Introduction
The prevalence of non-alcoholic fatty liver disease
(NAFLD) is increasing worldwide, and ~25% of adults worldwide
suffer from this disease (1). It is
estimated that in developed countries ~34% of children who are
obese may have NAFLD (2,3). Due to the lack of symptoms, NAFLD is
challenging to diagnose, and the progression of this disease is
unpredictable. Type 2 diabetes mellitus (T2DM) and cardiovascular
diseases are reported to be associated with NAFLD, and are the
leading causes of mortality in patients with NAFLD (4,5). Basic
and clinical studies have shown that NAFLD is detrimental to the
health of patients (6,7). Therefore, appropriate therapeutic
strategies and drugs are required for the treatment of NAFLD.
NAFLD is characterized by diffuse fatty acid (FA)
degeneration and excessive fat accumulation in the liver.
Therefore, NAFLD is often accompanied by increased central
adiposity and triglycerides (TGs), hyperlipidemia and low levels of
high-density lipoprotein (HDL), and this disease is now well
recognized as a type of metabolic syndrome (8,9). The
mechanisms resulting in the progression of hepatic steatosis are
complex, and include increased de novo FA generation
(lipogenesis), decreased β-oxidation and enhanced non-esterified FA
release from adipose tissue (lipolysis) (10,11).
Caloric restriction and exercise can improve NAFLD (12), but changing lifestyle can be
challenging for most patients with NAFLD. To the best of our
knowledge, with the exception of diet and lifestyle modifications,
no effective treatments for NAFLD are currently available (13). Therefore, the identification of
effective drugs and investigation of their protective mechanism in
the control of lipid levels is required for the treatment of
NAFLD.
Atorvastatin (Ato), a lipid-decreasing agent, is the
most commonly prescribed statin drug worldwide (14), and is used for the treatment of
hypercholesterolemia or mixed dyslipidemia. Mechanistically, Ato
exerts its protective roles by competitively inhibiting
3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, which
is known to suppress the mevalonate pathway and subsequently de
novo hepatic cholesterol (CHO) synthesis (15). Due to the wide application of Ato in
clinical settings, other therapeutic properties have been
identified in addition to its lipid-decreasing activity, and this
drug has been used in the treatment of various disorders, including
endothelial dysfunction, cardiovascular disease and depression
(16,17). However, studies investigating the
ability of Ato to prevent NAFLD are limited, and its molecular
mechanisms are not fully understood (18). Therefore, it is necessary to examine
the potential protective roles and underlying mechanisms of Ato in
the treatment of NAFLD in order to identify evidence supporting the
clinical application of this drug.
In the present study, golden hamsters were fed with
a high-fat diet (HFD) to induce NAFLD. The results suggested that
Ato effectively prevented the progression of NAFLD by promoting the
AMP-activated protein kinase (AMPK) signaling pathway. However,
following AMPK inhibition by Compound C in HepG2 cells, the
inhibitory effects of Ato on lipid accumulation were suppressed.
The results indicated that Ato may exhibit potential therapeutic
properties for the treatment of NAFLD, at least in part, by
promoting the AMPK signaling pathway and its downstream
targets.
Materials and methods
Experimental animals and treatment
protocols
Syrian hamsters received humane care according to
the Guidelines for the Experimental Laboratory Animal Committee of
the Chinese Academy of Medical Sciences and Peking Union Medical
College, and the experimental protocols were approved by the Ethics
Committee of the Chinese Academy of Medical Sciences and Peking
Union Medical College. A total of 24 male Golden Syrian hamsters
(age, 8 weeks; weight, 100±10 g) were purchased from Beijing Vital
River Laboratory Animal Technology Co., Ltd (Beijing, China).
Hamsters were housed in a temperature-controlled environment
(temperature, 22-2°C; humidity, 55–5%) with a 12-h light/dark cycle
and ad libitum access to food and water. To increase hepatic
lipid accumulation and create the NAFLD model, 16 hamsters were fed
with a HFD (20 kcal% protein, 20 kcal% carbohydrate and 60 kcal%
fat), while 8 hamsters were fed a normal diet (30 kcal% protein, 60
kcal% carbohydrate and 10 kcal% fat) and served as a control. The
diets were obtained from Beijing HFK Bioscience Co., Ltd.
After 2 weeks, 8 hamsters receiving the HFD were
administered 3 mg/kg/day Ato via gavage in a volume of 1 mg/ml
distilled water for 8 weeks to establish the Ato group (Fig. 1A). The other 8 hamsters receiving the
HFD (model group) and the 8 hamsters in the control group received
vehicle instead. The HFD and normal diets were continued during the
8-week treatment period. The daily state of the animals was
recorded and their body weight was measured weekly. After 8 weeks
of treatment, the animals were anesthetized using urethane (1.2
g/kg body weight), and blood samples were subsequently extracted
from the inferior vena cava for analysis. Under anesthesia, the
liver tissues were removed, weighed, snap-frozen in liquid nitrogen
and stored at −80°C. The coefficient of hepatic weight was
calculated as follows: Liver weight (g)/body weight (100 g).
Reagents
Ato (used in the in vivo experiments) was
purchased from Pfizer, Inc. CHO, TG, HDL and low-density
lipoprotein (LDL) determination kits were purchased from Nanjing
Jiancheng Bioengineering Institute. Ato, palmitate (PA) and
Compound C (used in the in vitro experiments) were purchased
from Sigma-Aldrich (Merck KGaA). TRIzol, PrimeScript RT Master mix
and SYBR Green Master mix were purchased from Takara Bio, Inc.
Cell culture and treatment
HepG2 liver cancer cells are a classical cell model
utilized in numerous studies to explore abnormalities of lipid
metabolism in the liver (17,19).
Thus, this cell line was selected for use in in vitro
experiments. The HepG2 cells (cat. no. 3111C0001CCC000035) were
obtained from the Cell Resource Center, Peking Union Medical
College (headquarters of the National Infrastructure of Cell Line
Resource, NSTI). The identity of the cell lines was authenticated
using STR profiling (CODIS; FBI). Cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin, in a 5% CO2 atmosphere at 37°C.
The cells were plated in six-well plates at 2×105
cells/well. The groups were as follows: i) Control group; ii) PA
treated group; HepG2 cells incubated with 250 µM PA at 37°C for 24
h; iii) PA + Ato group; HepG2 cells incubated with 250 µM PA and 10
µM at 37°C for 24 h; iv) PA + Ato + AMPK inhibitor group; Prior to
drug treatment, HepG2 cells pre-incubated with 10 µM Compound C at
37°C for 4 h.
Biochemical analysis
Serum lipid levels, namely CHO, TG, HDL and LDL,
were evaluated using the aforementioned commercial kits on an
automatic biochemical analyzer (Beckman Coulter, Inc.). Insulin and
C-reactive protein (CRP) levels were determined using an INS RIA
kit (cat. no. HY-10069) and CRP IRMA kit (cat. no. HY-10023),
respectively (Beijing Sino-UK Institute of Biological
Technology).
Hematoxylin and eosin (H&E)
staining
Livers were harvested from hamsters, fixed in 4%
paraformaldehyde for 24 h (room temperature), embedded in paraffin
and sectioned (thickness, 5 µm). The sections were stained with
H&E for 10 min (room temperature) and dehydrated. Sections were
examined under a light microscope (BX53; Olympus Corporation;
magnification, ×200).
Oil red staining
Oil red O staining was performed as previously
described (20). Briefly, frozen
liver sections (thickness, 5 µm) or HepG2 cells were stained with
oil red O working solutions for 30 min, washed with 60% isopropanol
and analyzed. ImageJ software (National Institutes of Health,
version no. 1.51j8) was used to quantify the results of the oil red
O staining of frozen liver sections. TGs were extracted from oil
red O-stained HepG2 cells with 100% isopropanol and were analyzed
by measuring the optical density at 490 nm.
Total RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR) analysis
Total RNA was extracted from tissues using TRIzol.
The purity and concentration of total RNA was measured using a
spectrophotometer (Nanodrop 3000; Thermo Fisher Scientific, Inc.).
