Introduction
The condition 3-methylglutaconic aciduria (3-MGA)
with deafness, encephalopathy and Leigh-like (MEGDEL) syndrome,
also known as type IV 3-MGA [Mendelian of Inheritance in Man (MIM)
#614739], is associated with a heterogeneous group of disorders,
including MGA, deafness, encephalopathy and Leigh-like lesions
(1) as observed on MRI scans,
progressive spasticity, dystonia (2-6),
brain atrophy and neonatal liver disease (4), followed by increasing levels of
enzymatic activity in the liver (7).
Type IV 3-MGA, in contrast to the other types (I, II, III and V),
is based on undefined clinical subtypes and the underlying
aetiology has yet to be elucidated, even though affected
individuals have been indicated to have heterogeneous genotypes,
similar to those in the other groups (8,9). Since
elevated levels of liver enzymes have been observed to be a
phenotype characteristic of MEGDEL syndrome, the term ‘MEGDEL’ was
suggested in order to include all of the phenotypic disorders that
occur most frequently in affected patients (4). MEGDEL syndrome is a rare, recessive
inherited disorder, the fundamental underlying causes of which are
genetic impairments attributable to multiple complexities,
including DNA demolition syndromes and mitochondrial DNA deletion
syndromes, as well as mitochondrial myopathy, encephalopathy,
lactic acidosis and stroke syndrome. Dysregulation of 3-MGA in all
five types has been identified on the basis of genetic pathogenic
variants. Type I 3-MGA (MIM #250950) has been reported to result
from a mutation in the AU RNA binding methylglutaconyl-CoA
hydratase gene, which causes a deficiency in 3-methylglutaconyl-CoA
hydratase, the enzyme involved in L-leucine degradation (7). Type II 3-MGA, also known as Barth
syndrome (10), is characterized by
oxidative phosphorylation dysfunction, neutropenia and
cardiomyopathy, which lead to defective cardiolipin remodelling
(8,11) due to a mutation in the tafazzin (TAZ)
gene on chromosome X (12,13). The TAZ gene encodes the TAZ protein,
the enzymatic function of which is to regulate cardiolipin
remodelling in order to maintain its level in the inner
mitochondrial membrane (11,14,15).
Type III 3-MGA, also known as Costeff syndrome, is a rare
neurogenic autosomal recessive inherited disorder (16,17) that
is caused by mutations in the outer mitochondrial membrane lipid
metabolism regulator OPA3 gene, whose function remains to be fully
elucidated (18). Patients with this
syndrome have been reported to exhibit optic atrophy, followed by
motor deficits and ataxia (16).
Type V 3-MGA, also known as dilated cardiomyopathy with ataxia
syndrome (19), is an autosomal
recessive disorder with a defect in the DnaJ heat shock protein
family (Hsp40) member C19 gene that leads to deficiencies in muscle
strength, anaemia and growth retardation (19,20).
MEGDEL syndrome is caused by biallelic pathogenic variants in the
Serine active site-containing 1 (SERAC1) gene, which is located on
chromosome 6q25.3. SERAC1 protein, with the phenotypic MIM number
614725, belongs to the PGAG-like protein family, containing 654
amino acids and comprising a serine lipase domain which is
hypothesized to fulfil a crucial role in protein and mitochondrial
function. Furthermore, SERAC1 has a role in intracellular
cholesterol trafficking and anomalous functioning of the protein
affects phospholipid mechanisms of action (3). The protein is assumed to be responsible
for phospholipid exchange at the mitochondrial interface and
endoplasmic reticulum, and defects in this process lead to the
phenotypic abnormalities that are characteristic of MEGDEL
syndrome.
A range of pathogenic variants that have been
observed in patients with MEGDEL syndrome have been identified and
confirmed by exome sequencing and Sanger sequencing (4,5,21). Through these studies, the majority of
the SERAC1 gene pathogenic variants have been categorized as
frameshift, nonsense or missense pathogenic variants within or
upstream of the lipase domain (3,4,8).
The aim of the present study was to investigate the
genes involved in the onset of MEGDEL syndrome and elucidate the
association between novel variants and phenotypic outcome of the
disease. Since the genetic complexity of MEGDEL syndrome has yet to
be completely elucidated, detailed studies are required to identify
further pathogenic variants that lead to the phenotypic
characteristics of the disorder. In the present study, biochemical
analysis and MRI scans were performed to confirm the primary
diagnosis with MEGDEL syndrome. Whole exome sequencing (WES)
analyses were performed to identify pathogenic variants associated
with the syndrome and the mutations were analysed with respect to
the patients' family to confirm the pattern of inheritance. The
results obtained from the present study suggested that this
mutation influences the severity of the disease, causing severe
developmental delays. Compared to previous studies using brain MRI
of patients with MEGDEL syndrome representing bilateral basal
ganglia alterations and dystonia, the present study observed a
greater impact of the mutation on the brain lobes. This resulted in
cerebellar atrophy, corpus callosum thinning and lateral ventricle
expansion as observed using MRI.
