury induced
by neonatal postasphyxial serum. Mol Cell Biochem. 388:85–94. 2014.
View Article : Google Scholar : PubMed/NCBI
|