Next, 1,000 ng total RNA was reverse transcribed using a
PrimeScript RT Master mix (37°C for 15 min, 85°C for 5 sec, then
hold at 4°C). The expression levels of adipose triglyceride lipase
(ATGL), lipase E, hormone sensitive type (HSL), pyruvate
dehydrogenase kinase 4 (PDK4), carnitine palmitoyltransferase 1A
(CPT1a), CPT1b, FA synthase (FASN), stearoyl-CoA desaturase (SCD1),
diacylglycerol O-acyltransferase 1 (DGAT1), peroxisome proliferator
activated receptor α (PPARα), PPARG coactivator 1 α (PPARGC1A) and
GAPDH were analyzed using the CFX-96 Touch Thermocycler with a SYBR
Green Master mix. The thermocycling conditions were as follows:
Incubation at 95°C for 30 sec, followed by 40 cycles of 95°C for 5
sec, 60°C for 30 sec and 72°C for 30 sec, then dissociation stage.
Primer sequences are presented in Table
I. The results were normalized to GAPDH and calculated using
the 2−ΔΔCq method (21).
 | Table I.Sequences of the primers used for
reverse transcription-quantitative PCR. |
Table I.
Sequences of the primers used for
reverse transcription-quantitative PCR.
| Gene name | Forward primer | Reverse primer |
|---|
| ATGL |
CACTTTAGCTCCAAGGATGA |
TGGTTCAGTAGGCCATTCCT |
| HSL |
GGTGACACTCGCAGAAGACAATA |
GCCGCCGTGCTGTCTCT |
| PDK4 |
CACAGTTGAGCACCAAGA |
AGCCTGACATAGAGTAGAGA |
| CPT1a |
CCACTGATGAAGGAAGGAG |
TAGTAGTTGCTGTTGACCAT |
| CPT1b |
CTCCACAAGCCAGATTCC |
ACTCACCGTCTCAGAACT |
| FASN |
CTTGTCCAGGTTCGTGAG |
TGTTGCTTCGGTGATGAG |
| SCD1 |
CTTCACCACATTCTTCATCG |
TCCCGTCTCCAGTTCTTT |
| DGAT1 |
GGCATCATACTCCATCATCT |
TCTCGGTAGTTCAGATTGTC |
| PPARα |
GAGAAAGCAAAACTGAAAGCAGAGA |
GAAGGGCGGGTTATTGCTG |
| PPARGC1A |
TGGATGAAGACGGATTGC |
GCGACTGTGGTTGTGTAT |
| GAPDH |
AACAGGGTGGTGGACCTCAT |
TGCTCTCAGTATCCTTGCTG |
Western blot analysis
Western blot assay was performed as previously
described (22). Briefly, proteins
were isolated from cells or tissue samples using RIPA buffer
containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5%
sodium deoxycholate and 0.1% SDS. The protein concentration was
measured using BCA Protein Assay Reagent (Thermo Fisher Scientific,
Inc.), and 50 µg of protein samples were loaded into 10% gel and
were separated by SDS-PAGE, and then transferred to a PVDF
membrane. After blocking with 5% non-fat milk in TBST for 2 h at
room temperature, the blots were incubated with the primary
antibodies at 4°C overnight, including AMPKα (CST; 1:1,000; cat.
no. 2532), phosphorylated (p-)AMPKα (CST; 1:1,000; cat. no. 2535),
Akt (CST; 1:1,000; cat. no. 9272), p-Akt (CST; 1:1,000; cat. no.
4060), PPARα (Abcam; 1:1,000; cat. no. ab61182), PGC1α (Abcam;
1:1,000; cat. no. ab54481) and GAPDH (Abcam; 1:2,000; cat. no.
ab8245). After washing with TBST, membranes were probed with
secondary antibody labeled with horseradish peroxidase (Thermo
Fisher Scientific, Inc.; 1;5,000; cat. no. 31460) for 1 h at room
temperature. The protein bands were detected using enhanced
chemiluminescent substrate (Thermo Fisher Scientific, Inc.; cat.
no. 34580) and quantified using Image Lab 3.0 (Bio-Rad
Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± SEM. The
statistical significance of differences among groups was assessed
by one-way ANOVA followed by the Student-Newman-Keuls test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Ato decreases the body weight and
serum lipid levels in golden hamsters
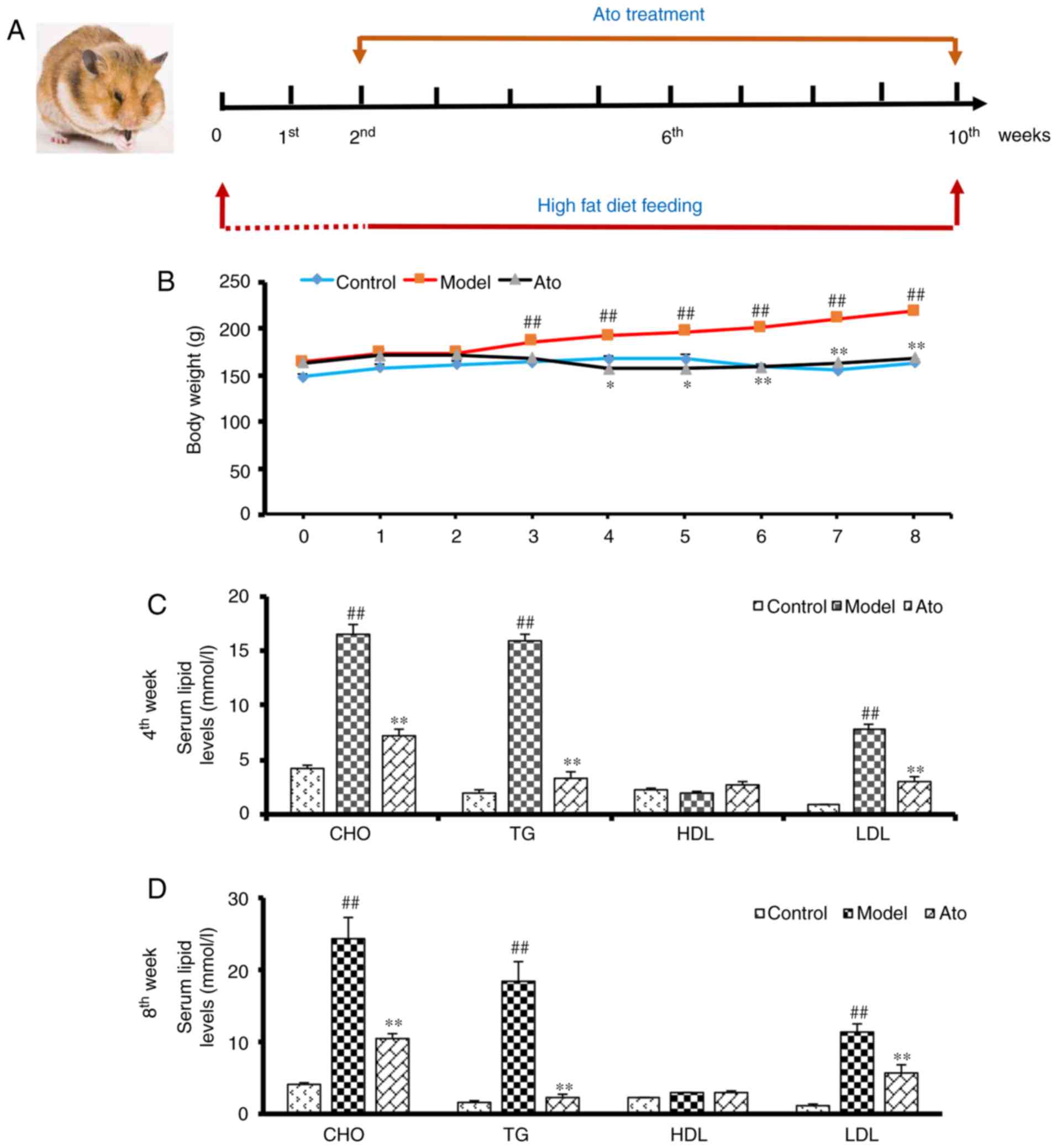
During the 8 weeks of treatment with Ato (Fig. 1A), the body weights of the golden
hamsters from each group were measured. Compared with the control
animals, HFD-fed hamsters showed increased body weight, which was
significantly attenuated by Ato treatment, indicating that Ato
suppressed HFD-induced weight gain (Fig.
1B).