Patients and methods
Patients
Two male siblings, born in 2013 and 2015, from
consanguineous Turkish parents were admitted to Biruni Hospital
(Istanbul, Turkey) with psychomotor retardation, spasticity,
dystonia and deafness.
Brain MRI scanning
MRI scans were performed for regular patient care
following admission to the hospital. Different pulse sequences were
used for the MRI procedure, and T1- and T2-weighted images were
available for the younger sibling.
WES by next-generation sequencing
(NGS) and subsequent bioinformatics analysis
WES was performed to analyse the coding exons and
the exon-intron boundaries of protein-coding genes. Genomic DNA
preparation, exome capture and Illumina® sequencing
(NextSeq500 platform) were performed according to the
manufacturer's instructions. In brief, genomic DNA (gDNA) was
extracted from the whole peripheral blood sample using an
Invitrogen® NextSeq500 iPrep PureLink gDNA blood kit
(Thermo Fisher Scientific, Inc.). The genomic library was prepared
using an Agilent SureSelect Target Enrichment system (Agilent
Technologies, Inc.). Enrichment of coding exons and flanking
intronic regions was performed using the Agilent SureSelect Human
All Exon V6 reagent (Agilent Technologies, Inc.), following the
manufacturer's protocol and as previously described (22), and sequencing was performed using an
Illumina® NextSeq500 system (Illumina, Inc.). Sequences
were mapped to the human genome (GRCh37/hg19) using the
Burrows-Wheeler Aligner (version 0.6.1; algorithm ‘BWA-SW’; default
parameters) to obtain targeted sequencing data. Variants with a
frequency >1% in the population were removed from the collected
data. Variants were subsequently annotated using Alamut®
Visual (a decision-support software dedicated to variant
diagnostics used by clinical and research molecular laboratories
worldwide; see https://www.interactive-biosoftware.com/alamut-visual/),
and the allele frequency was determined using the following
databases: The National Center for Biotechnology Information
database for nucleotide variations dbSNP (https://www.ncbi.nlm.nih.gov/snp), the exome
aggregation consortium (ExAC) and the 1000 Genomes Project
(https://www.internationalgenome.org/). Disease
causality was assessed using ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and exome
sequencing project (ESP) variants, ExAC variants and ESP
variants.
Sanger sequencing of the identified
mutation
DNA from the patients and the parents was isolated
from whole blood using the QIAamp DNA Blood Mini kit (Qiagen,
Inc.). Exon 10 of the SERAC1 gene were amplified using primer sets
(forwards, 5'-TCCAACCAAGAGCTAAGCAG-3'; and reverse, 5'-TGAA
CATATCATGAGGGGTAGAG-3') and MyTaq™ DNA Polymerases Mix (Bioline
Reagents Ltd.). PCR was performed with 40 ng of genomic DNA in 30
µl, with an initial denaturation at 94˚C for 3 min, 30 cycles of
94˚C for 30 sec, 55˚C for 45 sec and 72˚C for 2 min, followed by a
final extension at 72˚C for 10 min. The SERAC1 gene was sequenced
directly from purified PCR products using a BigDye Terminator Cycle
Sequencing kit (version 3.1; Applied Biosystems; Thermo Fisher
Scientific, Inc.) prior to analysis on an ABI 3130 automated DNA
sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Determination of biochemical
parameters
Standard biochemical and metabolic tests (see the
Results section for further details) were performed for the
patients during the first month of life (note that the second of
the siblings was admitted to the intensive care unit of the Biruni
Hospital 22 days following birth), and then subsequently at the
ages of 16 months and 3 years.
Computational analysis of protein
structure and severity of damage caused by mutation
Protein structure analysis was performed using
SWISS-MODEL exPASy (https://swissmodel.expasy.org/interactive) to
determine the effect of the mutation on the SERAC1 protein. In
addition, damage sensitivity of the mutation was analysed using
PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) to investigate
its role in the severity of the phenotypic characteristics. The
mutation taster program (www.mutationtaster.org/) was used to predict the
effects of the mutation on the protein.
Results
Patients data and biochemical
analysis
The proband, a male, was the second child of a
22-year-old mother who married a first-degree relative (cousin). He
was born in June 2013 with a neonatal weight of 2,900 g. The levels
of neonatal thyroid-stimulating hormone (TSH) and biotinidase were
normal and the phenylketonuria screening test was also negative on
the 5th day of life. When the infant was 22 days old; however, the
patient was admitted to an intensive care unit for 22 days due to
asphyxia after birth. The blood test performed on 20th day of age
revealed elevated levels of aspartate aminotransferase (AST),
bilirubin, chlorine, magnesium, sodium, potassium, creatine kinase,
lactate dehydrogenase (LDH) and TSH. At the time of the blood test,
the urea concentration was also within the normal range. At the age
of 16 months, the patient's biochemical analysis indicated an
elevated level of 3-MGA, which compelled physicians to perform
further investigations, including MRI scan. The patient was
indicated to have hearing loss at the age of 21 months and
strabismus was detected in the right eye. Furthermore, the clinical
biochemistry screen revealed that the levels of AST, alanine
aminotransferase, alkaline phosphatase and creatinine were above
the normal range. The biochemical test also revealed that the
lactate level was above the reference range. The TSH level,
however, was within the normal range during the same period.
