Introduction
Cardiac fibrosis is an important contributor to the
development of multiple cardiovascular diseases, such as myocardial
infarction and hypertension (1). It
is primarily characterized by an excessive accumulation of
extracellular matrix, including collagen I and collagen III
(2–5). Since myofibroblasts exhibit potent
proliferative and secretory activities, the transformation of
cardiac fibroblasts (CFs) to myofibroblasts is regarded as a
crucial step in the development and progression of cardiac fibrosis
(6–8). Despite significant advancements in the
treatment of cardiac fibrosis, the underlying molecular mechanisms
of cardiac fibroblast-to-myofibroblast transformation remain
undiscovered.
Accumulating evidence has suggested that miRNAs are
highly associated with the pathogenesis of various cardiovascular
diseases, including cardiac fibrosis (9,10).
Several miRNAs, such as miRNA (miR)-130, miR-328, miR-30a and
miR-29a, have been reported to function as potential antifibrotic
targets in cardiac fibrosis (11–14).
Notably, an emerging role has been proposed for miR-26 family
members in cardiovascular disease. For example, Wei et al
(15) reported that miR-26a
expression was markedly decreased in angiotensin II-induced
neonatal CFs, and overexpression of miR-26a suppressed the
expression of the connective tissue growth factor and collagen I.
Although miR-26b has been reported to inhibit the development of
hypertrophy (16), the exact role of
miR-26b in the pathogenesis of cardiac fibrosis is still
unclear.
Nuclear factor erythroid 2-related factor 2 (Nrf2)
is a key regulator of the antioxidative response (17,18).
Under normal conditions, Nrf2 remains in an inactive state and is
constitutively suppressed by Kelch-like ECH-associated protein 1
(Keap1). In response to stimulation, Nrf2 dissociates from Keap1,
translocates to the nucleus and activates the antioxidant response
element. Increasing evidence has demonstrated that oxidative stress
induces fibroblast activation and increases collagen production in
various organs, leading to pathological conditions such as liver,
pulmonary and cardiac fibrosis (19–21).
This evidence suggests that the Keap1/Nrf2 signaling pathway is a
potential candidate for antifibrotic therapy.
The present study demonstrated that miR-26b could
protect heart cells from isoproterenol (ISO)-induced cardiac
fibrosis via the Keap1/Nrf2 signaling pathway. The findings may
provide a novel understanding of the occurrence of cardiac fibrosis
and could be used for the development of a promising therapeutic
modality for the treatment of this disease.
Materials and methods
Animals and treatments
The experiments involving animals were approved by
the Animal Experimentation Ethics Committee of the People's
Hospital of Changshou (Chongqing, China). A total of 60 adult male
Sprague-Dawley rats (200–220 g) were purchased from the Laboratory
of the Animal Center of the Soochow University (Suzhou, China) and
randomly divided into the cardiac fibrosis model group (n=30) and
the saline group (n=30). The mice were maintained under conditions
of 50% relative humidity, a 12-h light/dark cycle and 22°C, and
received food and water ad libitum. To establish the cardiac
fibrosis group, ISO was injected subcutaneously for 10 days (5
mg/kg/day) (22–24). The Sprague-Dawley rats in the saline
group were given equal volumes of saline. The rats were sacrificed
by cervical dislocation after deep anesthesia with 2% isoflurane
(Baxter International, Inc.) and their hearts were isolated for the
subsequent experiments.
Cell culture and treatment
CFs were isolated from neonatal Sprague-Dawley rats.
A total of eight neonatal Sprague-Dawley rats (age, 1–3 days;
weight, 5–7 g) were obtained from the Laboratory of the Animal
Center of the Soochow University. The ventricles of the neonatal
rats were minced and digested with a mixed enzyme solution
(trypsin: Collagenase I; ratio, 2:1). Subsequently, the cells were
plated with DMEM (Sigma-Aldrich; Merck KGaA) containing 10% fetal
bovine serum (FBS; Thermo Fisher Scientific, Inc.) for 60 min at
37°C in humidified air with 5% CO2 until the CFs had
adhered to the wall of plate. The culture plates were washed twice
with PBS to detach the weakly attached and non-adherent cells and
then pure CFs were obtained. The extracted cells were cultured in
90% DMEM (HyClone; GE Healthcare Life Sciences) supplemented with
10% FBS (Thermo Fisher Scientific, Inc.) in a humidified incubator
at 37°C with 5% CO2. Second- or third-generation CFs
were used in these experiments.
Cell transfection
miR-26b mimics (5′-UUCAAGUAAUUCAGGAUAGGU-3′),
miR-26b inhibitor (5′-ACCUAUCCUGAAUUACUUGAA-3′) and their
respective negative controls miR-NC (5′-UUCUCCGAACGUGUCACGUUU-3′)
and inh-miR-NC (5′-ACGUGACACGUUCGGAGAAUU-3′) were synthesized by
Shanghai GenePharma Co., Ltd. Keap1 cDNA (GenePharma Co., Ltd.) was
cloned into pCDNA3.1 vectors (Invitrogen; Thermo Fisher Scientific,
Inc.) by Shanghai GenePharma Co., Ltd. in order to construct
corresponding Keap1 expression vector. The sequence of small
interfering RNA (siRNA) against Keap1 (siKeap1,
5′-GCGCCAAUGUUGACACGGA-3′) and the sequence of the scrambled siRNA
(siNC, 5′-GUACCUGACUAGUCGCAGAUU-3′) were synthesized by Shanghai
GenePharma Co., Ltd. CFs (5×104/ml) were transfected
with miR-26b mimics (20 nM) or miR-NC (20 nM), the miR-26b
inhibitor (20 nM) or inh-miR-NC (20 nM) and 20 nM vectors or 50 nM
siRNA. The transfection was conducted using
Lipofectamine® 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). At 48-h post-transfection, the
subsequent experiments were conducted.
MTT assay
The cell viability of CFs was measured by MTT assay.
CFs were seeded in a 96-well plate with 5×105
cells/well. After transfection, the cells were treated with ISO for
24 h. Subsequently, MTT solution was added into each well and the
cells were incubated for 3 h at 37°C. A total of 200 µl DMSO was
added to solubilize the MTT crystals, and the optical density was
measured at 570 nm.