Serum lipid levels were analyzed at the end of the
fourth and eighth weeks of treatment with Ato. After 4 weeks of
treatment, the serum levels of CHO, TG and LDL in the model group
were markedly increased by 285.33, 680.50 and 735.72%,
respectively, in comparison with the control group, indicating that
the hamsters in the model group exhibited hyperlipidemia. Following
4 weeks of Ato treatment, CHO, TG and LDL levels were decreased by
55.68, 79.16 and 60.78%, respectively compared with those in the
model group (Fig. 1C). After 8 weeks
of Ato treatment, in line with the results of the fourth week, Ato
significantly reduced the increased serum levels of CHO, TG and LDL
induced by the administration of HFD for 10 weeks (Fig. 1D).
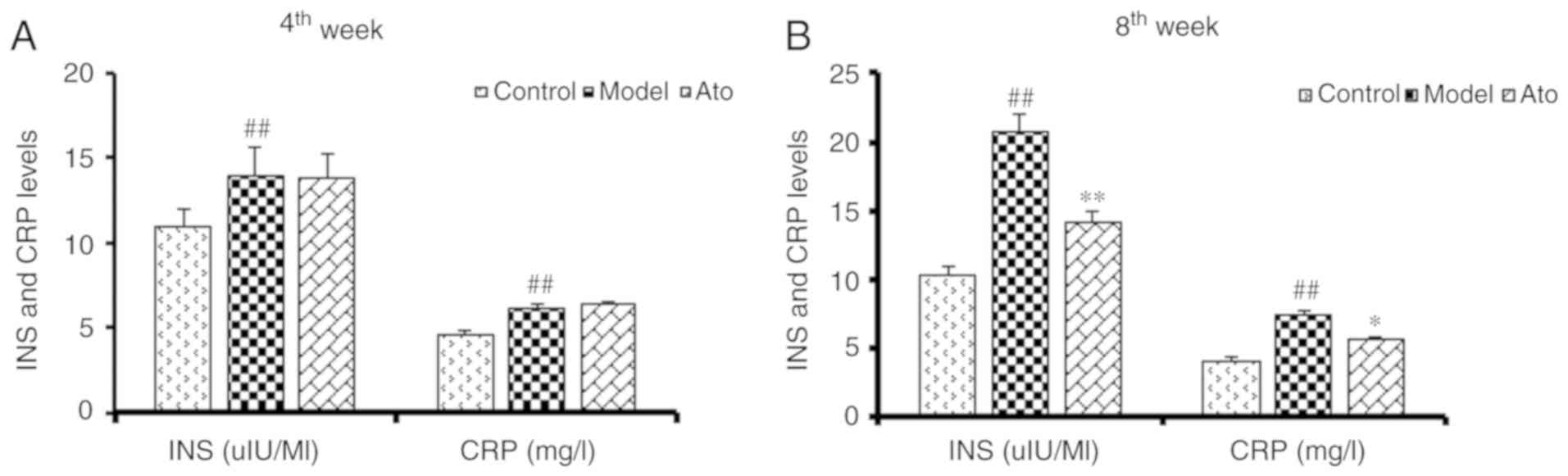
Ato decreases the serum levels of
insulin and CRP
Previous studies have shown that hyperinsulinemia
and increased CRP levels are associated with NAFLD (23,24). The
golden hamsters fed with HFD for 6 weeks exhibited increased serum
insulin and CRP levels (Fig. 2A).
After 10 weeks of HFD, the serum levels of insulin and CRP in the
model group were increased by 101.35 and 80.28% respectively, and
these increases were significantly attenuated following Ato
treatment (Fig. 2B).
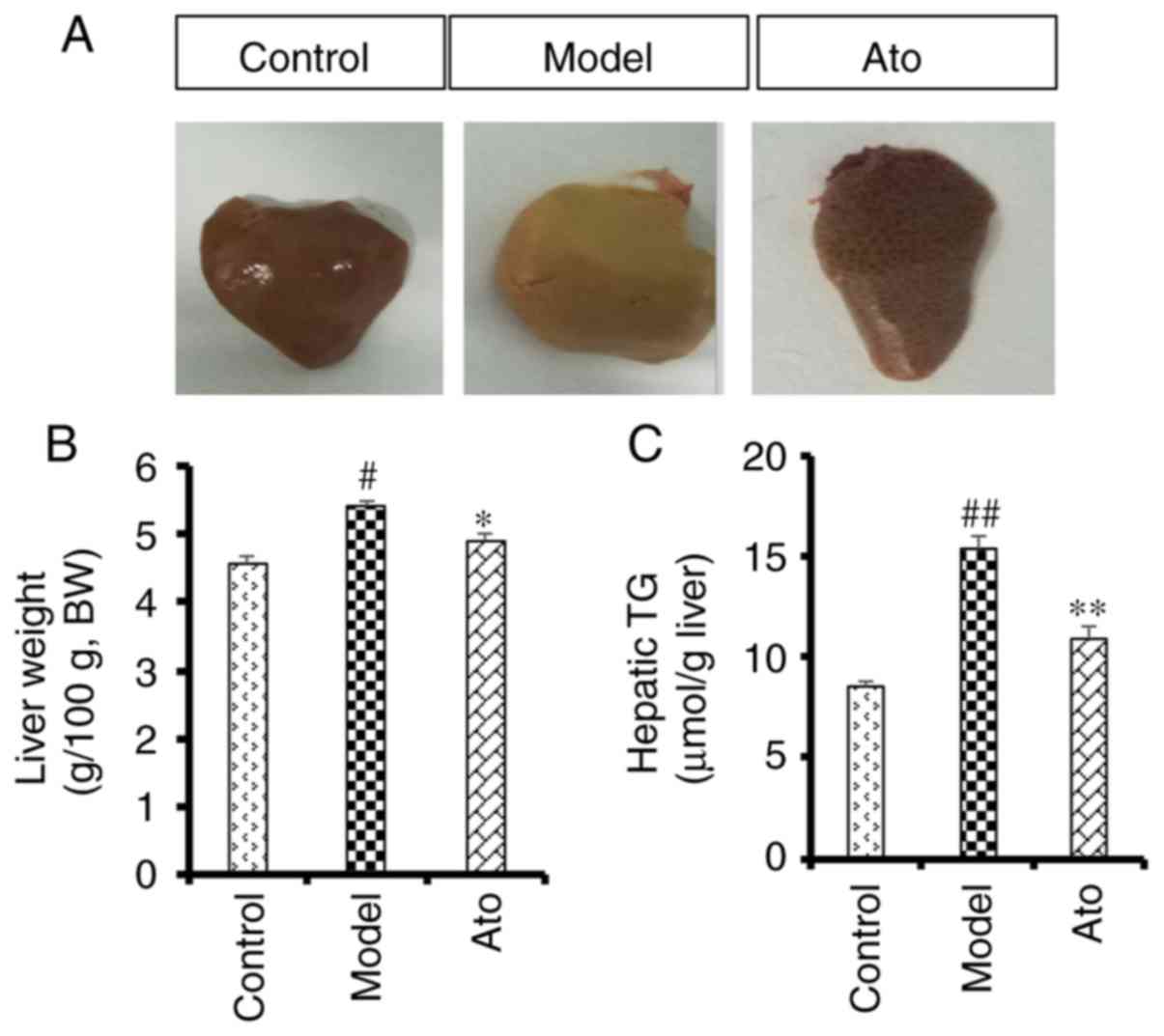
Ato reduces the accumulation of lipids
in the liver
After 10 weeks of HFD, macroscopic observation of
the liver tissue indicated an increased accumulation of lipids in
the model group compared with the control group (Fig. 3A), and this effect was alleviated
following treatment with Ato. Additionally, chronic HFD increased
liver weight (5.41 vs. 4.56/100 g body weight; Fig. 3B) and hepatic TG content (15.40 vs.
8.48 µmol/g; Fig. 3C) compared with
the control group. After 8 weeks of Ato treatment, the increased
liver weight and TG content were reduced by 9.20 and 29.22%,
respectively, indicating that Ato effectively inhibited fat
accumulation in the liver.