Essential minerals, including sodium, potassium, calcium,
magnesium, phosphorus and cholesterol, were revealed to have higher
values compared with the reference range. At the same time, the
biochemistry test results suggested that the uric acid, total
protein, albumin and urea levels were below the normal range. The
lactic acid level was close to the normal range (4.5-19.8), a
result that differed from the previous test, and the level of
vitamin B12 was above the reference range. The ferritin level was
also below the reference range. The results of the biochemical
analysis performed subsequently, when the patient was 3 years old,
revealed that glucose and lactate levels were above their normal
limits. An investigation was performed using tandem mass
spectrometry, which determined that the arginine level was above
the reference range. Biochemistry analyses also revealed that the
creatine level was low, whereas the levels of AST and LDH were
higher compared with the reference values. The analysis of the
patient's organic acids revealed that the levels of
3-hydroxypropionic acid, pyruvic acid, 3-hydroxybutyric acid, ethyl
malonic acid, adipic acid, 2-hydroxyglutaric acid, methylglutaric
acid and 3-methylglutaconic acid in the patient's urine sample were
higher than the reference levels (Table
SI).
Brain MRI for diagnosis of
encephalopathy and neurological analysis
Electroencephalography was performed on the patient
to determine the level of epileptic activity in the occipital
region. Muscle biopsy revealed the presence of myopathic fibres in
muscle tissue. The results of the echocardiogram were normal.
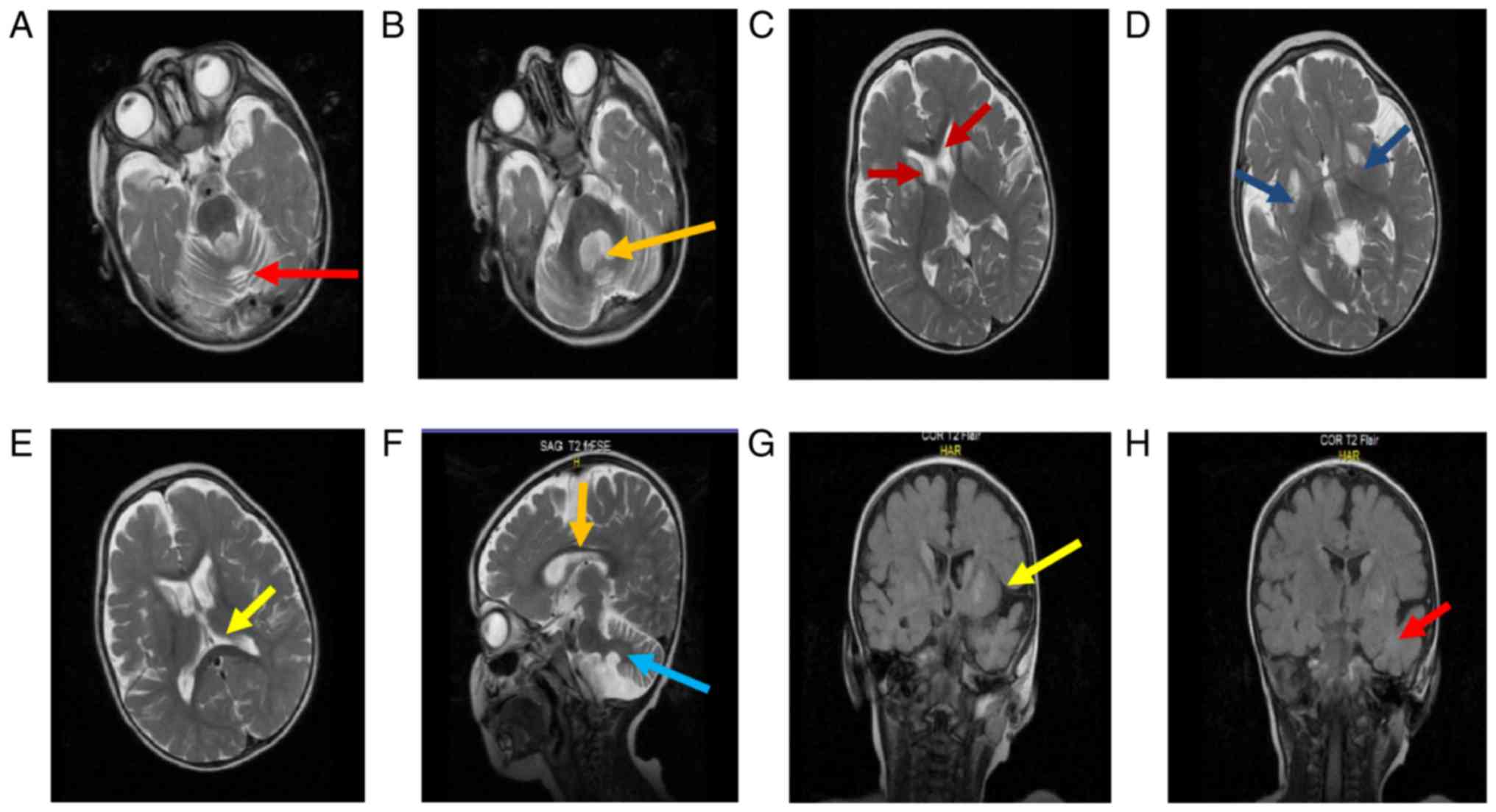
However, brain MRI scans of the patient revealed abnormalities in
different sections of the brain. Axial T2-weighted MRI revealed an
expansion of the cerebellar follicles and 4th ventricle, located in
the upper part of the medulla, causing cerebellar atrophy (Fig. 1A and B). In addition to the above observation,
increased intensities of the bilateral caudate nucleus (Fig. 1C) and bilateral symmetric signal
(Fig. 1D) were identified and
expansion of lateral ventricles had occurred (Fig. 1E), causing putamen volume loss.