Reverse transcription-quantitative
(RT-q)PCR analysis
Total RNA was extracted from cardiac tissues and
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The RNA was reverse transcribed into cDNA using
the Takara RT kit (Takara Biotechnology Co., Ltd.) at 37°C for 15
min. The expression levels of miR-26b, collagen I, α-smooth muscle
actin (α-SMA), Keap1 and Nrf2 were determined using the SYBR Green
PCR kit (Takara Biotechnology Co., Ltd.). GAPDH and U6 were used as
controls. The following amplification conditions were used:
Pre-denaturation at 95°C for 15 sec, denaturation at 94°C for 30
sec, annealing at 60°C for 20 sec, and extension at 72°C for 40 sec
for 40 cycles. Relative gene expression was calculated using the
2−ΔΔCq method (25). The
sequences of the primers were as follows: miR-26b forward,
5′-CCCAGTTCAAGTAATTCAGG-3′; and reverse, 5′-TTTGGCACTAGCACATT-3′;
collagen I forward, 5′-CAGAGCACGATGTCCTGAGA-3′; and reverse,
5′-GCAAATGTGAGCTTCTGTGC-3′; α-SMA forward,
5′-GGAGTGATGGTTGGAATGG-3′; and reverse, 5′-ATGATGCCGTGTTCTATCG-3′;
Keap1 forward, 5′-GGACGGCAACACTGATTC-3′; and reverse,
5′-TCGTCTCGATCTGGCTCATA-3′; Nrf2 forward,
5′-CACATCCAGACAGACACCAGT-3′; and reverse,
5′-CTACAAATGGGAATGTCTCTGC-3′; GAPDH forward,
5′-CAAGCTCATTTCCTGGTATGAC-3′; and reverse,
5′-CAGTGAGGGTCTCTCTCTTCCT-3′; U6 forward,
5′-CTCGCTTCGGCAGCACATATACTA-3′; and reverse,
5′-ACGAATTTGCGTGTCATCCTTGCG-3′.
Bioinformatics prediction and
luciferase reporter assay
The downstream target genes of miR-26b were
predicted using the TargetScan database (http://www.targetscan.org). Keap1 was identified as a
potential downstream target of miR-26b. 293T cells were obtained
from the American Type Culture Collection and cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
and 1% penicillin-streptomycin (Thermo Fisher Scientific, Inc.) at
37°C in a humidified atmosphere with 5% CO2. Luciferase
reporter assays were used to investigate the regulatory
relationship between miR-26b and Keap1. Site-directed mutagenesis
was used to create the mutant 3′-untranslated region (UTR) sequence
of Keap1. The wild-type and mutant-type sequences of Keap1 were
cloned into the pGL-luciferase plasmid (Promega, Corp.), while the
NC and miR-26b mimics sequences were co-transfected into 293T cells
using Lipofectamine® 2000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). After 48 h incubation
at 37°C, firefly and Renilla luciferase activities were
evaluated using the Dual-Luciferase Reporter Analysis kit (Promega
Corporation). Normalized relative light units represent firefly
luciferase activity/Renilla luciferase activity.
Western blot analysis
Cultured cells were lysed in RIPA buffer (Beyotime
Institute of Biotechnology) for total protein extraction. Protein
concentration was determined using a BCA assay (Beyotime Institute
of Biotechnology). A total of 10 µg protein/lane were separated by
SDS-PAGE (10% gel) and subsequently transferred to nitrocellulose
membranes. Following blocking with 5% bovine serum albumin (Beijing
Solarbio Science and Technology Co., Ltd.) for 1 h at room
