Introduction
Congenital heart disease (CHD) is one of the most
common birth defects in children. The mean prevalence of CHD from
1970 to 2017 stood at 8.224 per 1,000 globally (1). Furthermore, the estimated incidence of
sustained PH in all categories was reported at 5-8 cases per
million children per year and 26-33 per million children in the USA
(2). In particular, left-to-right
shunt CHD is the most common form of CHD. Due to long-term high
pulmonary blood flow, endothelial dysfunction and structural
remodeling ensues in the pulmonary arterioles from high vascular
shearing force, which ultimately leads to elevated arterial
pressure and resistance in the pulmonary vasculature (3). Pulmonary artery smooth muscle cells
(PASMCs) serve a vital role in vascular homeostasis, the regulation
of vascular tone and the regulation of pulmonary arterial pressure.
As one of the main transmembrane channels, calcium-activated
chloride channels (CaCCs) serve an important function in
transmembrane chloride ion homeostasis and vascular tone regulation
(4-6). As
PAH progresses, increased proliferation of smooth muscle cells,
endothelial cells and adventitial fibroblasts, coupled with reduced
PASMCs apoptosis contribute to the medial thickening and
neomuscularization of distal arterioles, ultimately resulting in
right ventricular hypertrophy and heart failure (7).
Transmembrane protein 16A (TMEM16A, also known as
ANO1) was first reported to encode CaCCs in 2008 (8-10).
TMEM16A is expressed in a variety of cells, including cardiac
fibroblasts (11), smooth muscle
cells (12) and sympathetic ganglion
cells (13). TMEM16A has been
revealed to be overexpressed in PASMCs in a number of different
pulmonary hypertension models (14,15). A
previous study from our laboratory at the Pediatric department, The
First Affiliated Hospital Of Guangxi Medical University (Nanning,
China) demonstrated that the CaCC inhibitor, niflumic acid,
attenuated pulmonary artery structural remodeling in PAH induced by
high pulmonary blood flow in vivo (16). However, the role and mechanism of
TMEM16A in PAH induced by high pulmonary blood flow remains
unclear. Therefore, the present study investigated the effects of
TMEM16A in the regulation of PASMCs in high pulmonary blood
flow-induced PAH.
Materials and methods
Animals and PAH models
All animal experimental procedures were performed in
accordance with the Guide to Care and Use of Experimental Animals
issued by the Ministry of Health of the People's Republic of China.
All experimental procedures were approved by The Animal and Human
Ethics Committee in Guangxi Medical University (Guangxi, China). A
total of 30 male Sprague Dawley rats (weight, 180-200 g; age, 6-8
weeks) were provided by the Animal Research Centre of Guangxi
Medical University (license no. SCXK 2009-0002). A total of 10 rats
were randomly assigned into three groups respectively: Control,
sham and shunt groups. Rats were anaesthetized by intraperitoneal
injection of sodium pentobarbital (0.25%; 40 mg/kg). According to
methods reported previously (16,17), an
abdominal aorta and inferior vena cava arteriovenous fistulization
was performed to establish PAH induced by high pulmonary blood flow
from the systemic circulation. Laparotomy was performed in the sham
and shunt groups, and the abdominal aorta was clamped for 3 min.
All rats had were housed in a specific pathogen free room with free
access to food and water, maintained at 22-24°C with 40%
humidity and 12 h light/dark cycle.
Right ventricular (RV) pressure (RVP),
right ventricular hypertrophy index (RVHI) and pulmonary structural
remodeling measurements
RVP, including systolic right ventricular pressure
(SRVP), diastolic right ventricular pressure (DRVP) and mean right
ventricular pressure (MRVP) were measured 11 weeks after surgery
using a cardiac function analyzer (MP160; Bipoac Systems, Inc.) as
previously described (16,17). Weights of the RV and left ventricle
(LV) with the septum (S) of the hearts were measured after
sacrifice. RVHI was calculated using the following formula:
RVHI=(RV)/(LV+S). Pulmonary arteriole tissue was isolated and fixed
in 10% paraformaldehyde at room temperature (RT) for 2 h,
dehydrated in a graded alcohol series, cleared with xylene and
embedded in paraffin. Tissue was then cut into 5 µm sections.
Hematoxylin and eosin (H&E) staining was performed to observe
pulmonary structural remodeling. All slides were imaged using a
video-linked light microscope (magnification x100; Olympus CX31;
Olympus Corporation) and analyzed using the ImageJ software
(version 1.49p; National Institutes of Health).
PASMC isolation and culture
Primary PASMCs from control, sham and shunt groups
were isolated and cultured for in vitro assessment as
previously described (16,17). Pulmonary arteriole tissue was removed
and immersed in ice-cold HEPES-buffered salt solution. The
endothelium was removed by rubbing the luminal surface using a
cotton swab. Tissue was then cut into 1x1 mm pieces and the
arterioles were digested at 37˚C for 20 min in HBSS containing 20
µmol Ca2+, 1,750 U/ml type I collagenase, 9.6 U/ml
papain, 20% fetal bovine serum (FBS; Thermo Fisher Scientific,
Inc.) and 1 mM dithiothreitol. A total of 3-4x104 cells
were seeded into a 25T culture flask and placed in a 5%
CO2 incubator, maintained at 37˚C. The culture medium
with DMEM containing 10% FBS was refreshed 5 days following culture
and ~80% confluence was typically achieved on day 9, where cell
passage was performed until the passage nine.
Immunocytochemical staining
PASMCs were trypsinized and suspended using a
pipette onto a circular coverslip in a 6-well plate. After rinsing
with PBS three times, PASMCs were fixed with 4% paraformaldehyde
for 15 min at RT. Following permeabilization in a 0.2% Triton X-100
solution, 1% bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA)
was used for blocking for 30 min at RT. The coverslips were
incubated with primary antibodies against α-actin (1:100, cat. no.
sc-32251; Santa Cruz Biotechnology, Inc.) for 8 h at RT. Secondary
antibodies were subsequently added in a dropwise manner, and the
mixture was incubated at room temperature for 30 min. The
coverslips were then incubated with diaminobenzidine,
counterstained with hematoxylin for 5 sec at RT and differentiated
using hydrochloric acid/alcohol before being sealed. A video-linked
light microscope (magnification, x100; Olympus CX31; Olympus
Corporation) was used for imaging the coverslips.