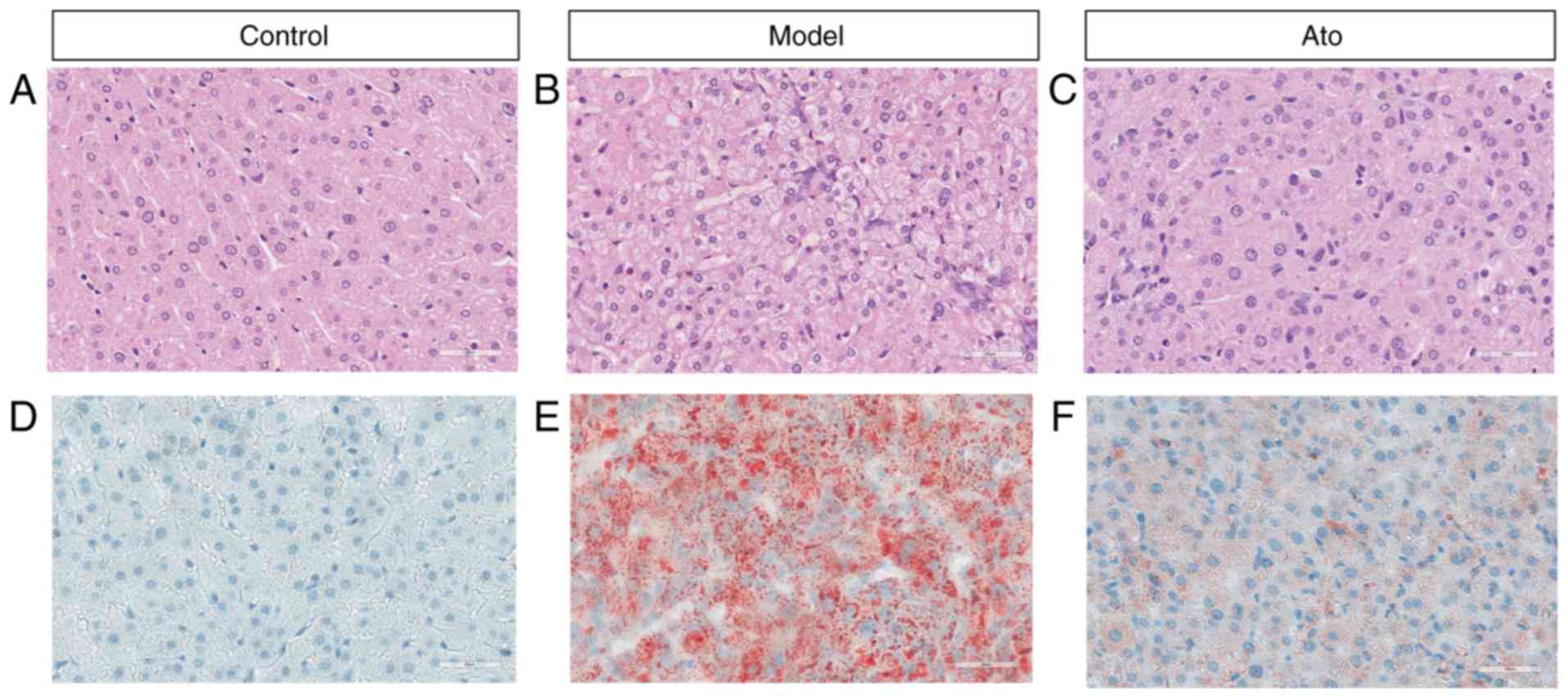
Possible pathological alterations were further
investigated. Results from H&E staining (Fig. 4A-C) showed that HFD increased hepatic
fat accumulation, as the majority of hepatocytes in the livers of
HFD-fed hamsters were increased in volume due to the accumulation
of fat. Numerous single large adipocytes exhibited displaced
nuclei, and ballooning degeneration was evident, and the
hepatocytes, which exhibited cytoplasmic vacuolation, were swollen
(Fig. 4B). Oil red O staining
results showed that Ato treatment for 8 weeks reduced the
accumulation of FAs in the liver compared with that in the model
group (Fig. 4D-F; Fig. S1). The present results suggested
that Ato exhibited the ability to inhibit the progression of
NAFLD.
Ato alleviates NAFLD by promoting the
expression of genes involved in lipolysis and inhibiting the
expression of genes involved in fat synthesis
HFD significantly decreased the hepatic mRNA levels
of ATGL and HSL in hamsters to 40 and 58%, respectively, compared
with the control group. In addition, HFD suppressed a subset of
genes involved in β oxidation, namely CPT1a, CPT1b and PDK4
(Fig. 5A). Ato reversed the
HFD-induced decrease in the hepatic mRNA levels of ATGL and HSL by
225% (P<0.01) and 122.41% (P=0.09), respectively, and
upregulated the reduced mRNA levels of CPT1a, CPT1b and PDK4
induced by HFD treatment, thus promoting adipose lipolysis.
Additionally, HFD increased the expression levels of FASN and DGAT1
compared with those in the control group, and these increases were
attenuated by Ato treatment (Fig.
5B). The present results suggested that Ato could increase fat
oxidation in the NAFLD model.
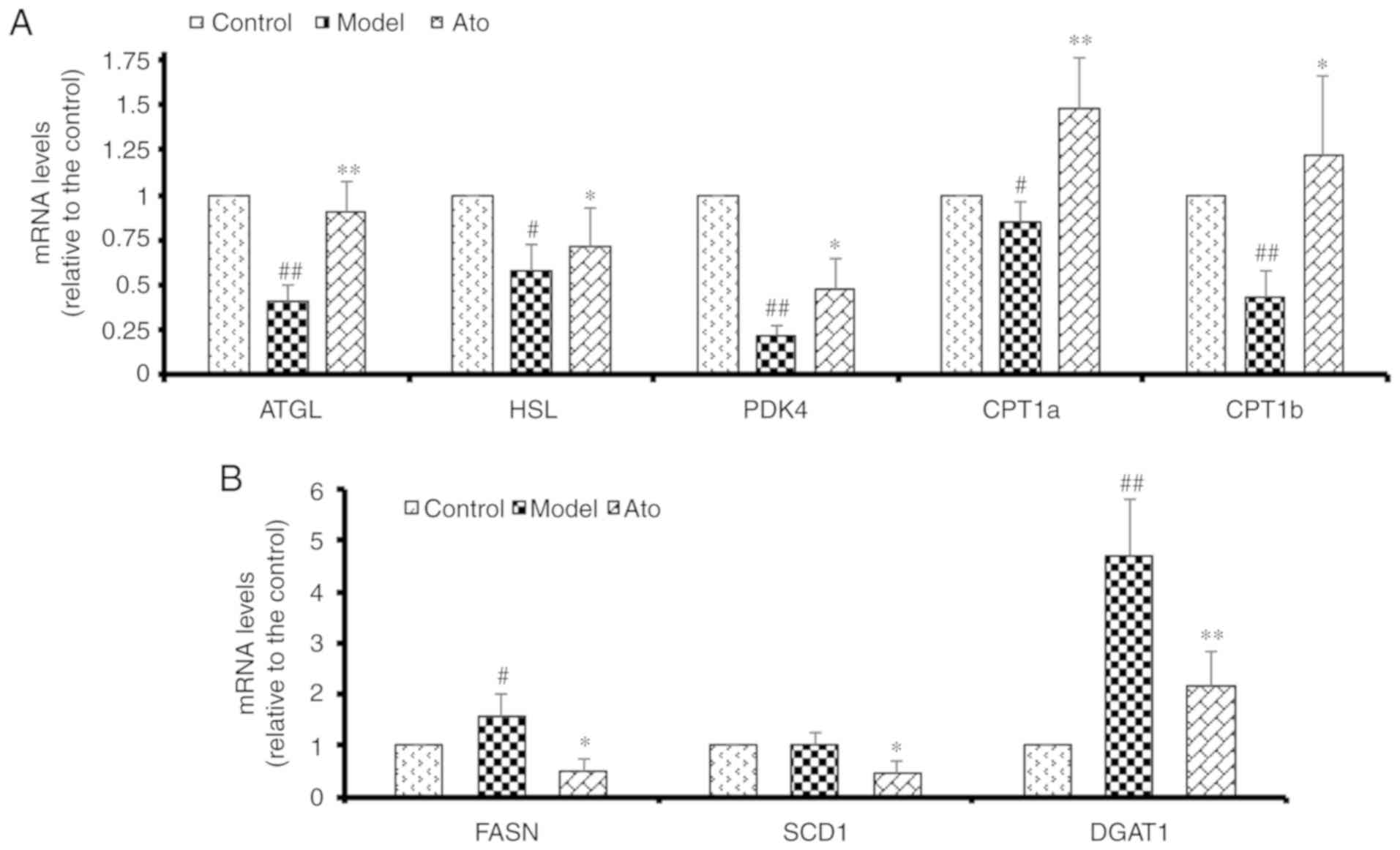 | Figure 5.Effect of Ato on the mRNA levels of
genes involved in lipid metabolism. (A) mRNA levels of genes
associated with lipolysis and β-oxidation. (B) mRNA levels of genes
associated with fatty acid synthesis. Data are presented as the
mean ± SEM (n=8/group). #P<0.05, ##P<0.01 vs. control group;
*P<0.05, **P<0.01 vs. model group. Ato, atorvastatin; ATGL,
adipose triglyceride lipase; HSL, lipase E, hormone sensitive type;
PDK4, pyruvate dehydrogenase kinase 4; CPT1a/CPT1b, carnitine
palmitoyltransferase 1A/B; FASN, fatty acid synthase; SCD1,
stearoyl-CoA desaturase; DGAT1, diacylglycerol O-acyltransferase
1. |
Ato inhibits lipid accumulation via
the AMPK-dependent upregulation of PPARα and PGC1α
AMPK is a key molecule involved in the regulation of
biological metabolism, and it has been reported to be associated
with diabetes and other metabolic diseases. Accumulating evidence
has shown that statins can activate the AMPK pathway (25,26).