temperature, the proteins were incubated with primary antibodies,
namely anti-collagen I (cat. no. ab34710), anti-α-SMA (cat. no.
ab5694) and anti-β-actin (cat. no. ab8227) (all 1:1,000; all from
Abcam), at 4°C overnight. Following washing, the membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies (goat anti-mouse IgG; cat. no. ab205719; and goat
anti-rabbit IgG; cat. no. ab205718) (both 1:1,000; both from Abcam)
for 2 h at room temperature. The proteins were detected using an
enhanced chemiluminescence detection system (ProteinSimple) and
analyzed using Image-Pro® Plus software (version 6.0;
Media Cybernetics, Inc.).
Statistical analysis
Statistical analyses were performed using SPSS
software (version 18.0; SPSS, Inc.). All data are presented as the
mean ± standard deviation of at least three independent
experiments. The group differences were determined by either
Student's t-test or one-way ANOVA. Multiple comparisons between the
groups was performed using the Student-Newman-Keuls method.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Upregulation of collagen I and α-SMA
in ISO-treated cardiac tissues
ISO has been reported to increase the expression of
pro-inflammatory cytokines [interleukin (IL)-1, IL-6 and IL-17]
(26). The release of cytokines
contributes to cardiac fibrosis via upregulation of matrix
metalloproteinase expression in CFs (26). In the present study, Sprague-Dawley
rats were treated with ISO to induce cardiac fibrosis.
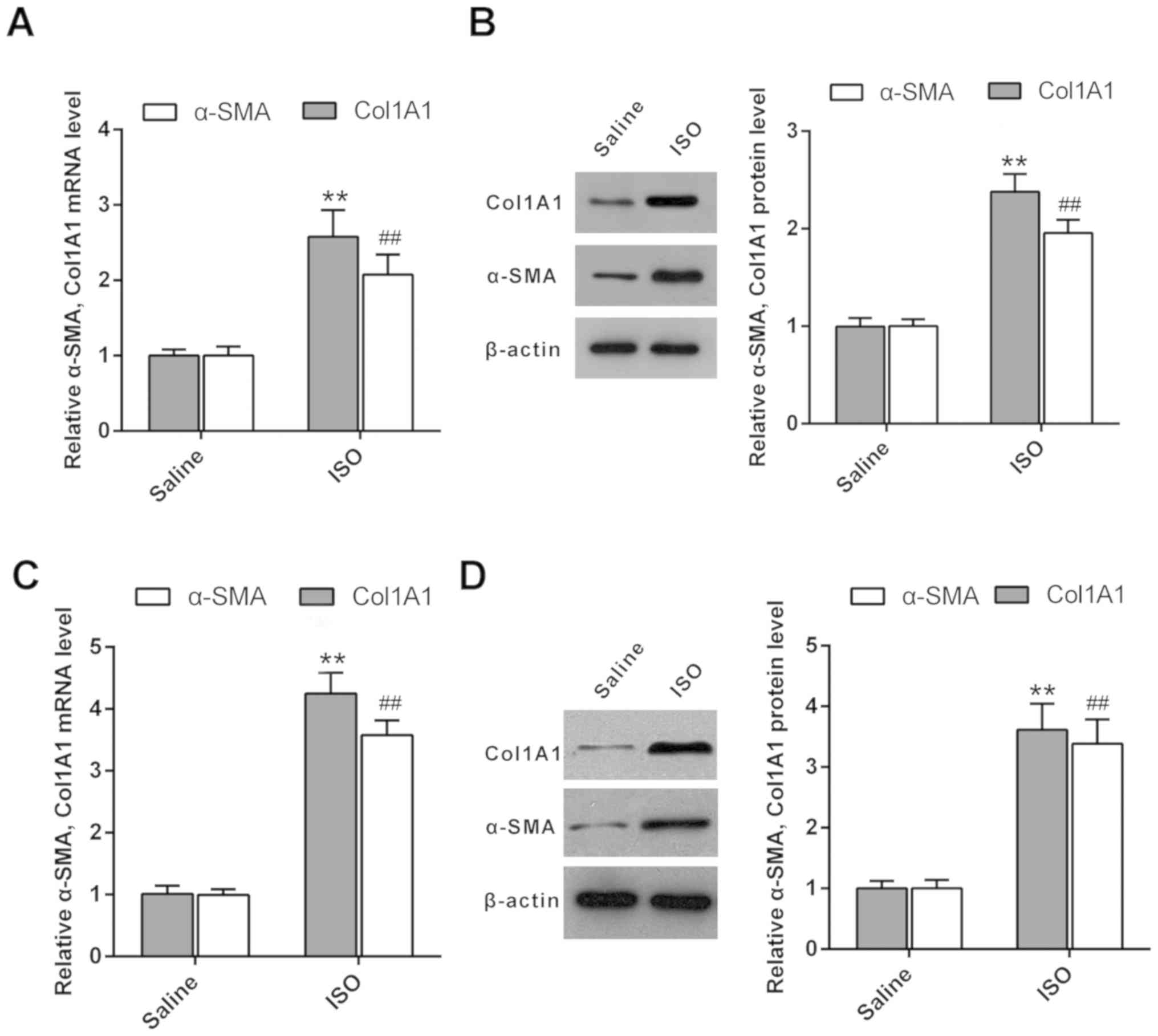
Subsequently, RT-qPCR was used to evaluate the mRNA expression
levels of collagen I and α-SMA in cardiac tissues. The mRNA
expression levels of collagen I and α-SMA were increased in
ISO-treated rats compared with those of the untreated rats
(Fig. 1A). Furthermore, western blot
analysis indicated that the protein expression levels of collagen I
and α-SMA were increased in ISO-treated rat cardiac tissues
compared with those noted in the saline group (Fig. 1B). In addition, ISO was used to treat
CFs to induce cardiac fibrosis. RT-qPCR and western blot analysis
demonstrated that the expression levels of collagen I and α-SMA
were upregulated in ISO-treated CFs compared with those of the
control groups (Fig. 1C and D). In
summary, the results indicated that collagen I and α-SMA were
overexpressed in the ISO-treated cardiac fibrosis model.
Expression of miR-26b and Keap1 in
ISO-treated cardiac tissue and CFs
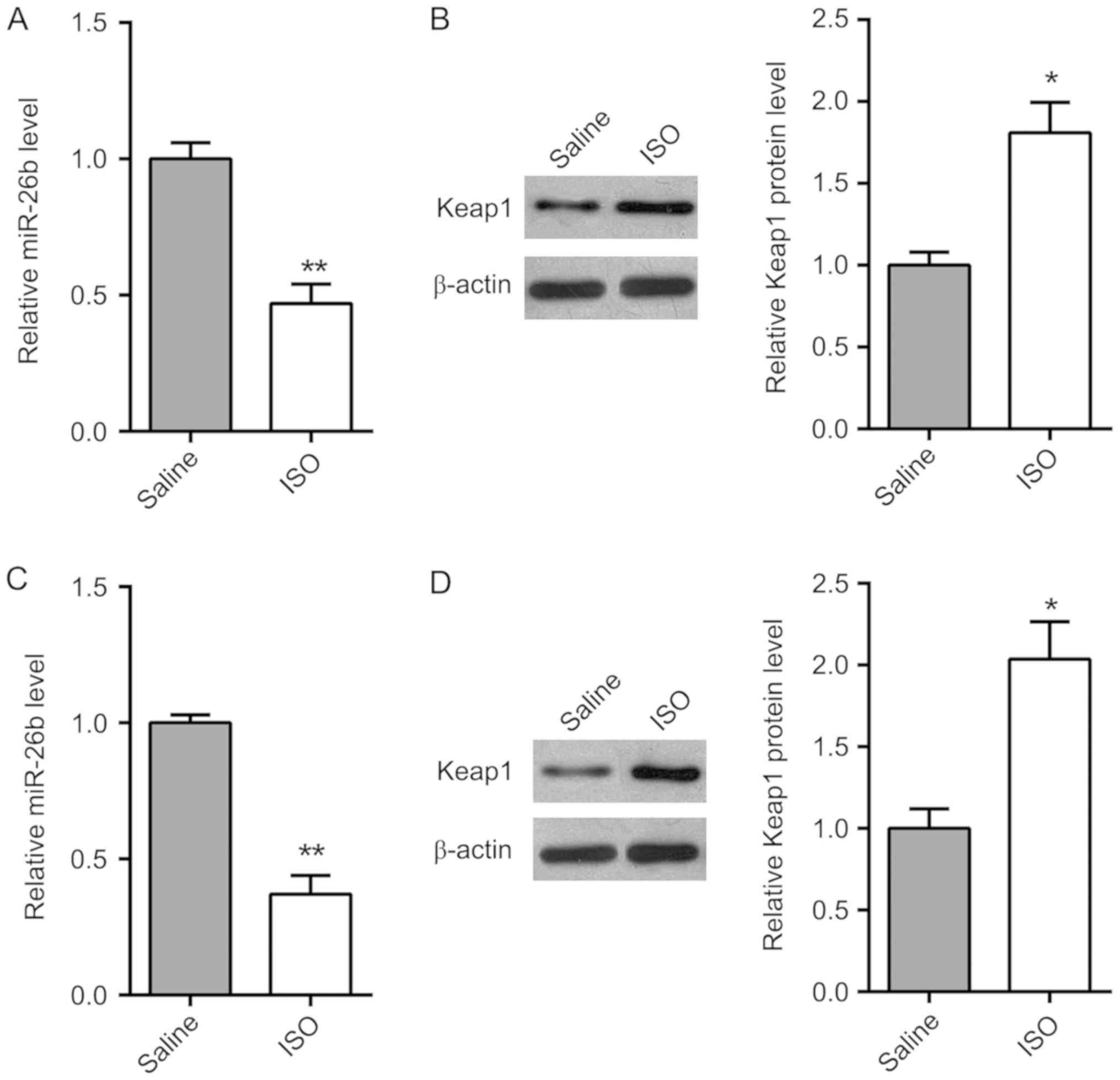
The expression of miR-26b and Keap1 in ISO-treated
cardiac tissues and normal tissues was determined by RT-qPCR. As
shown in Fig. 2A and B, the
expression of miR-26b was downregulated in ISO-treated cardiac
tissues compared with saline-treated group, while the expression of
Keap1 was upregulated in ISO-treated cardiac tissues. Furthermore,
the expression of miR-26b and Keap1 was analyzed in vitro
(Fig. 2C and D). Consistent with the
results in vivo, the expression level of miR-26b was also
decreased and the expression level of Keap1 was increased in
ISO-treated CFs compared with the saline group.