Lentiviral siRNA infection of
PASMCs
Lentiviruses (LV-TMEM16A-siRNA and control siRNA)
enconding green fluorescence protein (GFP) were purchased from
Shanghai Jikai Gene Chemical Technology Co., Ltd., and PASMCs were
transfected as previously described (17). Puromycin was used for cell screening.
PASMCs from the shunt group were digested with 0.25% trypsin and
suspended at a density of 2x104/ml prior to seeding into
a 6-well plate on day 1 after culture. Lentiviral particles
prepared in a 2 ml suspension were then added after 24 h to achieve
a multiplicity of infection of 80 with the final concentration of
Polybrene maintained at 5 µg/ml. The culture solution was changed
after 36 h and cells were cultured in a 37˚C, 5% CO2
incubator for a further 3 days before subsequent experiments. For
in vitro studies, PASMCs were divided into control, shunt,
shunt + siRNA-TMEM16A and shunt + siRNA-negative control
groups.
Flow cytometry
When the cells grew to cover ~85% of the flask, they
were harvested, centrifuged (5 min, 175 x g) at RT and washed twice
with 3 ml PBS. 70% ethanol was used for cell fixation for 1 h at
4˚C. Cells were then centrifuged at 850 x g for 5 min at RT and
washed twice with 3 ml PBS. Propidium iodide (PI; 500 µl) was added
for staining according to the manufacturer's protocol (cat. no.
550825; BD Biosciences). The proportions of cells in
G0/G1 phase, S and G2/M phases
were measured using flow cytometry (Guava®
easyCyte™ 6-2L; Merck KGaA) and the
guavaSoft™ software (version 3.1.1; Merck KGaA).
Western blotting
Proteins were homogenized or lysed in ice-cold RIPA
lysis buffer (Santa Cruz Biotechnology, Inc.). The concentration of
the protein was examined using Bradford protein assay kit (Bio-Rad
Laboratories, Inc.). In total, 20 µg proteins were separated by 10%
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred to nitrocellulose membranes (Bio-Rad
Laboratories, Inc.). The membranes were blocked in Tris-buffered
saline with 5% non-fat milk and 0.5% BSA for 1 h at RT as
previously described (17).
Membranes with corresponding proteins were incubated with primary
antibodies against TMEM16A (1:1,000; cat. no. ab53213; Abcam),
cyclin D1 (1:1,000; cat. no. 2978; Cell Signaling Technology,
Inc.), cyclin-dependent kinase 2 (CDK2; 1:1,000; cat. no. 2546;
Cell Signaling Technology, Inc.), cyclin-dependent kinase inhibitor
P27 (p27KIP; 1:1,000; cat. no. 3686; Cell Signaling Technology,
Inc.), cyclin E1 (1:1,000; cat. no. 20808; Cell Signaling
Technology, Inc.) and β-actin (1:5,000, cat. no. 21338; SAB
Biotherapeutics, Inc.) overnight at 4˚C. The membranes were then
incubated with a horseradish peroxidase-conjugated goat anti-rabbit
IgG secondary antibody (1:5,000; cat. no. L3012, SAB
Biotherapeutics, Inc.) for 1.5 h at RT, and washed four times using
0.1% Tween-20 solution. Blots were visualized with
SuperSignalTM West Femto Maximum Sensitivity Substrate
(Thermo Fisher Scientific, Inc.) and quantified with Quantity 5.2
software System (Bio-Rad Laboratories, Inc.).
Statistical analysis
SPSS 22.0 software (IBM Corp.) was used to analyze
the data. Data are expressed as the mean ± standard deviation. A
two-sample t-test was used for comparisons between two groups.
One-way ANOVA was used for comparisons between groups followed by
Turkey's multiple comparisons. All hypotheses were tested
bilaterally. P<0.05 was considered to indicate a statistically
significant difference.
Results
Establishment of high blood
flow-induced PAH and structural remodeling
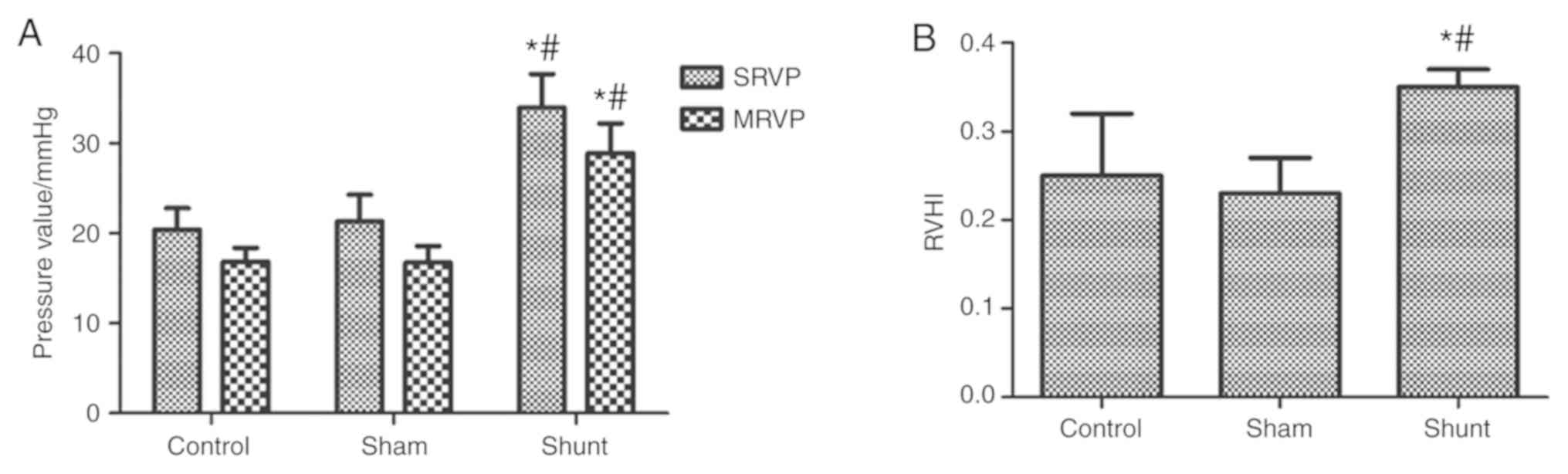
After 11 weeks of shunting, SRVP and MRVP were
significantly higher in the shunt group compared with the sham
group, whereas no statistically significant differences were
observed between the control and sham groups (Fig. 1A). Similarly, RVHI in the shunt group
was significantly higher compared with the control and sham groups,
but no significant differences were observed in the RVHI between
the control and sham groups (Fig.