Therefore, the present study aimed to determine whether Ato exerts
its protective roles by regulating AMPK (Fig. 6). Western blotting results showed
that the p-AMPK/AMPK and p-Akt/Akt ratios in the livers of HFD-fed
hamsters were decreased to 56 and 31%, respectively, of those in
the control group (Fig. 6A). Ato
treatment for 8 weeks significantly upregulated the p-AMPK/AMPK and
p-Akt/Akt ratios in the liver by 1.91- and 2.42-fold, respectively,
compared with those in the model group (Fig. 6B). In addition, Ato was found to
suppress the lipid accumulation in HepG2 cells induced by treatment
with 200 µM PA for 24 h (Fig. 6D).
In line with the present in vivo results, the
phosphorylation ratio of AMPK was decreased in PA-treated HepG2
cells, but was upregulated following treatment with 20 µM Ato for
24 h (Fig. 6E and F). Treatment with
10 µM Compound C, an AMPK inhibitor, for 24 h significantly
suppressed the protective effects of Ato as an inhibitor of lipid
accumulation in PA-treated HepG2 cells. Collectively, the present
results indicate that AMPK is one of the main targets of Ato
involved in the preventive effects of Ato on NAFLD.
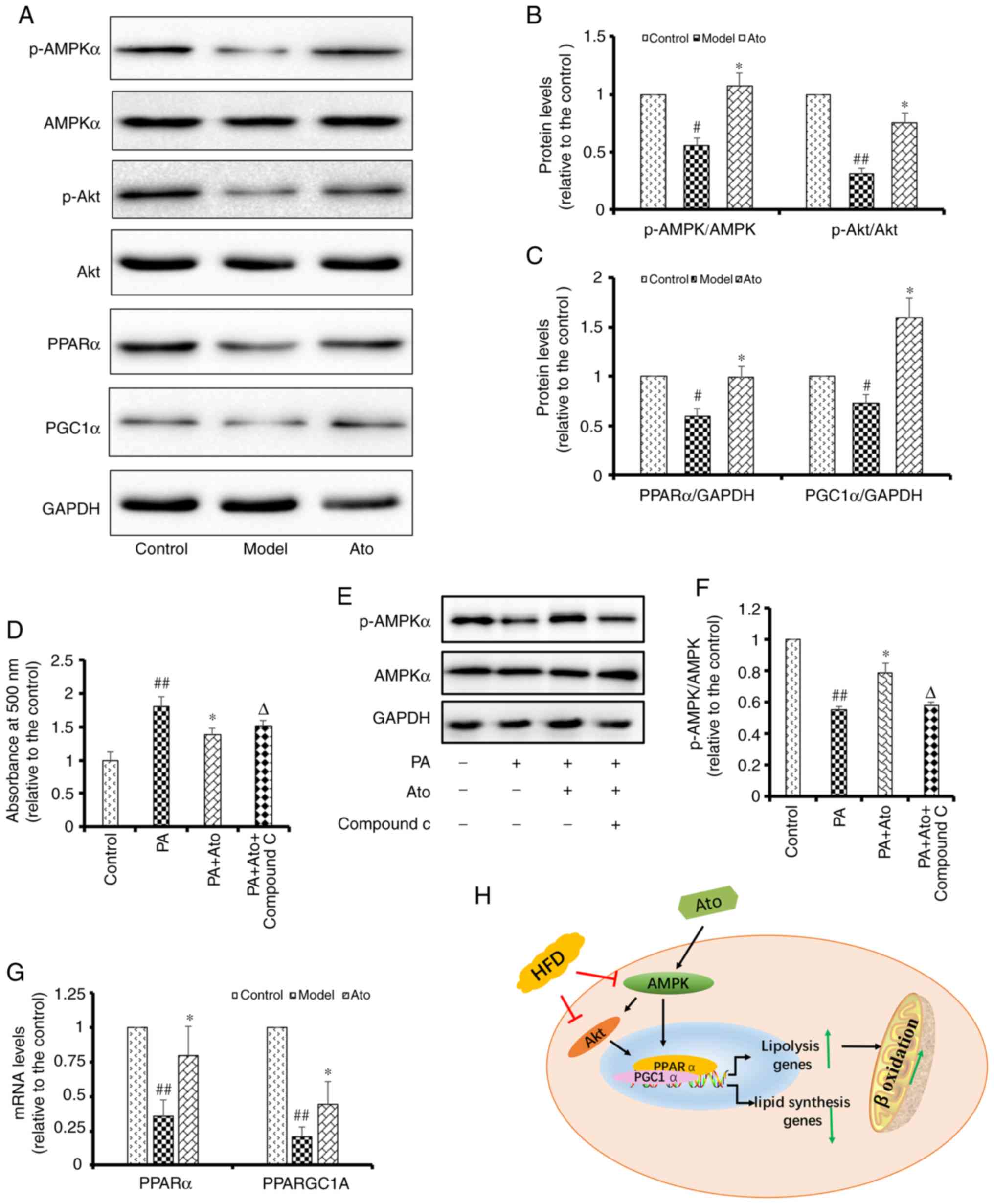 | Figure 6.Ato inhibits lipid accumulation by
promoting the AMPK signaling pathway. (A) Representative images of
p-AMPK, AMPK, p-Akt, Akt, PPARα and PGC1α western blots. (B)
Relative p-AMPK/AMPK, p-Akt/Akt and (C) PPARα/GAPDH and PGC1α/GAPDH
protein levels are presented. #P<0.05, ##P<0.01 vs. control
group; *P<0.05 vs. model group. (D) Lipid accumulation in HepG2
cells, quantified by the absorbance value of oil red O reagent at
490 nm. Data are presented as the mean ± SEM (n=6). (E)
Representative images of p-AMPK and AMPK in PA-treated HepG2 cells
with or without Ato and Compound C treatment. (F) Relative
p-AMPK/AMPK protein levels. Data are presented as the mean ± SEM
(n=3). ##P<0.01 vs. control group; *P<0.05 vs. PA group;
ΔP<0.05 vs. PA + Ato group. (G) mRNA levels of PPARα
and PPARGC1A in each group. Data are presented as the mean ± SEM
(n=3/group). ##P<0.01 vs. control group; *P<0.05 vs. model
group. (H) Schematic illustration showing the suggested mechanisms
underlying the protective effect of Ato against non-alcoholic fatty
liver disease. Ato, atorvastatin; p-, phosphorylated; PPARα,
peroxisome proliferator activated receptor α; AMPK, AMP-activated
protein kinase; PGC1α, peroxisome proliferator-activated receptor-γ
coactivator 1 α; PPARGC1A, PPARG coactivator 1 α; PA,
palmitate. |
Numerous previous studies have indicated that PPARα
is downstream of AMPK and Akt, and that its expression level is
regulated by Akt (27–29). Therefore, the mRNA and protein
expression levels of PPARα and PPARGC1A were investigated in the
liver tissues of the hamsters in the present study. Western
blotting results showed that the protein expression levels of PPARα
and PGC1α were decreased to 59.75 and 72.26%, respectively,
compared with those in the control group (P<0.05). Treatment
with Ato for 8 weeks upregulated the protein expression levels of
PPARα and PGC1α by 1.66- and 2.20-fold, respectively, compared with
those in the model group (P<0.05; Fig. 6C). Consistent with the western
blotting results, the RT-qPCR results showed that Ato significantly
promoted the mRNA expression levels of PPARα and PPARGC1A
(P<0.05; Fig. 6G). Collectively,
the present results indicate that Ato exerted its protective
effects in preventing NAFLD via the AMPK-dependent upregulation of
PPARα and PGC1α.