Hypoplasia of the inferior vermis, observed in the sagittal
T2-weighted MRI scan, resulted in thinning and atrophy of the
corpus callosum (brain stem; Fig.
1F). Fluid-attenuated inversion recovery analysis of the
bilateral basal ganglia and temporal lobe revealed a signal
intensity that may be attributed to the involvement of white matter
(Fig. 1G and H).
Genetic analysis Determination of
pathogenic variants in the patient using NGS
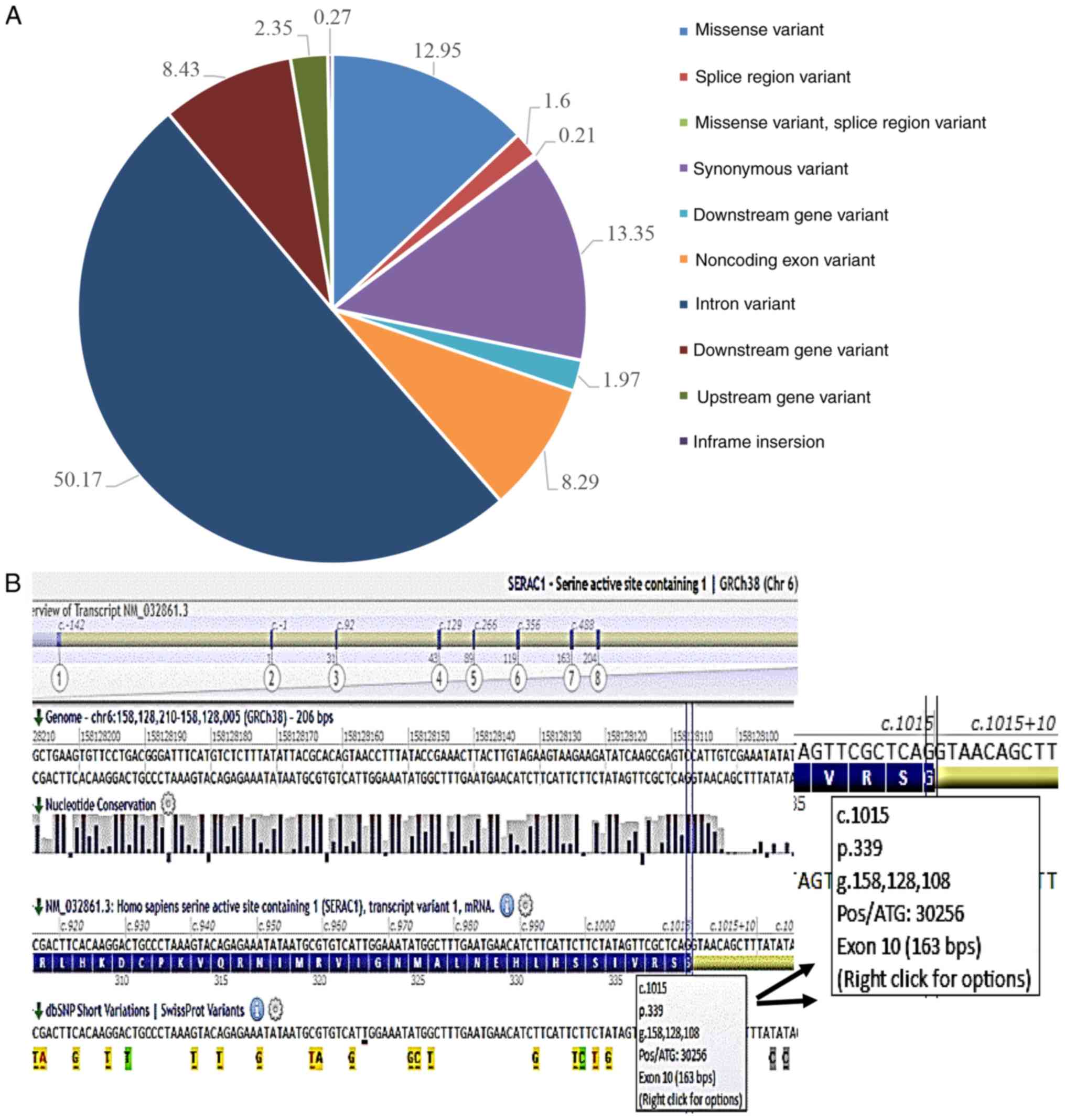
WES revealed 13,520 genomic variants, which were
filtered from >100 reads. Analysis of the distribution of types
and frequencies of sequence variants revealed that 50.17% were
intron variants, 12.95% were missense variants, 13.35% were
synonymous variants, 18.43% were downstream gene variants, 8.29%
were non-coding exon variants, 2.35% were upstream gene variants,
1.16% were splice region variants, 0.27% had in-frame insertions
and 0.21% were missense variant splice region variants (Fig. 2A). Amongst the variants, certain
pathogenic variants were identified, including NOC2-like nucleolar
associated transcriptional repressor [NOC2L; single nucleotide
variation (SNV), NM_015658.3:c.1443+455_1443+457delCTG], DFFB
(NM_004402.2:c.782+58_782+59 insCGGCCC), cyclin D kinase (CDK) 11B
(NM_033487.1:c.-167+273T>C), nephrocystin 4 (NPHP4;
NM_015102.3:c.3131G>A, NP_055917.1:p.Arg1044His) and SERAC1. WES
detected 13 different pathogenic variants in the SERAC1 gene, 11 of
which were indicated to be associated with the disease (Table I). Analysis of the 2 variants that
were detected in the SERAC1 gene using Alamut® Visual
software suggested that mutation-associated pathogenicity linked to
the disorder was present in a homozygous mutation at c.1015G>C
(p.Gly339Arg) (Fig. 2B).
 | Table IVariants detected in the serine active
site-containing 1 gene using whole-exome sequencing. |
Table I
Variants detected in the serine active
site-containing 1 gene using whole-exome sequencing.
| Mutation type | Reference sequence
mRNA (NM) | Mutation
location | Mutation type | Reference SNP
(rs) | Variant type |