miR-26b suppresses the cell viability
of CFs
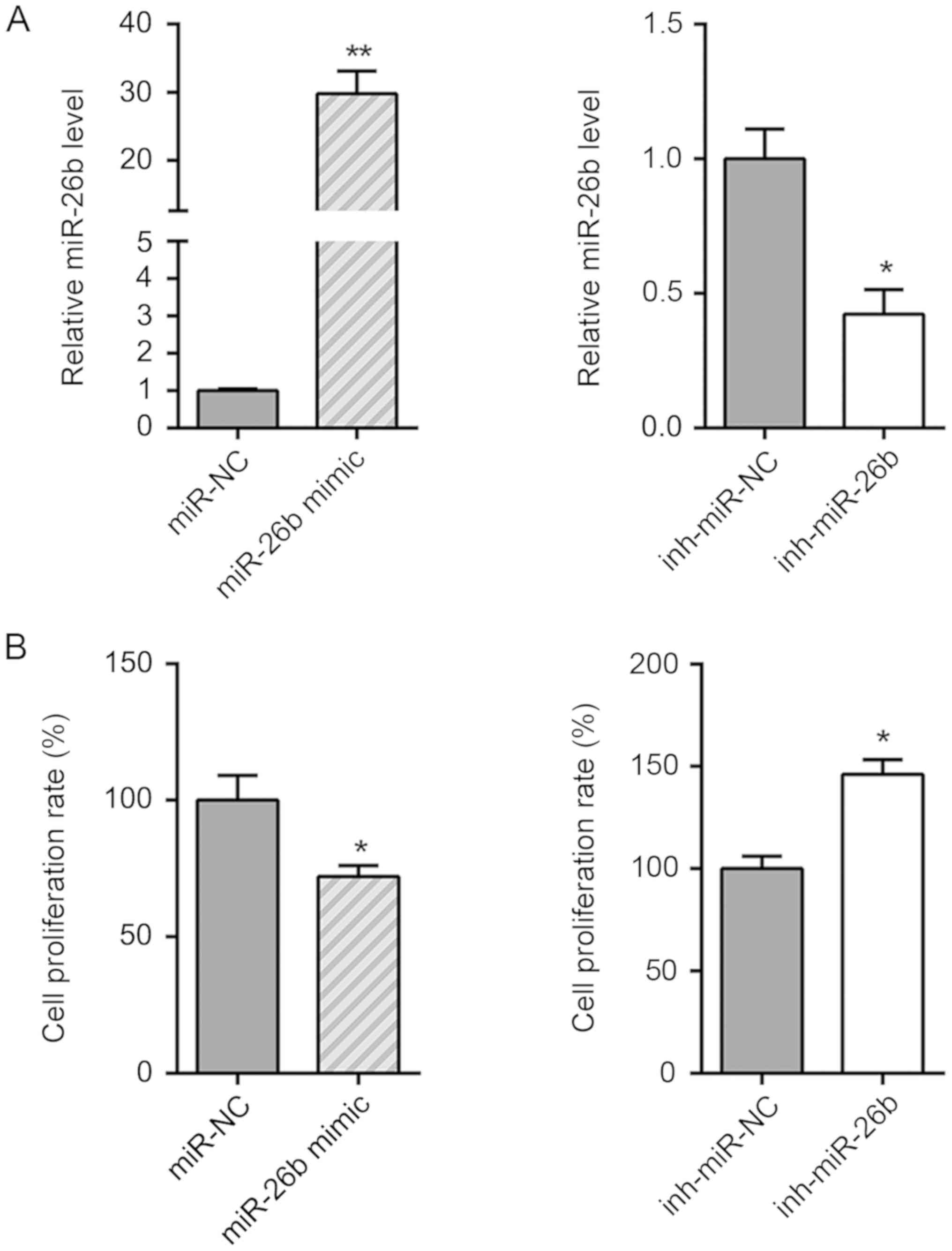
To investigate the precise role of miR-26b in
ISO-induced cardiac fibrosis, ISO-treated CFs were transfected with
miR-26b mimics to overexpress miR-26b, and with the miR-26b
inhibitor to decrease miR-26b expression. The transfection
efficiency was confirmed by RT-qPCR (Fig. 3A). miR-26b overexpression
significantly inhibited the cell viability of ISO-treated CFs,
while miR-26b inhibition enhanced the growth of ISO-treated CFs
(Fig. 3B). Furthermore, western blot
analysis indicated that miR-26b overexpression decreased the
protein levels of collagen I and α-SMA compared with those of the
miR-NC group. In contrast to these findings, miR-26b inhibition
caused an increase in the expression levels of collagen I and α-SMA
proteins (Fig. 3C). Taken
collectively, the data indicated that miR-26b inhibited the
transformation of CFs into myofibroblasts.
Keap1 is a target of miR-26b
The analysis using the bioinformatics prediction
database TargetScan demonstrated that Keap1 was a potential target
of miR-26b (Fig. 4A). To determine
whether miR-26b could directly interact with Keap1, a dual
luciferase reporter assay was performed. The results indicated that
miR-26b mimics significantly reduced the luciferase activity of
wild-type Keap1, whereas no effect was noted in the cells with the
mutant 3′-UTR of Keap1 (Fig. 4B).
Moreover, the data indicated that miR-26b mimics significantly
reduced Keap1 expression, while the miR-26b inhibitor increased the
expression levels of Keap1 (Fig. 4C and
D). These results demonstrated that miR-26b could directly
target Keap1 to inhibit its expression.