Discussion
Statins are the most commonly prescribed drugs for
the attenuation of hypercholesterolemia by blocking HMG-CoA
reductase (30). Recent clinical
studies have shown that statins can reduce hepatic lipid
accumulation, thereby alleviating NAFLD (31), but the mechanisms underlying this
effect are poorly understood. Animal models of NAFLD induced by HFD
(32,33) have been widely used to examine the
pathogenesis of NAFLD and to investigate new treatment strategies
(34). HFD-induced liver disease, in
contrast with rifampicin-isoniazid-induced hepatotoxicity (35), not only leads to chronic inflammation
and liver fibrosis, but also disrupts endoplasmic reticulum calcium
homeostasis (36–38). In the present study, hamsters were
given a HFD for 10 weeks and developed hepatic steatosis with high
levels of TG, TCH, insulin and CRP, phenocopying the clinical
features of human NAFLD. Following Ato treatment, the increased
body weight and elevated serum levels of TCH, TG, LDL, insulin and
CRP identified in HFD-fed hamsters were decreased. Additionally,
the relative liver weight of HFD-fed hamsters treated with Ato was
markedly decreased compared with that of the model group.
Morphologically, the livers of hamsters in the model group
exhibited numerous large lipid droplets and various defects
compared with the control group. However, the livers of HFD-fed
hamsters treated with Ato exhibited fewer lipid droplets and an
improved liver morphology, indicating that Ato exhibited beneficial
effects in suppressing lipid accumulation and attenuating the
disruption of liver structure.
The prevalence of NAFLD in China is increasing. In
2014, a meta-analysis of epidemiologic studies revealed that the
overall prevalence of NAFLD was ~20% in China (39). NAFLD has been identified to be
associated with obesity, dyslipidemia, insulin resistance and T2DM
(40). Due to the increase in
metabolic risk factors, patients suffering from NAFLD are at higher
risk of developing cardiovascular diseases, which are associated
with increased morbidity and mortality (41). NAFLD contributes to the development
of T2DM, non-alcoholic steatohepatitis and hepatocellular carcinoma
(42). However, the number of
targeted drugs to treat NAFLD remain limited. In clinical settings,
statins are widely used to reduce the serum levels of lipids, which
is considered to suppress NAFLD. However, the number of studies
investigating the protective effects of Ato on NAFLD remain limited
and its underlying molecular mechanisms are unclear. Therefore, to
investigate these questions, an animal model of NAFLD was
constructed in the present study, in which golden hamsters were fed
a HFD to induce NAFLD. Ato treatment not only decreased the serum
levels of TG, TCH, LDL, CRP and insulin, but also attenuated lipid
accumulation and pathological alterations, indicating that Ato
exhibited the potential to treat NAFLD.
The pathogenesis of NAFLD is complex and involves
numerous signaling pathways, including the PI3K/Akt, Toll-like
receptor (TLR), apoptosis and mTOR pathways (43,44).
Deficient PI3K activity leads to an increase in intracellular
FA-derived metabolites (45). TLR-4
can activate X-box binding protein-1 and thereby promote the
progression of NAFLD (46).
Increases in active caspase 2, active caspase 3 and apoptosis have
been observed in the livers of patients with NAFLD (47). Metabolic abnormality of lipids in the
liver may cause insulin resistance, inflammation and apoptosis
(48–50).
AMPK regulates numerous metabolic pathways and may
have therapeutic potential in the treatment of obesity, insulin
resistance, T2DM and NAFLD. Activation of AMPK can inhibit acetyl
CoA carboxylase (ACC), which plays crucial roles in lipogenesis.
AMPK also regulates FA metabolism in the liver via the regulation
of total mitochondrial content and function. Recent studies have
indicated that statins may be able to alleviate numerous diseases
by regulating the AMPK-mediated pathway, including neurotoxicity,
myocardial fibrosis and endoplasmic reticulum stress (51–53).
These previous studies suggest that Ato may protect against NAFLD
by increasing the activity of the AMPK signaling pathway.
As an energy sensor that maintains cellular energy
homeostasis, AMPK has been reported to positively regulate FA
oxidation by activating PPARα and PGC1α (54). Additionally, there is evidence to
indicate that AMPK activates the PI3K/Akt pathway by inhibiting the
phosphorylation of insulin receptor substrate-1
(Ser636/639) (55–57).
Hinoi et al (58) found that
the PI3K/Akt pathway could indirectly promote the nuclear
translocation of PGC1α, which serves crucial roles in FA oxidation.
Consistent with a previous study (59), the livers of HFD-fed hamsters in the
present study exhibited a significant reduction in AMPK
phosphorylation levels compared with the control group. Akt
phosphorylation, as well as PPARα and PGC1α protein expression
levels were also reduced in the model group compared with the
control. However, Ato treatment for 8 weeks significantly reversed
the decreased levels of p-AMPK, p-Akt, PPARα and PGC1α, indicating
that Ato is able to regulate the expression of AMPK and its
downstream targets.
Activation of FA β-oxidation facilitates the
metabolism of lipids in the liver. PPARα has been indicated to
stimulate FA β-oxidation by increasing the expression level of PDK4
(60). Additionally, PPARα controls
the constitutive expression of mitochondrial FA β-oxidation enzymes
(61) and increases the level of
uncoupling protein 2 in mitochondria (62). Since Ato upregulated PPARα via AMPK
and its downstream targets, the present study also investigated the
mRNA levels of several genes involved in lipid metabolism,
including lipogenesis and β-oxidation. The results revealed that
the HFD-induced inhibition of ATGL, HSL, PDK4, CPT1a and CPT1b
expression levels could be effectively attenuated by Ato treatment.
Collectively, the present data indicate that Ato may promote lipid
metabolism via the AMPK-dependent upregulation of PPARα, PGC1α and
their target genes.
However, the present study presents certain
limitations. The impact the Ato on control mice was not explored in
this study. Although Ato may protect against NAFLD by activating
AMPK and its target genes, it may have multiple targets in addition
to AMPK and HMG-CoA reductase. Furthermore, in vitro
experiments are required to investigate whether the protective
effects of Ato are neutralized following the knockdown of AMPK
using small interfering RNA or adenoviral means. Future studies
conducted by the present team will aim to investigate this
aspect.
In conclusion, the results of the present study
suggest that Ato could effectively prevent the progression of NAFLD
in a hamster model, as evidenced by its ability to attenuate the
HFD-induced increases in serum levels of lipids, insulin and CRP,
and by the reduction of lipid accumulation in the liver.
Mechanistically, the results also indicate that Ato inhibited fat
accumulation in the liver via the AMPK-dependent upregulation of
PPARα, PGC1α and their target genes, which are associated with
β-oxidation. In conclusion, the present study suggests the
potential of Ato as a therapeutic drug for the clinical treatment
of NAFLD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by The National Science
Foundation for Young Scientists of China (grant no. 81803803).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BZ, GS and XS designed the study. BZ, CZ, XZ, NL and
ZD performed the experiments. CZ analyzed the data and BZ wrote the
manuscript. All authors discussed, edited and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The animal protocol was approved by The Experimental
Laboratory Animal Committee of Chinese Academy of Medical Sciences
and Peking Union Medical College.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Younossi ZM, Koenig AB, Abdelatif D, Fazel
Y, Henry L and Wymer M: Global epidemiology of nonalcoholic fatty
liver disease-Meta-analytic assessment of prevalence, incidence,
and outcomes. Hepatology. 64:73–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Anderson EL, Howe LD, Jones HE, Higgins
JP, Lawlor DA and Fraser A: The prevalence of non-alcoholic fatty
liver disease in children and adolescents: A systematic review and
meta-analysis. PLoS One. 10:e01409082015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alisi A, Feldstein AE, Villani A, Raponi M
and Nobili V: Pediatric nonalcoholic fatty liver disease: A
multidisciplinary approach. Nat Rev Gastroenterol Hepatol.