|---|
| SNV | NM_032861.3 | c.1403+537C>G | Homozygous | rs977794 | Intron variant |
| SNV | NM_032861.3 |
c.1016-2838A>G | Homozygous | rs12523959 | Intron variant |
| SNVa | NM_032861.3 |
c.1015G>Cp.Gly339Arg | Homozygous | ? | Missense variant,
splice region variant |
| SNV | NM_032861.3 | c.738+6201C>T | Homozygous | rs12197370 | Intron variant |
| SNV | NM_032861.3 | c.738+4454A>C | Homozygous | rs844140 | Intron variant |
| SNV | NM_032861.3 | c.356-216C>T | Homozygous | rs9365928 | Intron variant |
| SNV | NM_032861.3 | c.355+300A>G | Homozygous | rs9356398 | Intron variant |
| SNV | NM_032861.3 | c.355+297C>T | Homozygous | rs9364823 | Intron variant |
| SNV | NM_032861.3 | c.355+11A>G | Homozygous | rs9356399 | Intron variant |
| SNV | NM_032861.3 | c.249C>T | Homozygous | rs6929274 | Synonymous
variant |
| SNV | NM_032861.3 | c.129-34C>G | Homozygous | rs6929520 | Intron variant |
| SNV | NM_032861.3 | c.92-334G>T | Heterozygous | ? | Intron variant |
| SNV | NM_032861.3 | c.-1-4619G>A | Homozygous | rs76958658 | Intron variant |
Identification of novel pathogenic
variants in SERAC1
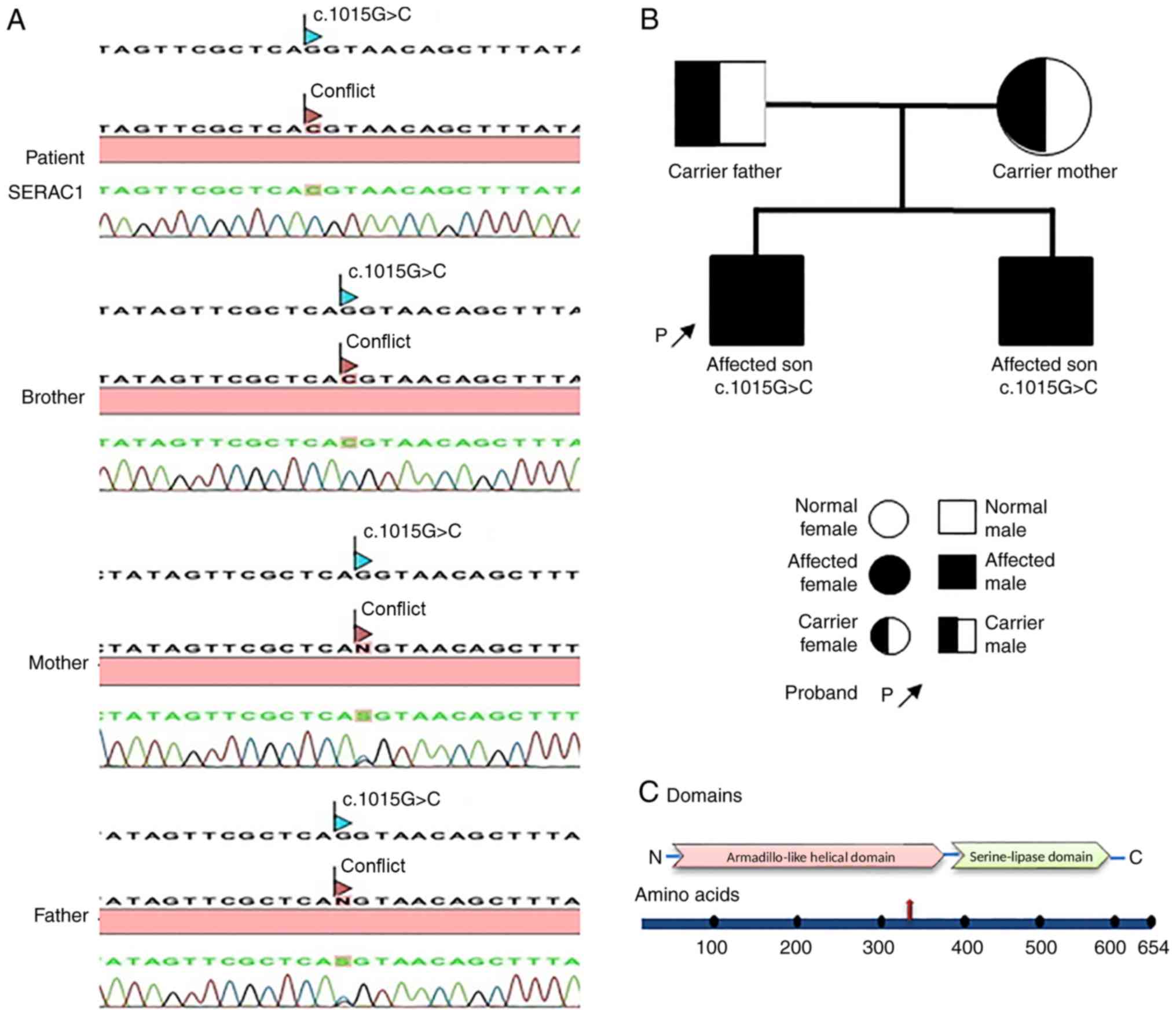
As the WES results revealed a novel SNV c.1015G>C
(p.Gly339Arg) mutation located on exon 10 of the SERAC1 gene in the
proband, confirmation analysis was performed on the patient to
determine the presence of the mutation. Patient genotype
confirmation and familial co-segregation analysis were performed.
All coding exons and exon-intron boundaries of the SERAC1 gene were
amplified using primer sets designed by a previous study (9). In order to confirm the presence of the
novel c.1015G>C (p.Gly339Arg) mutation located on exon 10 of the
SERAC1 gene and its inheritance pattern, co-segregation analysis
was performed on the proband, the proband's sibling and the parents
by Sanger sequencing. The results confirmed the pathogenic variants
to be homozygous in the proband and the proband's affected brother,
who inherited one mutant allele from each heterozygous parent
(Fig. 3A). Using sequencing data and