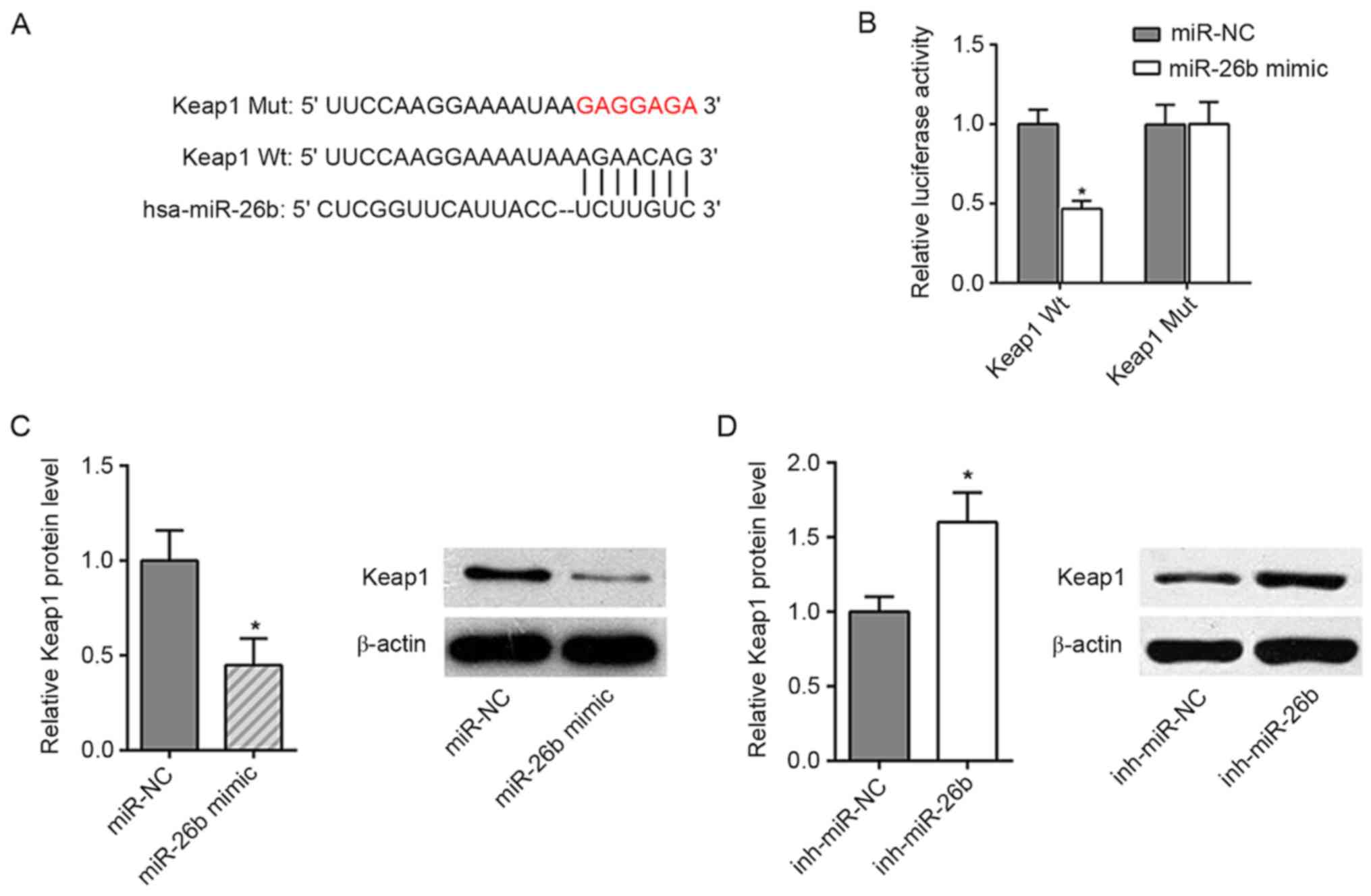 | Figure 4.Keap1 is one of the targets of
miR-26b. (A) Bioinformatics prediction of the binding site for
miR-26b in Keap1. (B) The luciferase reporter assay confirmed that
miR-26b was able to bind to Wt and not Mut Keap1 in 293T cells. (C)
RT-qPCR analysis showed the expression levels of miR-26b and Keap1
in ISO-treated CFs (NC and miR-26b mimics). (D) RT-qPCR analysis
showed the expression levels of miR-26b and Keap1 in ISO-treated
CFs (NC and miR-26b inhibitor). *P<0.05 vs. respective control.
Mut, mutant; Wt, wild-type; NC, negative control; miR, microRNA;
Keap1, Kelch-like ECH-associated protein 1; inh, inhibitor;
RT-qPCR, reverse transcription-quantitative PCR; CF, cardiac
fibroblast; ISO, isoproterenol; NC, negative control. |
miR-26b-mediated inhibitory effects on
ISO-induced cardiac fibrosis are abrogated by Keap1
To examine whether the effect of miR-26b on
ISO-induced cardiac fibrosis is regulated by Keap1, two cell groups
of ISO-treated CFs were prepared, one that was transfected with
miR-NC, miR-26b mimics, miR-26b+pcDNA or miR-26b mimics+Keap1, and
another that was transfected with inh-miR-NC, miR-26b inhibitor,
inh-miR-26b+siNC or inh-miR-26b+siKeap1 (Fig. 5A and B). The data indicated that
Keap1 overexpression significantly reduced the inhibitory effect of
miR-26b on cell viability (Fig. 5C).
Conversely, Keap1 silencing attenuated the cell growth of CFs
transfected with the miR-26b inhibitor (Fig. 5D). Moreover, western blot analysis
demonstrated that miR-26b mimics decreased the protein levels of
collagen I and α-SMA in ISO-treated CFs, indicating that miR-26b
inhibited ISO-induced cardiac fibrosis; these effects were
abrogated by Keap1 overexpression (Fig.
5E). The opposite effect was noted in the ISO-treated CFs
co-transfected with inh-miR-26b and siKeap1 (Fig. 5F). In summary, the results
demonstrated that Keap1 restoration or silencing abrogated the
effects caused by miR-26b mimics or the miR-26b inhibitor in
ISO-treated CFs.