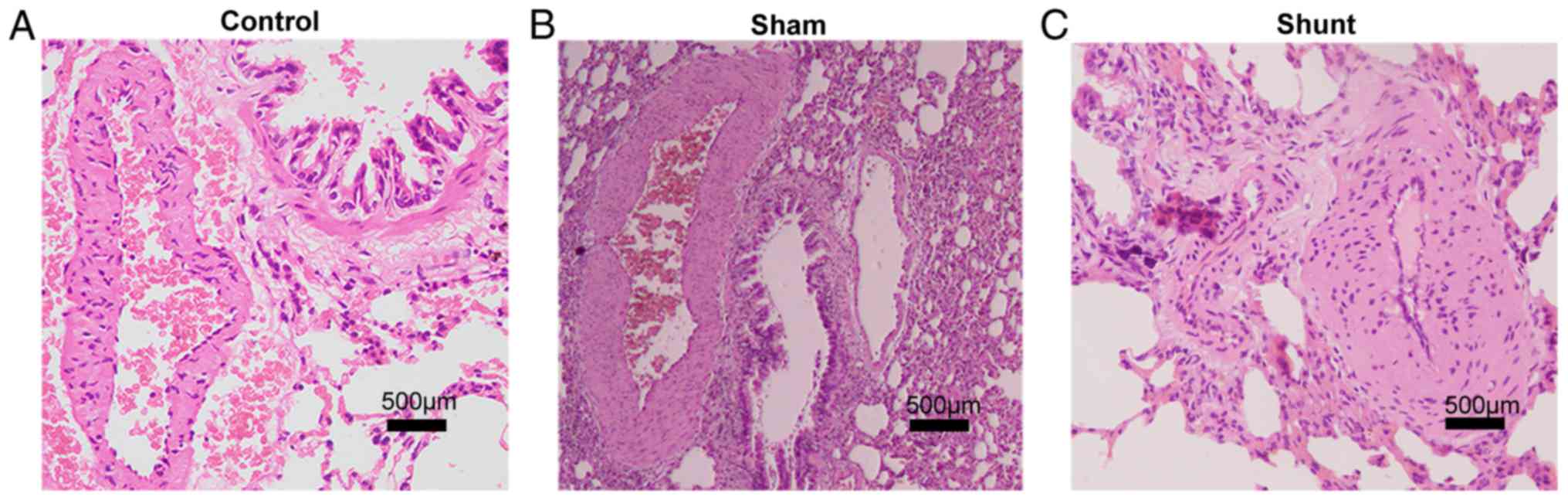
1B). Histological staining demonstrated marked thickening of
the pulmonary arteriole walls in the shunt group that was
accompanied with narrowing of the lumen, which was not observed in
the control and sham groups (Fig.
2).
Characteristics and transfection of
lentiviral TMEM16A-siRNA transfection in PASMCs
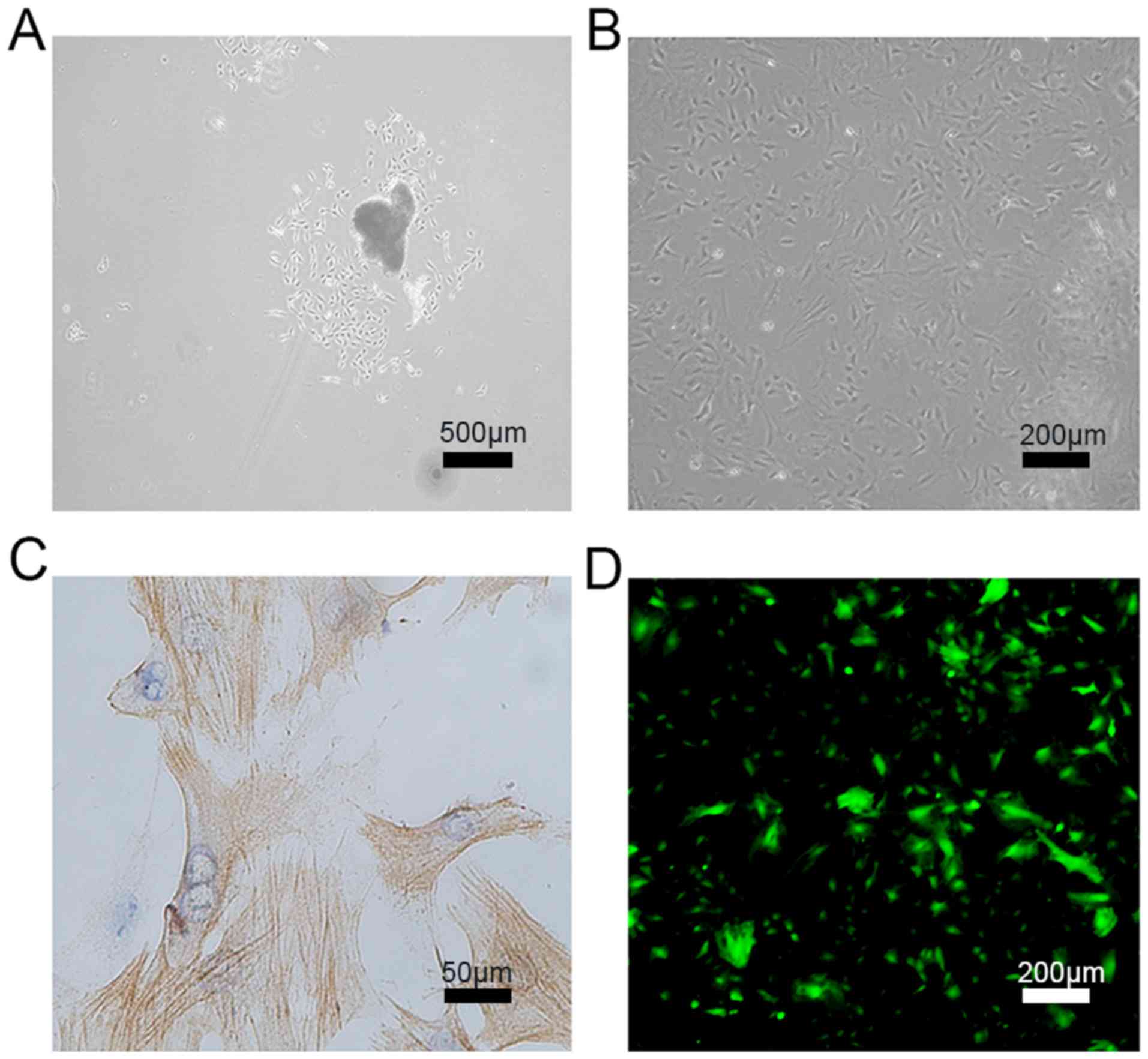
Cultured PASMCs in all three groups (Control, sham
and shunt) grew and aggregated at 3-5 days after seeding (Fig. 3A), and colonies formed on ~day 10.
Typically, spindle-shaped cells start to divide and extend on the
culture plate in a dispersed pattern, with some colonies forming
clusters (Fig. 3B).
Immunocytochemical staining confirmed the expression of α-smooth
muscle actin based on the specific myofilament structure of PASMCs
(Fig. 3C). A lentiviral vector
specifically expressing TMEM16A-siRNA or the corresponding scramble