information collected from family members, the hereditary pattern
of MEGDEL syndrome in the family was illustrated in a pedigree
chart that was performed on the patients' and the family's blood
samples. The parents were both found to be a carrier while the
children both manifested the disease and were affected (Fig. 3B). Analysis of the mutation location
on the SERAC1 gene revealed the approximate position of the
mutation to be upstream in a serine-lipase domain (Fig. 3C), which was downstream of the
Armadillo-like helical domain. The mutation was predicted to be a
missense variant. However, the data that were collected from the
Alamut analysis revealed the position of the mutation to be at the
border of exon 10-intron 10 of the SERAC1 gene, and from this
analysis, it was predicted to be a splice-site variant. The
mutation, identified by PolyPhen2, was considered to be a causative
factor of abnormality in intracellular cholesterol trafficking. The
results of the bioinformatics protein structure analysis by
SWISS-MODEL exPASy revealed changes in the conformation of the
protein and in its binding sites, suggesting that the detected
mutation was located in the upstream lipase domain, and may cause
impaired protein-protein interaction (Fig. S1A and B). The analysis of the G339R substitution
by damage sensitivity program suggested that the mutation is likely
to be damaging, with a score of 1.000. Furthermore, the mutation
taster program suggested that the genetic impairment may cause
alterations in the function of the SERAC1 protein and is
disease-causing.
Discussion
Previous case studies identified pathogenic variants
associated with the phenotypes of MEGDEL syndrome. In the present
study, the characteristics of the disease were detected in both
siblings, including the proband. The present study investigated the
case of two siblings with 3-methylglutaconic aciduria and type IV
3-MGA with severe phenotypic characteristics of the disease in
order to study the responsible genetic alteration in depth
(2,4,21). To
date, ~70 cases of MEGDEL syndrome have been reported worldwide.