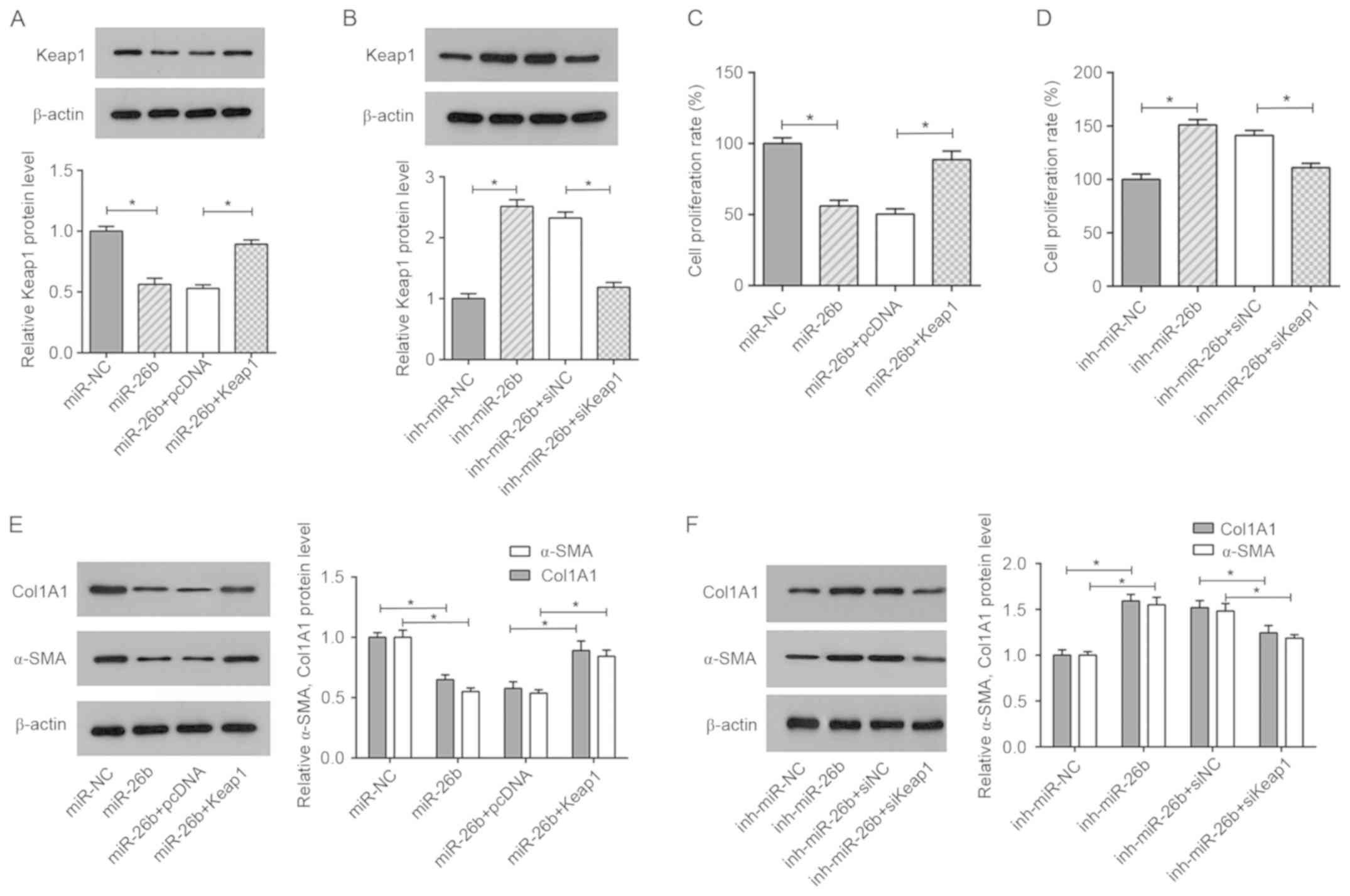 | Figure 5.Restoration of Keap1 abolishes the
inhibitory effect of miR-26b on ISO-treated CFs. (A) Western blot
analysis showing the relative expression levels of Keap1 in
ISO-treated CFs transfected with miR-NC, miR-26b, miR-26b+pcDNA and
miR-26b+Keap1. (B) Western blot analysis showing the relative
expression levels of Keap1 in ISO-treated CFs transfected with
inh-miR-NC, inh-miR-26b, inh-miR-26b+siNC and inh-miR-26b+siKeap1.
(C) Cell viability of ISO-treated CFs transfected with miR-NC,
miR-26b, miR-26b+pcDNA and miR-26b+Keap1, as determined by MTT
assay. (D) Cell viability of ISO-treated CFs transfected with
inh-miR-NC, inh-miR-26b, inh-miR-26b+siNC and inh-miR-26b+siKeap1
as determined by MTT assay. (E) The protein levels of collagen I
and α-SMA were detected in ISO-treated CFs transfected with miR-NC,
miR-26b, miR-26b+pcDNA and miR-26b+Keap1 by western blotting. (F)
The protein levels of collagen I and α-SMA were detected by western
blotting in ISO-treated CFs transfected with inh-miR-NC,
inh-miR-26b, inh-miR-26b+siNC and inh-miR-26b+siKeap1. *P<0.05.
Keap1, Kelch-like ECH-associated protein 1; NC, negative control;
α-SMA, α-smooth muscle actin inh, inhibitor; ISO, isoproterenol;
miR, microRNA; CF, cardiac fibroblast; si, small interfering;
Col1A1, collagen I. |
miR-26b inhibits ISO-induced cardiac
fibrosis via the Keap1/Nrf2 signaling pathway
Previous studies have shown that Nrf2 plays an
important role in myofibroblast dedifferentiation and its activity
is under the control of Keap1 (27).
Therefore, it was assumed that miR-26b and Keap1 may exert their
roles in cardiac fibrosis via activation of Nrf2. The data
demonstrated that the upregulation of miR-26b increased the
expression levels of Nrf2 in ISO-treated CFs, while this effect was
abolished by Keap1 overexpression (Fig.
6A). In contrast to these findings, treatment of the cells with
the miR-26b inhibitor caused a downregulation in the expression
levels of Nrf2, and Keap1 silencing alleviated the inhibitory
effect of miR-26b inhibitor on the expression of Nrf2 (Fig. 6B). In summary, these data
demonstrated that miR-26b caused an upregulation of Nrf2 expression
levels by targeting Keap1 in ISO-treated CFs.
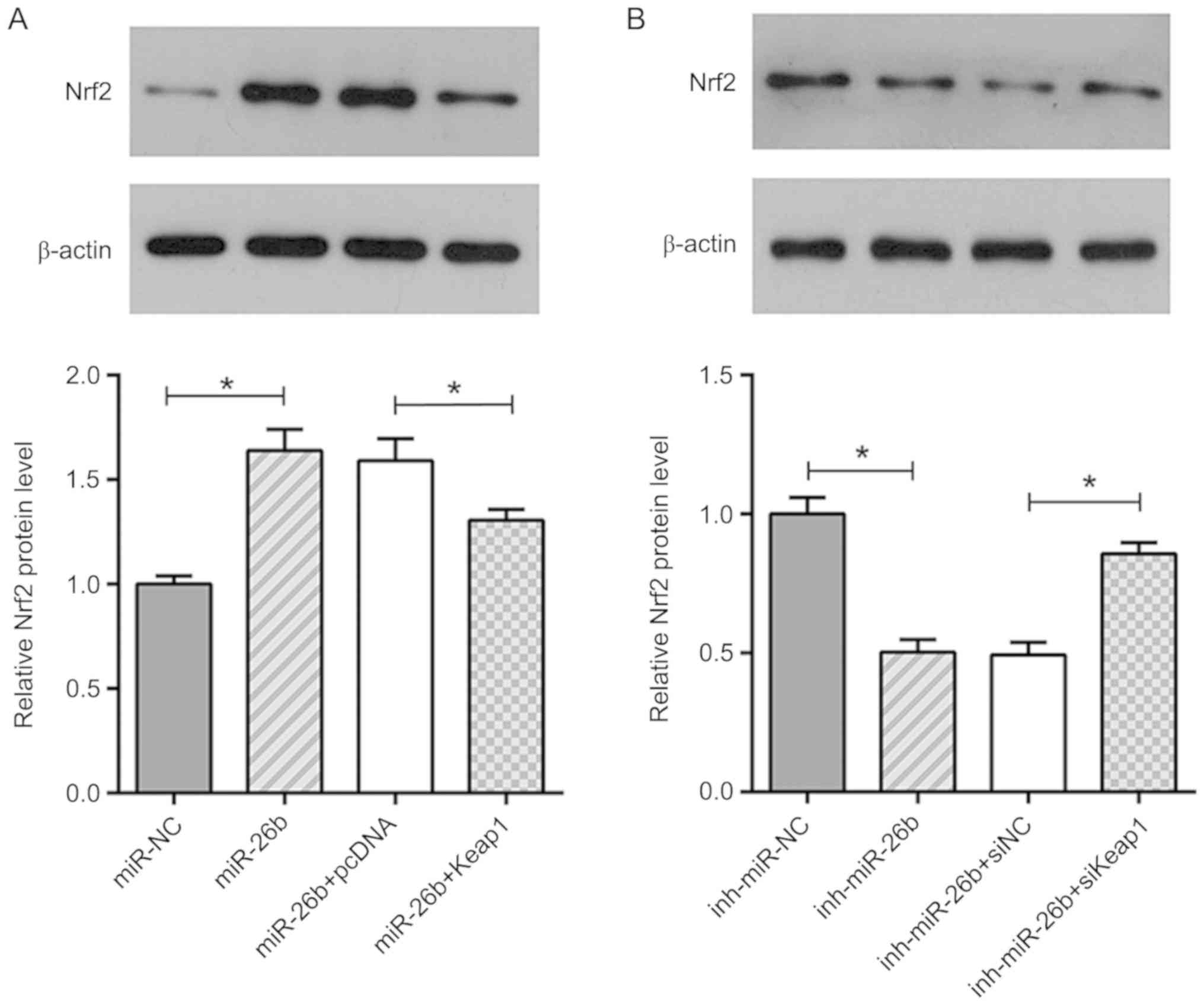 | Figure 6.miR-26b targets Keap1 to activate
Nrf2. (A) Western blot analysis showing the relative expression
levels of Nrf2 in ISO-treated CFs transfected with miR-NC, miR-26b,
miR-26b+pcDNA and miR-26b+Keap1. (B) Western blot analysis showing
the relative expression levels of Nrf2 in ISO-treated CFs
transfected with inh-miR-NC, inh-miR-26b, inh-miR-26b+siNC and
inh-miR-26b+siKeap1. *P<0.05. miR, microRNA; Keap1, Kelch-like
ECH-associated protein 1; NC, negative control; Nrf2, nuclear
factor erythroid 2-related factor 2; inh, inhibitor; si, small
interfering; ISO, isoproterenol. |
Discussion
Abnormal activation and cell viability of CFs is a
pivotal step in the occurrence and development of cardiac fibrosis.
Therefore, identifying approaches that inhibit CF activation may be
an effective therapeutic strategy to prevent cardiac fibrosis. The
present study demonstrated that miR-26b suppressed the activation
and cell viability of ISO-treated CFs by upregulating Nrf2
expression and targeting Keap1.
It has been thoroughly documented that miRNAs are
involved in the development and progression of cardiac fibrosis.
For example, Zhou et al (28)
reported that miR-21 could promote cardiac
fibroblast-to-myofibroblast transition by downregulating the
expression of Jagged1. Moreover, several studies have indicated
that the miR-26 family plays a central role in the development of
cardiovascular disease by controlling critical signaling pathways
or downstream gene targets. For example, miR-26a inhibits
pathological and physiological angiogenesis by targeting bone
morphogenic protein/SMAD1 signaling in endothelial cells (29). Han et al (16) reported that miR-26b inhibited the
development of cardiac hypertrophy by inhibiting the expression of
GATA binding protein 4. However, the functional role of miR-26b,
which is a member of the miR-26 family, has not been extensively