control vector was transfected into PASMCs from the shunt group.
Cells were screened using 3 mg/l puromycin and GFP fluorescence was
observed using an inverted fluorescence phase contrast microscope
(Fig. 3D). Flow cytometry was also
used to measure lentiviral transfection efficiency, which was found
to be >80% (data not shown).
TMEM16A expression is upregulated in
PASMCs following PAH induction
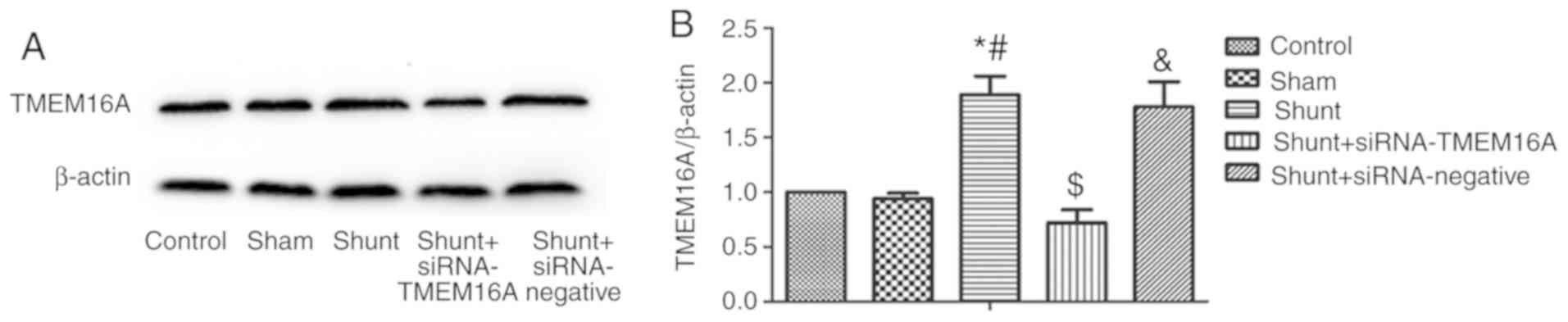
The expression of TMEM16A in PASMCs was
significantly higher in the shunt group compared with the control
and sham groups, whereas no significant differences were observed
between the control and sham groups (Fig. 4A and B). Transfection of PASMCs with lentiviral
TMEM16A-siRNA significantly reduced TMEM16A expression in the shunt
group compared with non-transfected cells from the same group, but
not in cells transfected with the scramble control siRNA (Fig. 4A and B).