Since the clinical profiles of the affected siblings were entirely
compatible with the characteristics of MEGDEL syndrome, the SERAC1
gene was selected as a candidate for mutation screening amongst
genes, including NOC2L, DFFB, CDK11B and NPHP4, with certain
pathogenic variants. The detection of a novel homozygous frameshift
mutation in the SERAC1 gene confirmed the diagnosis of the syndrome
and indicated the association of this gene with the phenotypic
characteristics of MEGDEL syndrome.
Previous studies detected mutations in the
Armadillo-like helical domain, serine-lipase domain and
transmembrane domain of the SERAC1 protein, indicating the
significance of each domain's function for protein stability
(1,3). The present study demonstrated that the
location of the patient's mutation was upstream of the lipase
domain and the localization of the mutation markedly increased
damage sensitivity, causing SERAC1 protein dysfunction. Therefore,
it may be inferred that this impairment has a crucial role in the
onset of symptoms. Furthermore, the information obtained from the
presence of a conserved lipase domain having the consensus lipase
motif of GXSXG provides evidence highlighting the function of the
serine lipase domain in lipid metabolism (3). Although MEGDEL syndrome is a
phospholipid-remodelling disorder, it may also be classified as a
disorder of intracellular cholesterol trafficking.
To summarize, the detailed investigation of the
present study confirmed a novel mutation in the SERAC1 gene in the
affected pedigree. This mutation is suggested to cause protein
conformational changes that consequently result in protein
dysfunction, which may lead to abnormalities in intracellular
cholesterol trafficking. The present case has demonstrated several
phenotypic characteristics of MEGDEL syndrome with a novel SERAC1
mutation. The 3-dimensional protein structure analysis revealed
that the detected mutation may cause conformational changes in
protein and binding sites, affecting protein-protein interaction.
Furthermore, the other novel genetic alterations detected in the
present case study may be responsible for the intense phenotypic
characteristics of MEGDEL syndrome and require further
investigation, which may lead to novel insight regarding the
association of these pathogenic variants with the syndrome.
Supplementary Material
Conformational changes of the SERAC1
protein structure. (A) The 3-dimensional structure of the wild-type
SERAC1 protein and (B) accessibility of its binding site at
position 339 with glycine, which demonstrates marked conformational
changes where glycine is substituted with arginine. The arrows
indicate the site where the mutation occurs. Serine-lipase domain
controls intracellular cholesterol trafficking and the mutation is
shown to be located near the domain at the border of exon 10-intron
10. SERAC1, serine active site-containing 1.
Biochemical analysis of the patient
with 3-methylglutaconic aciduria with deafness, encephalopathy and
Leigh-like syndrome.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MA performed the molecular genetic studies, and
bioinformatics analysis of WES; participated in the design of the
study; and drafted the manuscript. NK helped with the design of the
study, performed data analysis and wrote the paper. ST helped with
the data analysis. HB analysed MRI images. AY was involved in the
collection of the patient's medical data and clinical diagnosis.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Ethics approval was obtained from the Biruni
University Ethics Committee (approval no. 2017/10-11). All
procedures followed were in accordance with the ethical standards
of the Helsinki Declaration of 1975, as revised in 2013.
Patient consent for publication
Informed consent was obtained from parents in order
to publish the clinical data as well as images that were included
in the study.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Roeben B, Schüle R, Ruf S, Bender B,
Alhaddad B, Benkert T, Meitinger T, Reich S, Böhringer J, Langhans
CD, et al: SERAC1 deficiency causes complicated HSP: Evidence from
a novel splice mutation in a large family. J Med Genet. 55:39–47.
2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wortmann S, Rodenburg RJ, Huizing M,
Loupatty FJ, de Koning T, Kluijtmans LA, Engelke U, Wevers R,
Smeitink JA and Morava E: Association of 3-methylglutaconic
aciduria with sensori-neural deafness, encephalopathy, and
Leigh-like syndrome (MEGDEL association) in four patients with a
disorder of the oxidative phosphorylation. Mol Genet Metab.
88:47–52. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wortmann SB, Vaz FM, Gardeitchik T,
Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, Kulik W, Lammens M,
Christin C, Kluijtmans LA, et al: Mutations in the phospholipid
remodeling gene SERAC1 impair mitochondrial function and
intracellular cholesterol trafficking and cause dystonia and
deafness. Nat Genet. 44:797–802. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Sarig O, Goldsher D, Nousbeck J,
Fuchs-Telem D, Cohen-Katsenelson K, Iancu TC, Manov I, Saada A,
Sprecher E and Mandel H: Infantile mitochondrial hepatopathy is a
cardinal feature of MEGDEL syndrome (3-methylglutaconic aciduria
type IV with sensorineural deafness, encephalopathy and Leigh-like
syndrome) caused by novel mutations in SERAC1. Am J Med Genet A.