9:152–161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ekstedt M, Hagström H, Nasr P, Fredrikson
M, Stål P, Kechagias S and Hultcrantz R: Fibrosis stage is the
strongest predictor for disease-specific mortality in NAFLD after
up to 33 years of follow-up. Hepatology. 61:1547–1554. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Angulo P, Kleiner DE, Dam-Larsen S, Adams
LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC,
Lafferty HD, Stahler A, et al: Liver fibrosis, but no other
histologic features, is associated with long-term outcomes of
patients with nonalcoholic fatty liver disease. Gastroenterology.
149:389–397.e310. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tanaka S, Hikita H, Tatsumi T, Sakamori R,
Nozaki Y, Sakane S, Shiode Y, Nakabori T, Saito Y, Hiramatsu N, et
al: Rubicon inhibits autophagy and accelerates hepatocyte apoptosis
and lipid accumulation in nonalcoholic fatty liver disease in mice.
Hepatology. 64:1994–2014. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aller R, Fernández-Rodríguez C, Lo Iacono
O, Bañares R, Abad J, Carrión JA, García-Monzón C, Caballería J,
Berenguer M, Rodríguez-Perálvarez M, et al: Consensus document.
Management of non-alcoholic fatty liver disease (NAFLD). Clinical
practice guideline. Gastroenterol Hepatol. 41:328–349. 2018.(In
English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marchesini G, Brizi M, Bianchi G,
Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S,
Forlani G and Melchionda N: Nonalcoholic fatty liver disease: A
feature of the metabolic syndrome. Diabetes. 50:1844–1850. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamasaki T and Tomita K: Relationship
between hyperuricemia and metabolic syndrome. Nihon Rinsho.
66:766–770. 2008.(In Japanese). PubMed/NCBI
|
|
10
|
Kim JY, Garcia-Carbonell R, Yamachika S,
Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR and Karin M: ER
stress drives lipogenesis and steatohepatitis via caspase-2
activation of S1P. Cell. 175:133–145.e15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park JG, Xu X, Cho S, Hur KY, Lee MS,
Kersten S and Lee AH: CREBH-FGF21 axis improves hepatic steatosis
by suppressing adipose tissue lipolysis. Sci Rep. 6:279382016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stavropoulos K, Imprialos K, Pittaras A,
Faselis C, Narayan P and Kokkinos P: Lifestyle modifications in
non-alcoholic fatty liver disease and non-alcoholic
steatohepatitis. Curr Vasc Pharmacol. 16:239–245. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mathews SE, Kumar RB and Shukla AP:
Nonalcoholic steatohepatitis, obesity, and cardiac dysfunction.
Curr Opin Endocrinol Diabetes Obes. 25:315–320. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye YC, Zhao XL and Zhang SY: Use of
atorvastatin in lipid disorders and cardiovascular disease in
Chinese patients. Chin Med J (Engl). 128:259–266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bakker-Arkema RG, Davidson MH, Goldstein
RJ, Davignon J, Isaacsohn JL, Weiss SR, Keilson LM, Brown WV,
Miller VT, Shurzinske LJ and Black DM: Efficacy and safety of a new
HMG-CoA reductase inhibitor, atorvastatin, in patients with
hypertriglyceridemia. JAMA. 275:128–133. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amarenco P, Bogousslavsky J, Callahan A
III, Goldstein LB, Hennerici M, Rudolph AE, Sillesen H, Simunovic
L, Szarek M, Welch KM, et al: High-dose atorvastatin after stroke
or transient ischemic attack. N Engl J Med. 355:549–559. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Taniguti EH, Ferreira YS, Stupp IJV,
Fraga-Junior EB, Doneda DL, Lopes L, Rios-Santos F, Lima E, Buss
ZS, Viola GG and Vandresen-Filho S: Atorvastatin prevents
lipopolysaccharide-induced depressive-like behaviour in mice. Brain
Res Bull. 146:279–286. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cioboată R, Găman A, Traşcă D, Ungureanu
A, Docea AO, Tomescu P, Gherghina F, Arsene AL, Badiu C, Tsatsakis
AM, et al: Pharmacological management of non-alcoholic fatty liver
disease: Atorvastatin versus pentoxifylline. Exp Ther Med.
13:2375–2381. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brenachot X, Ramadori G, Ioris RM,
Veyrat-Durebex C, Altirriba J, Aras E, Ljubicic S, Kohno D,
Fabbiano S, Clement S, et al: Hepatic protein tyrosine phosphatase
receptor gamma links obesity-induced inflammation to insulin
resistance. Nat Commun. 8:18202017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lerat H, Honda M, Beard MR, Loesch K, Sun
J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA and Lemon SM:
Steatosis and liver cancer in transgenic mice expressing the
structural and nonstructural proteins of hepatitis C virus.
Gastroenterology. 122:352–365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li M, Tang Y, Wu L, Mo F, Wang X, Li H, Qi
R, Zhang H, Srivastava A and Ling C: The hepatocyte-specific
HNF4alpha/miR-122 pathway contributes to iron overload-mediated
hepatic inflammation. Blood. 130:1041–1051. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nuñez-Durán E, Aghajan M, Amrutkar M, Sütt
S, Cansby E, Booten SL, Watt A, Ståhlman M, Stefan N, Häring HU, et
al: Serine/threonine protein kinase 25 antisense oligonucleotide
treatment reverses glucose intolerance, insulin resistance, and
nonalcoholic fatty liver disease in mice. Hepatol Commun. 2:69–83.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee J, Yoon K, Ryu S, Chang Y and Kim HR:
High-normal levels of hs-CRP predict the development of
non-alcoholic fatty liver in healthy men. PLoS One.
12:e01726662017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang JC, Li XX, Sun X, Li GY, Sun JL, Ye
YP, Cong LL, Li WM, Lu SY, Feng J, Liu PJ, et al: Activation of
AMPK by simvastatin inhibited breast tumor angiogenesis via
impeding HIF-1alpha-induced pro-angiogenic factor. Cancer Sci.
109:1627–1637. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han JS, Sung JH and Lee SK: Inhibition of
cholesterol synthesis in HepG2 cells by GINST-decreasing HMG-CoA
reductase expression via AMP-activated protein kinase. J Food Sci.
82:2700–2705. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dihingia A, Bordoloi J, Dutta P, Kalita J
and Manna P: Hexane-Isopropanolic Extract of Tungrymbai, a
North-East Indian fermented soybean food prevents hepatic steatosis
via regulating AMPK-mediated SREBP/FAS/ACC/HMGCR and
PPARalpha/CPT1A/UCP2 pathways. Sci Rep. 8:100212018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gorowska-Wojtowicz E, Dutka P, Kudrycka M,
Pawlicki P, Milon A, Plachno BJ, Tworzydlo W, Pardyak L, Kaminska
A, Hejmej A, et al: Regulation of steroidogenic function of mouse
Leydig cells: G-coupled membrane estrogen receptor and peroxisome
proliferator-activated receptor partnership. J Physiol Pharmacol.
69:2018.PubMed/NCBI
|
|
29
|
Muthukumaran P, Thiyagarajan G, Arun Babu
R and Lakshmi BS: Raffinose from Costus speciosus attenuates lipid
synthesis through modulation of PPARs/SREBP1c and improves insulin
sensitivity through PI3K/AKT. Chem Biol Interact. 284:80–89. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sirtori CR: The pharmacology of statins.
Pharmacol Res. 88:3–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Athyros VG, Boutari C, Stavropoulos K,
Anagnostis P, Imprialos KP, Doumas M and Karagiannis A: Statins: An
under-appreciated asset for the prevention and the treatment of
NAFLD or NASH and the related cardiovascular risk. Curr Vasc
Pharmacol. 16:246–253. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xue L, Lu X, He J, Zhang T, Wu X, Zhang Y,
Wang N, An Z, Xu J and Geng Y: Serum CK 18-M30 reflect liver
pathological severity during NAFLD progression in a rat model.
Pathol Res Pract. 214:1778–1786. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou D, Pan Q, Xin FZ, Zhang RN, He CX,
Chen GY, Liu C, Chen YW and Fan JG: Sodium butyrate attenuates
high-fat diet-induced steatohepatitis in mice by improving gut
microbiota and gastrointestinal barrier. World J Gastroenterol.