TMEM16A may regulate cell cycle
progression in PASMC following PAH induction
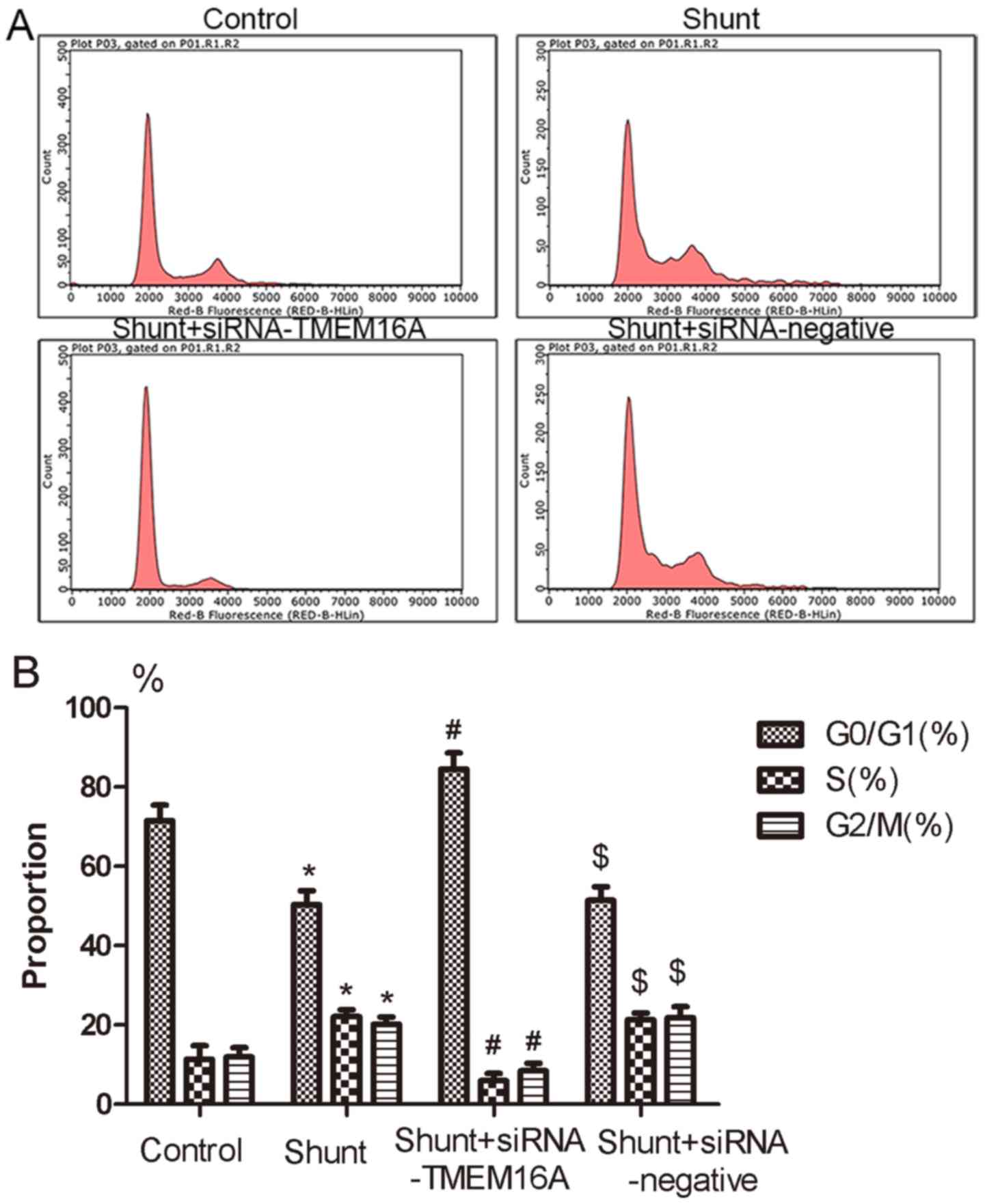
To examine the TMEM16A-mediated regulation of PASMC
cell cycle progression, the ratios of cells in
G0/G1, S and G2/M phases were
measured via flow cytometry. The ratio of cells in the
G0/G1 phase was significantly decreased in
PASMCs in the shunt group compared with the control group, and
TMEM16A-siRNA abolished this increase (Fig. 5A and B). The ratios of S and G2/M in
PASMCs from the shunt group was significantly increased compared
with the control group, which was also reversed by TMEM16A-siRNA
transfection but not by the negative control siRNA (Fig. 5A and B). These data suggest that TMEM16A may be
involved in cell cycle progression regulation in PAH induction.
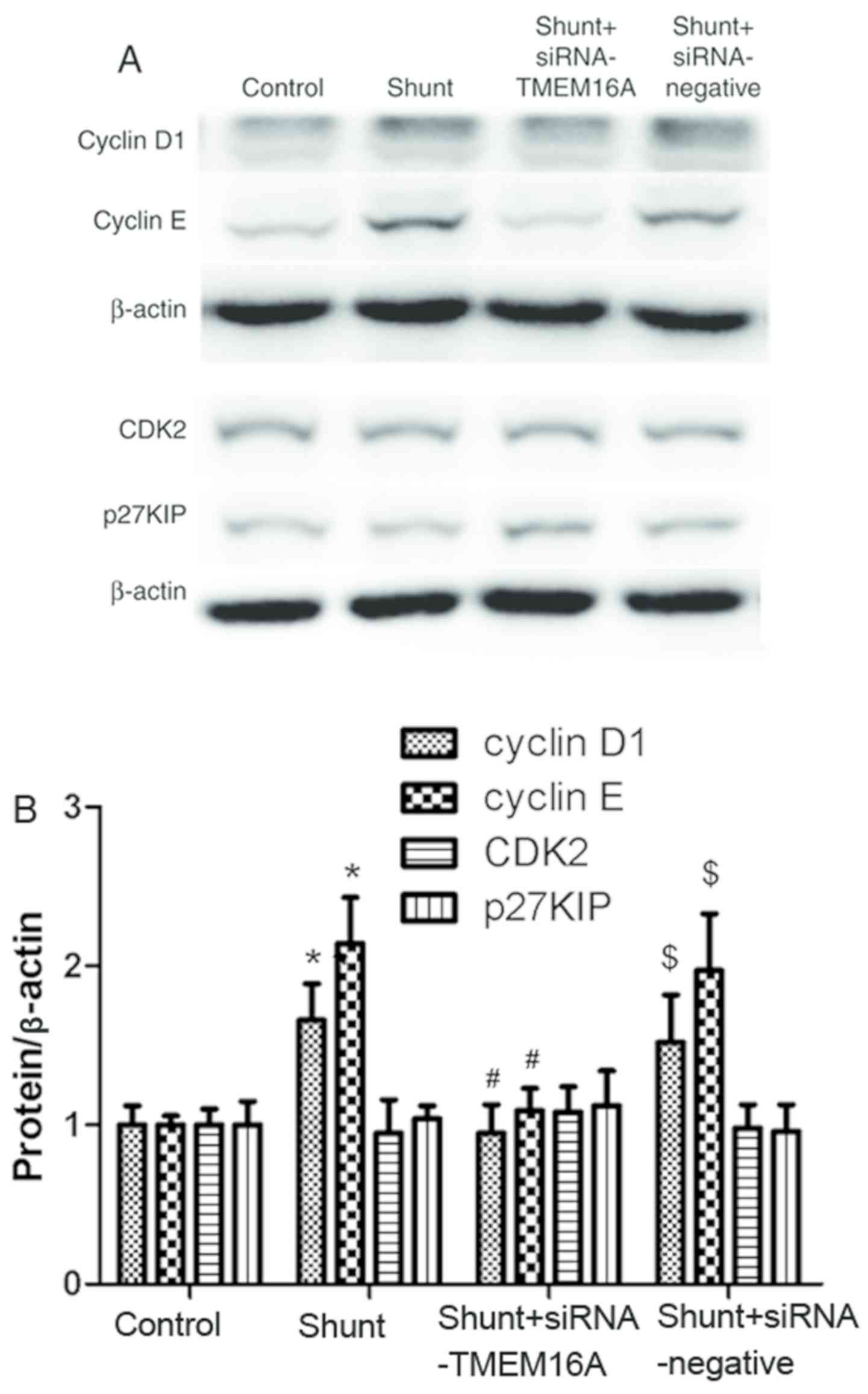
TMEM16A contributes to the regulation
of proteins associated with the PASMC cell cycle
To clarify the potential mechanism of TMEM16A in PAH
induced by high blood flow, the expression of proteins associated
with cell cycle regulation, including cyclin D1, cyclin E, CDK2 and
p27KIP, in PASMCs from the different experimental groups was
detected. The expression of cyclin D1 and cyclin E in the shunt
group was significantly higher compared with the control group,
which was reversed by TMEM16A-siRNA transfection. There were no
significant differences in CDK2 or p27KIP expression between the
control and shunt groups (Fig. 6).
Taken together, the results indicated that TMEM16A might contribute
to the regulation of proteins associated with cell cycle
progression in PASMCs.
Discussion
High pulmonary blood flow-induced PAH is one of the
most common causes of PAH during childhood, the mechanism of which
has not been fully elucidated. Under aberrant levels of mechanical
shear stress caused by excessive blood flow, pulmonary arteriole
endothelial and smooth muscle cells undergo progressive
dysfunction. A previous study (16,18) in
our laboratory in the Pediatric department, The First Affiliated
Hospital of Guangxi Medical University (Nanning, China)
demonstrated that CaCCs contribute to elevated pulmonary artery
pressure and structural remodeling in PAH induced by high pulmonary
flow. TMEM16A was reported as a candidate for the molecular basis
of CaCCs in 2008 (8-10),
and there is accumulating evidence revealing that TMEM16A is
involved in a variety of physical and pathophysiological processes
(15,19-22).
However, the mechanism of TMEM16A regulation of PASMCs in PAH
induced by high blood flow remains unclear. The present study
established an animal model of high blood flow-induced PAH, which
increased TMEM16A expression accompanied with pulmonary structural
remodeling. In particular, TMEM16A may contribute to PAH induced by