161A:2204–2215. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Maas RR, Iwanicka-Pronicka K, Kalkan Ucar
S, Alhaddad B, AlSayed M, Al-Owain MA, Al-Zaidan HI,
Balasubramaniam S, Barić I, Bubshait DK, et al: Progressive
deafness-dystonia due to SERAC1 mutations: A study of 67 cases. Ann
Neurol. 82:1004–1015. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Radha Rama Devi A and Lingappa L: Novel
mutations in SERAC1 gene in two Indian patients presenting with
dystonia and intellectual disability. Eur J Med Genet. 61:100–103.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
IJlst L1. Loupatty FJ, Ruiter JP, Duran M,
Lehnert W and Wanders RJ: 3-Methylglutaconic aciduria type I is
caused by pathogenic variants in AUH. Am J Hum Genet. 71:1463–1466.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Wortmann SB, Kluijtmans LA, Engelke UF,
Wevers RA and Morava E: The 3-methylglutaconic acidurias: What's
new? J Inherit Metab Dis. 35:13–22. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wortmann SB, Rodenburg RJT, Jonckheere A,
de Vries MC, Huizing M, Heldt K, van den Heuvel LP, Wendel U,
Kluijtmans LA, Engelke UF, et al: Biochemical and genetic analysis
of 3-methylglutaconic aciduria type IV: A diagnostic strategy.
Brain. 132:136–146. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Houtkooper RH, Turkenburg M, Poll-The BT,
Karall D, Pérez-Cerdá C, Morrone A, Malvagia S, Wanders RJ, Kulik W
and Vaz FM: The enigmatic role of tafazzin in cardiolipin
metabolism. Biochim Biophys Acta. 1788:2003–2014. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xu Y, Kelley RI, Blanck TJ and Schlame M:
Remodeling of cardiolipin by phospholipid transacylation. J Biol
Chem. 278:51380–51385. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bione S, D'Adamo P, Maestrini E, Gedeon
AK, Bolhuis PA and Toniolo D: A novel X-linked gene, G4.5. is
responsible for Barth syndrome. Nat Genet. 12:385–389.
1996.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Whited K, Baile MG, Currier P and Claypool
SM: Seven functional classes of Barth syndrome mutation. Hum Mol
Genet. 22:483–492. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Schlame M, Kelley RI, Feigenbaum A, Towbin
JA, Heerdt PM, Schieble T, Wanders RJ, DiMauro S and Blanck TJ:
Phospholipid abnormalities in children with Barth syndrome. J Am
Coll Cardiol. 42:1994–1999. 2003.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Vreken P, Valianpour F, Nijtmans LG,
Grivell LA, Plecko B, Wanders RJ and Barth PG: Defective remodeling
of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem
Biophys Res Commun. 279:378–382. 2000.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Costeff H, Elpeleg O, Apter N, Divry P and
Gadoth N: 3-Methylglutaconic aciduria in ‘optic atrophy plus’. Ann
Neurol. 33:103–104. 1993.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Elpeleg ON, Costeff H, Joseph A, Shental
Y, Weitz R and Gibson KM: 3-Methylglutaconic aciduria in the
Iraqi-Jewish ‘optic atrophy plus’ (Costeff) syndrome. Dev Med Child
Neurol. 36:167–172. 1994.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nystuen A, Costeff H, Elpeleg ON, Apter N,
Bonné-Tamir B, Mohrenweiser H, Haider N, Stone EM and Sheffield VC:
Iraqi-Jewish kindreds with optic atrophy plus (3-methylglutaconic
aciduria type 3) demonstrate linkage disequilibrium with the CTG
repeat in the 3' untranslated region of the myotonic dystrophy
protein kinase gene. Hum Mol Genet. 6:563–569. 1997.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Davey KM, Parboosingh JS, McLeod DR, Chan
A, Casey R, Ferreira P, Snyder FF, Bridge PJ and Bernier FP:
Mutation of DNAJC19, a human homologue of yeast inner mitochondrial
membrane co-chaperones, causes DCMA syndrome, a novel autosomal
recessive Barth syndrome-like condition. J Med Genet. 43:385–393.
2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ojala T, Polinati P, Manninen T, Hiippala
A, Rajantie J, Karikoski R, Suomalainen A and Tyni T: New mutation
of mitochondrial DNAJC19 causing dilated and noncompaction
cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr
Res. 72:432–437. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Tort F, García-Silva MT, Ferrer-Cortès X,
Navarro-Sastre A, Garcia-Villoria J, Coll MJ, Vidal E,
Jiménez-Almazán J, Dopazo J, Briones P, et al: Exome sequencing
identifies a new mutation in SERAC1 in a patient with
3-methylglutaconic aciduria. Mol Genet Metab. 110:73–77.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bonnefond A, Philippe J, Durand E,
Dechaume A, Huyvaert M, Montagne L, Marre M, Balkau B, Fajardy I,
Vambergue A, et al: Whole-exome sequencing and high throughput
genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS One.
7(e37423)2012.PubMed/NCBI View Article : Google Scholar
|