studied. The present study demonstrated that miR-26b was
significantly downregulated in ISO-treated cardiac tissues and CFs.
Furthermore, we investigated the effects of miR-26b on ISO-treated
CFs. The results indicated that miR-26b mimics inhibited cell
viability and decreased the protein levels of collagen I and α-SMA,
while the miR-26b inhibitor increased the cell growth rate and the
expression levels of collagen I and the α-SMA proteins. The data
suggested that miR-26b exerts a suppressive role on ISO-induced
cardiac fibrosis.
The induction of oxidative stress contributes to the
occurrence and development of cardiac fibrosis. The
Nrf2-antioxidant response element signaling pathway acts as a
critical cellular defense mechanism that antagonizes oxidative
stress (30,31). Keap1 is an important regulator and
repressor of this signaling pathway (30,31). For
example, Civantos et al (32)
reported that sitagliptin ameliorated oxidative stress in diabetic
kidney rat tissues through downregulation of miR-200a, which
further regulated the Keap1/Nrf2 signaling pathway. Moreover, Yang
et al (33) reported that
Nrf2 could protect against hepatic stellate fibrosis by
functionally activating the transcription of antioxidant response
genes. In the present study, bioinformatics analysis and luciferase
reporter assays demonstrated that Keap1 could directly interact
with miR-26b, and that its overexpression abrogated the inhibitory
effect of miR-26b on ISO-treated CFs, as determined by increased
cell viability and expression of fibrosis-related indices in
ISO-treated CFs. Moreover, it was demonstrated that the targeting
of Keap1 by miR-26b caused an upregulation of Nrf2 expression.
Therefore, the data suggested that miR-26b could regulate the
Keap1/Nrf2 signaling pathway to inhibit ISO-induced cardiac
fibrosis.
Overall, the present study demonstrated for the
first time, to the best of our knowledge, that miR-26b could
attenuate ISO-induced cardiac fibrosis by activating Nrf2 and by
interacting with Keap1. These findings may provide additional
evidence for the development of a novel therapeutic strategy for
cardiac fibrosis and for understanding the pathogenesis of this
disease.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
SX and ZZ designed the present study. ZZ collected
the samples, and ZZ and JL performed all the experiments. SX and JL
analyzed the data and prepared the figures. SX drafted the initial
manuscript. ZZ reviewed and revised the initial manuscript. All
authors approved the final published version of this
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the People's Hospital of Changshou. The experiments
involving animals were approved by the Animal Experimentation
Ethics Committee of the People's Hospital of Changshou.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Molkentin JD, Bugg D, Ghearing N, Dorn LE,
Kim P, Sargent MA, Gunaje J, Otsu K and Davis J:
Fibroblast-specific genetic manipulation of p38 mitogen-activated
protein kinase in vivo reveals its central regulatory role in
fibrosis. Circulation. 136:549–561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Segura AM, Frazier OH and Buja LM:
Fibrosis and heart failure. Heart Fail Rev. 19:173–185. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Edgley AJ, Krum H and Kelly DJ: Targeting
fibrosis for the treatment of heart failure: A role for
transforming growth factor-β. Cardiovasc Ther. 30:e30–e40. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Espira L and Czubryt MP: Emerging concepts
in cardiac matrix biology. Can J Physiol Pharmacol. 87:996–1008.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan D, Takawale A, Lee J and Kassiri Z:
Cardiac fibroblasts, fibrosis and extracellular matrix remodeling
in heart disease. Fibrogenesis Tissue Repair. 5:152012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barnes JL and Gorin Y: Myofibroblast
differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney
Int. 79:944–956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weber KT, Sun Y, Tyagi SC and Cleutjens
JP: Collagen network of the myocardium: Function, structural
remodeling and regulatory mechanisms. J Mol Cell Cardiol.