high blood flow by regulating the cell cycle progression of
PASMCs.
Although TMEM16A has been extensively studied in
cardiovascular diseases (15,19-21,23-25),
conflicting results have been reported. Whilst certain studies have
determined that the TMEM16A protein activated CaCCs in PASMC
proliferation and spontaneous hypertension (15,19,20),
others have indicated that TMEM16A served an inhibitory role in the
regulation of PASMC cell proliferation (21,25).
During the process of cerebral vascular remodeling of hypertension
(21), the reduced expression of
TMEM16A promoted G1-S phase cell cycle progression in
cerebral arterial smooth muscle cells, indicating that TMEM16A is a
negative regulator of cell proliferation in vascular smooth muscle
cells. Expression of TMEM16A in the pulmonary arterial smooth
muscle of rats has been demonstrated to be upregulated in
monocrotaline-induced pulmonary hypertension (26) and chronic hypoxia-induced pulmonary
hypertension (16).
TMEM16A serves a vital role in the regulation of
cell proliferation. The effect of TMEM16A on cell proliferation has
varied in previous studies, and the reason for these contradictory
results have not reached a unified conclusion. In an angiotensin II
treatment-induced hypertension model, TMEM16A expression in basilar
smooth muscle cells (BASMCs) was reduced (21), which was accompanied by an increased
BASMC proliferation. Increased TMEM16A expression also promoted
portal vein smooth muscle cell (PVSMC) proliferation in
vitro (22), which is consistent
with the results of the present study. TMEM16A was reported to be
downregulated in PVSMCs from a rat model of portal hypertension,
which resulted in increased PVSMC proliferation, indicating that
angiotensin II may be a candidate regulatory factor for PASMC
proliferation mediated by TMEM16A (22). In congenital hypertensive rats
(20), hypoxia-induced and
monocrotaline-induced pulmonary hypertension models (14,15), the
overexpression of TMEM16A promoted the proliferation of PASMCs. The
present study determined that TMEM16A expression was upregulated in
PASMCs from PAH induced by high blood flow.
Previous experiments have demonstrated that TMEM16A
is closely associated with the expression of some cyclins (27,28). In
the present study it was determined that the expression of cyclin E
and cyclin D1 increased in PASMCs from high blood flow-induced PAH.
Cyclin D1 promotes cell cycle progression from the G1
phase to the S phase, whereas cyclin E is essential for the normal
development of the cell cycle (27).
The complex of cyclin E and CDK2 (Cyclin E-CDK2) serves a crucial
role in the process of entering the S phase from the G1
phase, and it is a key kinase complex regulator of the cell cycle.