23:60–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ou TH, Tung YT, Yang TH and Chien YW:
Melatonin improves fatty liver syndrome by inhibiting the
lipogenesis pathway in hamsters with high-fat diet-induced
hyperlipidemia. Nutrients. 11:E7482019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nwidu LL and Teme RE: Hot aqueous leaf
extract of Lasianthera africana (Icacinaceae) attenuates
rifampicin-isoniazid-induced hepatotoxicity. J Integr Med.
16:263–272. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wires ES, Trychta KA, Bäck S, Sulima A,
Rice KC and Harvey BK: High fat diet disrupts endoplasmic reticulum
calcium homeostasis in the rat liver. J Hepatol. 67:1009–1017.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Y, Qian Q and Yu J: Carbenoxolone
ameliorates insulin sensitivity in obese mice induced by high fat
diet via regulating the IkappaB-α/NF-κB pathway and NLRP3
inflammasome. Biomed Pharmacother. 115:1088682019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Goto R, Kamimura K, Shinagawa-Kobayashi Y,
Sakai N, Nagoya T, Niwa Y, Ko M, Ogawa K, Inoue R, Yokoo T, et al:
Inhibition of sodium glucose cotransporter 2 (SGLT2) delays liver
fibrosis in a medaka model of nonalcoholic steatohepatitis (NASH).
FEBS Open Bio. 9:643–652. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li Z, Xue J, Chen P, Chen L, Yan S and Liu
L: Prevalence of nonalcoholic fatty liver disease in mainland of
China: A meta-analysis of published studies. J Gastroenterol
Hepatol. 29:42–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Caballería L, Pera G, Auladell MA, Torán
P, Muñoz L, Miranda D, Alumà A, Casas JD, Sánchez C, Gil D, et al:
Prevalence and factors associated with the presence of nonalcoholic
fatty liver disease in an adult population in Spain. Eur J
Gastroenterol Hepatol. 22:24–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Söderberg C, Stål P, Askling J, Glaumann
H, Lindberg G, Marmur J and Hultcrantz R: Decreased survival of
subjects with elevated liver function tests during a 28-year
follow-up. Hepatology. 51:595–602. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wong CR, Nguyen MH and Lim JK:
Hepatocellular carcinoma in patients with non-alcoholic fatty liver
disease. World J Gastroenterol. 22:8294–8303. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang X: Down-regulation of lncRNA-NEAT1
alleviated the non-alcoholic fatty liver disease via mTOR/S6K1
signaling pathway. J Cell Biochem. 119:1567–1574. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu H, Li J, Tillman B, Morgan TR, French
BA and French SW: TLR3/4 signaling is mediated via the
NFkappaB-CXCR4/7 pathway in human alcoholic hepatitis and
non-alcoholic steatohepatitis which formed Mallory-Denk bodies. Exp
Mol Pathol. 97:234–240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim JK, Fillmore JJ, Chen Y, Yu C, Moore
IK, Pypaert M, Lutz EP, Kako Y, Velez-Carrasco W, Goldberg IJ, et
al: Tissue-specific overexpression of lipoprotein lipase causes
tissue-specific insulin resistance. Proc Natl Acad Sci USA.
98:7522–7527. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y,
Man K, Lo CM, Li X and Xu A: Toll-like receptor-4 mediates
obesity-induced non-alcoholic steatohepatitis through activation of
X-box binding protein-1 in mice. Gut. 61:1058–1067. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ferreira DM, Castro RE, Machado MV,
Evangelista T, Silvestre A, Costa A, Coutinho J, Carepa F,
Cortez-Pinto H and Rodrigues CM: Apoptosis and insulin resistance
in liver and peripheral tissues of morbidly obese patients is
associated with different stages of non-alcoholic fatty liver
disease. Diabetologia. 54:1788–1798. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Smith GI, Shankaran M, Yoshino M,
Schweitzer GG, Chondronikola M, Beals JW, Okunade AL, Patterson BW,
Nyangau E, Field T, et al: Insulin resistance drives hepatic de
novo lipogenesis in nonalcoholic fatty liver disease. J Clin
Invest. 134165Dec 5–2019.(Epub ahead of print). PubMed/NCBI
|
|
49
|
Wang Q, Ou Y, Hu G, Wen C, Yue S, Chen C,
Xu L, Xie J, Dai H, Xiao H, et al: Naringenin attenuates
nonalocholic fatty liver disease by downregulating NLRP3/NF-kappaB
pathway in mice. Br J Pharmacol. Nov 23–2019.(Epub ahead of print).
View Article : Google Scholar
|
|
50
|
Kanda T, Matsuoka S, Yamazaki M, Shibata
T, Nirei K, Takahashi H, Kaneko T, Fujisawa M, Higuchi T, Nakamura
H, et al: Apoptosis and non-alcoholic fatty liver diseases. World J
Gastroenterol. 24:2661–2672. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li HH, Lin CL and Huang CN:
Neuroprotective effects of statins against amyloid β-induced
neurotoxicity. Neural Regen Res. 13:198–206. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hermida N, Markl A, Hamelet J, Van Assche
T, Vanderper A, Herijgers P, van Bilsen M, Hilfiker-Kleiner D,
Noppe G, Beauloye C, et al: HMGCoA reductase inhibition reverses
myocardial fibrosis and diastolic dysfunction through AMP-activated
protein kinase activation in a mouse model of metabolic syndrome.
Cardiovasc Res. 99:44–54. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jia F, Wu C, Chen Z and Lu G: Atorvastatin
inhibits homocysteine-induced endoplasmic reticulum stress through
activation of AMP-activated protein kinase. Cardiovasc Ther.
30:317–325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ,
Oh KS, Koh EH, Won JC, Kim MS, Oh GT, et al: AMPK activation
increases fatty acid oxidation in skeletal muscle by activating
PPARalpha and PGC-1. Biochem Biophys Res Commun. 340:291–295. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tzatsos A and Kandror KV: Nutrients
suppress phosphatidylinositol 3-kinase/Akt signaling via
raptor-dependent mTOR-mediated insulin receptor substrate 1
phosphorylation. Mol Cell Biol. 26:63–76. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chopra I, Li HF, Wang H and Webster KA:
Phosphorylation of the insulin receptor by AMP-activated protein
kinase (AMPK) promotes ligand-independent activation of the insulin
signalling pathway in rodent muscle. Diabetologia. 55:783–794.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zheng T, Yang X, Wu D, Xing S, Bian F, Li
W, Chi J, Bai X, Wu G, Chen X, et al: Salidroside ameliorates
insulin resistance through activation of a mitochondria-associated
AMPK/PI3K/Akt/GSK3beta pathway. Br J Pharmacol. 172:3284–3301.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hinoi E, Iezaki T, Fujita H, Watanabe T,
Odaka Y, Ozaki K and Yoneda Y: PI3K/Akt is involved in brown
adipogenesis mediated by growth differentiation factor-5 in
association with activation of the Smad pathway. Biochem Biophys
Res Commun. 450:255–260. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Docrat TF, Nagiah S, Krishnan A, Naidoo DB
and Chuturgoon AA: Atorvastatin induces MicroRNA-145 expression in
HEPG2 cells via regulation of the PI3K/AKT signalling pathway. Chem
Biol Interact. 287:32–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ferrari A, Longo R, Fiorino E, Silva R,
Mitro N, Cermenati G, Gilardi F, Desvergne B, Andolfo A, Magagnotti
C, et al: HDAC3 is a molecular brake of the metabolic switch
supporting white adipose tissue browning. Nat Commun. 8:932017.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Aoyama T, Peters JM, Iritani N, Nakajima
T, Furihata K, Hashimoto T and Gonzalez FJ: Altered constitutive
expression of fatty acid-metabolizing enzymes in mice lacking the
peroxisome proliferator-activated receptor alpha (PPARalpha). J
Biol Chem. 273:5678–5684. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Patterson AD, Shah YM, Matsubara T, Krausz
KW and Gonzalez FJ: Peroxisome proliferator-activated receptor
alpha induction of uncoupling protein 2 protects against
acetaminophen-induced liver toxicity. Hepatology. 56:281–290. 2012.
View Article : Google Scholar : PubMed/NCBI
|