26:279–292. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Swynghedauw B: Molecular mechanisms of
myocardial remodeling. Physiol Rev. 79:215–262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Romaine SP, Tomaszewski M, Condorelli G
and Samani NJ: MicroRNAs in cardiovascular disease: An introduction
for clinicians. Heart. 101:921–928. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gurha P: MicroRNAs in cardiovascular
disease. Curr Opin Cardiol. 31:249–254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Bounds KR, Chatterjee P and Gupta S:
MicroRNA-130a, a potential antifibrotic target in cardiac fibrosis.
J Am Heart Assoc. 6(pii): e0067632017.PubMed/NCBI
|
|
12
|
Du W, Liang H, Gao X, Li X, Zhang Y, Pan
Z, Li C, Wang Y, Liu Y, Yuan W, et al: MicroRNA-328, a potential
anti-fibrotic target in cardiac interstitial fibrosis. Cell Physiol
Biochem. 39:827–836. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen L, Ji Q, Zhu H, Ren Y, Fan Z and Tian
N: miR-30a attenuates cardiac fibrosis in rats with myocardial
infarction by inhibiting CTGF. Exp Ther Med. 15:4318–4324.
2018.PubMed/NCBI
|
|
14
|
Qin RH, Tao H, Ni SH, Shi P, Dai C and Shi
KH: microRNA-29a inhibits cardiac fibrosis in Sprague-Dawley rats
by downregulating the expression of DNMT3A. Anatol J Cardiol.
20:198–205. 2018.PubMed/NCBI
|
|
15
|
Wei C, Kim IK, Kumar S, Jayasinghe S, Hong
N, Castoldi G, Catalucci D, Jones WK and Gupta S: NF-κB mediated
miR-26a regulation in cardiac fibrosis. J Cell Physiol.
228:1433–1442. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han M, Yang Z, Sayed D, He M, Gao S, Lin
L, Yoon S and Abdellatif M: GATA4 expression is primarily regulated
via a miR-26b-dependent post-transcriptional mechanism during
cardiac hypertrophy. Cardiovasc Res. 93:645–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Niture SK, Khatri R and Jaiswal AK:
Regulation of Nrf2-an update. Free Radic Biol Med. 66:36–44. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baird L and Dinkova-Kostova AT: The
cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol.
85:241–272. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi H, Shi A, Dong L, Lu X, Wang Y, Zhao
J, Dai F and Guo X: Chlorogenic acid protects against liver
fibrosis in vivo and in vitro through inhibition of oxidative
stress. Clin Nutr. 35:1366–1373. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cheresh P, Kim SJ, Tulasiram S and Kamp
DW: Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta.
1832:1028–1040. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang LP, Fan SJ, Li SM, Wang XJ, Gao JL
and Yang XH: Oxidative stress promotes myocardial fibrosis by
upregulating KCa3.1 channel expression in AGT-REN double transgenic
hypertensive mice. Pflugers Arch. 469:1061–1071. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu Y, Liu Y, Pan Y, Lu C, Xu H, Wang X,
Liu T, Feng K and Tang Y: MicroRNA-135a inhibits cardiac fibrosis
induced by isoproterenol via TRPM7 channel. Biomed Pharmacother.
104:252–260. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tao H, Zhang JG, Qin RH, Dai C, Shi P,
Yang JJ, Deng ZY and Shi KH: LncRNA GAS5 controls cardiac
fibroblast activation and fibrosis by targeting miR-21 via
PTEN/MMP-2 signaling pathway. Toxicology. 386:11–18. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu J, Wang QY, Zhou Y, Lu XC, Liu YH, Wu
Y, Guo Q, Ma YT and Tang YQ: AstragalosideIV against cardiac
fibrosis by inhibiting TRPM7 channel. Phytomedicine. 30:10–17.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Periasamy S, Chen SY and Liu MY: The study
of ISO induced heart failure rat model, Exp Mol Pathol.
2010;88:299-304. Exp Mol Pathol. 90:842011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Artaud-Macari E, Goven D, Brayer S, Hamimi
A, Besnard V, Marchal-Somme J, Ali ZE, Crestani B, Kerdine-Römer S,
Boutten A and Bonay M: Nuclear factor erythroid 2-related factor 2
nuclear translocation induces myofibroblastic dedifferentiation in
idiopathic pulmonary fibrosis. Antioxid Redox Signal. 18:66–79.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou XL, Xu H, Liu ZB, Wu QC, Zhu RR and
Liu JC: miR-21 promotes cardiac fibroblast-to-myofibroblast
transformation and myocardial fibrosis by targeting Jagged1. J Cell
Mol Med. May 28–2018.(Epub ahead of print).
|
|
29
|
Icli B, Wara AK, Moslehi J, Sun X, Plovie
E, Cahill M, Marchini JF, Schissler A, Padera RF, Shi J, et al:
MicroRNA-26a regulates pathological and physiological angiogenesis
by targeting BMP/SMAD1 signaling. Circ Res. 113:1231–1241. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Köhler UA, Kurinna S, Schwitter D, Marti
A, Schäfer M, Hellerbrand C, Speicher T and Werner S: Activated
Nrf2 impairs liver regeneration in mice by activation of genes
involved in cell-cycle control and apoptosis. Hepatology.
60:670–678. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gambhir L, Checker R, Thoh M, Patwardhan
RS, Sharma D, Kumar M and Sandur SK: 1,4-Naphthoquinone, a
pro-oxidant, suppresses immune responses via KEAP-1
glutathionylation. Biochem Pharmacol. 88:95–105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Civantos E, Bosch E, Ramirez E, Zhenyukh
O, Egido J, Lorenzo O and Mas S: Sitagliptin ameliorates oxidative
stress in experimental diabetic nephropathy by diminishing the
miR-200a/Keap-1/Nrf2 antioxidant pathway. Diabetes Metab Syndr
Obes. 10:207–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang JJ, Tao H, Hu W, Liu LP, Shi KH, Deng
ZY and Li J: MicroRNA-200a controls Nrf2 activation by target Keap1
in hepatic stellate cell proliferation and fibrosis. Cell Signal.
26:2381–2389. 2014. View Article : Google Scholar : PubMed/NCBI
|