The overexpression of cyclin D1 may shorten the time required to
enter S phase and accelerate the G1/S transition
process, leading to cell proliferation (28). The siRNA-TMEM16A lentivirus was used
to infect PASMCs isolated from rats that underwent PAH injury,
where the decrease in cyclin D1 expression lead to cell cycle
arrest at the G0-G1 phase, which inhibited
cell proliferation and pulmonary vascular remodeling (29). The present study demonstrated
increased expressions of cyclin D1 and cyclin E in PASMCs from PAH
models, which was consistent with the results of a previous study
(21). The p27 KIP family protein is
a regulator of the cyclinE-CDK2 complex that inhibits cell
proliferation by inhibiting cyclin E expression (30). As a negative regulatory factor of the
cyclin E-CDK2 complex, the expression of p27 KIP was not affected
by PAH, which is consistent with a previous study by Wang et
al (21). In present study,
although the expression of cyclin E in the PASMCs from shunt group
increased compared with control group, the expression of CDK2
complexed to cyclin E in the shunt group did not change
significantly compared with control PASMCs. The potential reason
for this observation may be that the upregulation of TMEM16A during
high pulmonary blood flow-induced PASMCs injury affected the
expression of cyclin E and cyclin D1 via another paracrine
mechanism without affecting the expression of p27 KIP and CDK2. It
is also possible that the expression of CDK2 and p27 KIP remained
relatively stable inside the cells but existed in an inactive form
or were in complex with other proteins.
The present study has certain limitations. The
recorded right heart pressure was mild and was limited to an early
stage of high blood flow-induced PAH. In addition, studies
addressing the TMEM16A-mediated regulation of protein expression
that is associated with cell cycle progression, particularly the
effect of TMEM16A knockout in vivo, are urgently required.
Finally, specific signaling pathways, including ERK/cyclin
D1(31) and/or cyclin D1-MARK
(32-34),
serve as potential targets for future studies.
In conclusion, TMEM16A may be involved in high
pulmonary blood flow-induced PAH by regulating cell cycle
progression in PASMCs. TMEM16A may therefore serves as a novel
therapeutic target for high pulmonary flow-induced PAH.
Acknowledgements
Not applicable.
Funding
The study was supported in part by the Natural
Science Foundation of China (grant no. NSFC 81160040); Guangxi
Natural Science Foundation (grant no. 2013GXNSFAA019177); Natural
Science Foundation of Zhejiang Province (grant no. LY16H020010);
Medicine Health Science and Technology Plan of Zhejiang Province
(grant no. 2017194804) and Wenzhou Science and Technology Bureau,
P.R. China (grant no. Y20160021).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LS and KW performed experiments and wrote the
manuscript. DL, SQ, JH, YZ and YP contributed to data
interpretation and discussion. YP conceived and directed the
project, and edited the manuscript. YP is the guarantor of this
project and, as such, has full access to all the data in the study
and take responsibility for the integrity of the data and the
accuracy of the data analysis. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of Guangxi Medical University (Nanning, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu Y, Chen S, Zühlke L, Black GC, Choy
MK, Li N and Keavney BD: Global birth prevalence of congenital
heart defects 1970-2017: Updated systematic review and
meta-analysis of 260 studies. Int J Epidemiol. 48:455–463.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li L, Jick S, Breitenstein S, Hernandez G,
Michel A and Vizcaya D: Pulmonary arterial hypertension in the USA:
An epidemiological study in a large insured pediatric population.
Pulm Circ. 7:126–136. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Rubin LJ: Primary pulmonary hypertension.
N Engl J Med. 336:111–117. 1997.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Huang F, Wong X and Jan LY: International
Union of Basic and Clinical Pharmacology. LXXXV: Calcium-activated
chloride channels. Pharmacol Rev. 64:1–15. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hübner CA, Schroeder BC and Ehmke H:
Regulation of vascular tone and arterial blood pressure: Role of
chloride transport in vascular smooth muscle. Pflugers Arch.
467:605–614. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang Z, Zhang Z, Xu Y, Li Y and Ye T:
Relationship of intracellular free Ca2+ concentration
and calcium-activated chloride channels of pulmonary artery smooth
muscle cells in rats under hypoxic conditions. J Huazhong Univ Sci
Technolog Med Sci. 26:172–174, 191. 2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zaiman AL, Damico R, Thoms-Chesley A,
Files DC, Kesari P, Johnston L, Swaim M, Mozammel S, Myers AC,
Halushka M, et al: A critical role for the protein apoptosis
repressor with caspase recruitment domain in hypoxia-induced
pulmonary hypertension. Circulation. 124:2533–2542. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Caputo A, Caci E, Ferrera L, Pedemonte N,
Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O and
Galietta LJ: TMEM16A, a membrane protein associated with
calcium-dependent chloride channel activity. Science. 322:590–594.
2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Schroeder BC, Cheng T, Jan YN and Jan LY:
Expression cloning of TMEM16A as a calcium-activated chloride
channel subunit. Cell. 134:1019–1029. 2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yang YD, Cho H, Koo JY, Tak MH, Cho Y,
Shim WS, Park SP, Lee J, Lee B, Kim BM, et al: TMEM16A confers
receptor-activated calcium-dependent chloride conductance. Nature.
455:1210–1215. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tian XQ, Ma KT, Wang XW, Wang Y, Guo ZK
and Si JQ: Effects of the calcium-activated chloride channel
inhibitors T16Ainh-A01 and CaCCinh-A01 on cardiac fibroblast
function. Cell Physiol Biochem. 49:706–716. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hyuga S, Danielsson J, Vink J, Fu XW,
Wapner R and Gallos G: Functional comparison of anoctamin 1
antagonists on human uterine smooth muscle contractility and
excitability. J Smooth Muscle Res. 54:28–42. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Martinez-Pinna J, Soriano S, Tuduri E,
Nadal A and de Castro F: A calcium-dependent chloride current
increases repetitive firing in mouse sympathetic neurons. Front
Physiol. 9(508)2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Revermann M, Schloss M, Mieth A, Babelova
A, Schröder K, Neofitidou S, Buerkl J, Kirschning T, Schermuly RT,
Hofstetter C and Brandes RP: Levosimendan attenuates pulmonary
vascular remodeling. Intensive Care Med. 37:1368–1377.
2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sun H, Xia Y, Paudel O, Yang XR and Sham
JS: Chronic hypoxia-induced upregulation of Ca2+-activated Cl-
channel in pulmonary arterial myocytes: A mechanism contributing to
enhanced vasoreactivity. J Physiol. 590:3507–3521. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang K, Ma J, Pang Y, Lao J, Pan X, Tang
Q, Zhang F, Su D, Qin S and Shrestha AP: Niflumic acid attenuated
pulmonary artery tone and vascular structural remodeling of
pulmonary arterial hypertension induced by high pulmonary blood
flow in vivo. J Cardiovasc Pharmacol. 66:383–391. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lao J, Pang Y, Wang K, Liu D, Su D, Zhang
F, Pan X and Li S: Inhibition of survivin promotes pulmonary
arterial smooth muscle cell apoptosis in high blood flow-induced
pulmonary arterial hypertension in rats. Int J Clin Exp Pathol.
9:6821–6834. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang K, Chen C, Ma J, Lao J and Pang Y:
Contribution of calcium-activated chloride channel to elevated
pulmonary artery pressure in pulmonary arterial hypertension
induced by high pulmonary blood flow. Int J Clin Exp Pathol.
8:146–154. 2015.PubMed/NCBI
|
|
19
|
Manoury B, Tamuleviciute A and Tammaro P:
TMEM16A/anoctamin 1 protein mediates calcium-activated chloride
currents in pulmonary arterial smooth muscle cells. J Physiol.
588:2305–2314. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wang B, Li C, Huai R and Qu Z:
Overexpression of ANO1/TMEM16A, an arterial
Ca2+-activated Cl- channel, contributes to spontaneous
hypertension. J Mol Cell Cardiol. 82:22–32. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang M, Yang H, Zheng LY, Zhang Z, Tang
YB, Wang GL, Du YH, Lv XF, Liu J, Zhou JG and Guan YY:
Downregulation of TMEM16A calcium-activated chloride channel
contributes to cerebrovascular remodeling during hypertension by
promoting basilar smooth muscle cell proliferation. Circulation.
125:697–707. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zeng X, Huang P, Chen M, Liu S, Wu N, Wang
F and Zhang J: TMEM16A regulates portal vein smooth muscle cell
proliferation in portal hypertension. Exp Ther Med. 15:1062–1068.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Dai WJ, Qiu J, Sun J, Ma CL, Huang N,
Jiang Y, Zeng J, Ren BC, Li WC and Li YH: Downregulation of
microRNA-9 reduces inflammatory response and fibroblast
proliferation in mice with idiopathic pulmonary fibrosis through
the ANO1-mediated TGF-β-Smad3 pathway. J Cell Physiol.
234:2552–2565. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wu H, Wang H, Guan S, Zhang J, Chen Q,
Wang X, Ma K, Zhao P, Zhao H, Yao W, et al: Cell-specific
regulation of proliferation by Ano1/TMEM16A in breast cancer with
different ER, PR, and HER2 status. Oncotarget. 8:84996–85013.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang XH, Zheng B, Yang Z, He M, Yue LY,
Zhang RN, Zhang M, Zhang W, Zhang X and Wen JK: TMEM16A and
myocardin form a positive feedback loop that is disrupted by KLF5
during Ang II-induced vascular remodeling. Hypertension.
66:412–421. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Forrest AS, Joyce TC, Huebner ML, Ayon RJ,
Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, et al:
Increased TMEM16A-encoded calcium-activated chloride channel
activity is associated with pulmonary hypertension. Am J Physiol
Cell Physiol. 303:C1229–C1243. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Deng L, Yang J, Chen H, Ma B, Pan K, Su C,
Xu F and Zhang J: Knockdown of TMEM16A suppressed MAPK and
inhibited cell proliferation and migration in hepatocellular
carcinoma. Onco Targets Ther. 9:325–333. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu J, Liu Y, Ren Y, Kang L and Zhang L:
Transmembrane protein with unknown function 16A overexpression
promotes glioma formation through the nuclear factor-κB signaling
pathway. Mol Med Rep. 9:1068–1074. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zeng DX, Xu GP, Lei W, Wang R, Wang CG and
Huang JA: Suppression of cyclin D1 by plasmid-based short hairpin
RNA ameliorated experimental pulmonary vascular remodeling.
Microvasc Res. 90:144–149. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu P and Chen L: Inhibition of sonic
hedgehog signaling blocks cell migration and growth but induces
apoptosis via suppression of FOXQ1 in natural killer/T-cell
lymphoma. Leuk Res. 64:1–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li T, Song T, Ni L, Yang G, Song X, Wu L,
Liu B and Liu C: The p-ERK-p-c-Jun-cyclinD1 pathway is involved in
proliferation of smooth muscle cells after exposure to cigarette
smoke extract. Biochem Biophys Res Commun. 453:316–320.
2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Qin L, Yang YB, Yang YX, Gong YZ, Li XL,
Li GY, Luo HD, Xie XJ, Zheng XL and Liao DF: Inhibition of smooth
muscle cell proliferation by ezetimibe via the cyclin D1-MAPK
pathway. J Pharmacol Sci. 125:283–291. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Duvvuri U, Shiwarski DJ, Xiao D, Bertrand
C, Huang X, Edinger RS, Rock JR, Harfe BD, Henson BJ, Kunzelmann K,
et al: TMEM16A induces MAPK and contributes directly to
tumorigenesis and cancer progression. Cancer Res. 72:3270–3281.
2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Song Y, Gao J, Guan L, Chen X, Gao J and
Wang K: Inhibition of ANO1/TMEM16A induces apoptosis in human
prostate carcinoma cells by activating TNF-α signaling. Cell Death
Dis. 9(703)2018.PubMed/NCBI View Article : Google Scholar
|