Introduction
The close association between incidence and
mortality indicates an extremely poor outcome in patients with
pancreatic cancer. Gemcitabine alone or gemcitabine-based
combination with other chemotherapeutic drugs is still the
front-line standard chemotherapy of advanced pancreatic cancer
(1). However, reversing the observed
chemoresistance and enhancing the chemosensitivity of cancer cells
to gemcitabine remains a challenge in improving the prognosis of
patients with cancer (1,2).
The concept of cancer stem cells (CSCs) is used to
define a tiny number of malignant cells with the characteristics of
self-renewal, multi-potency and tumor formation (3). The theory regarding CSCs suggests that
these stem-like cancer cells possess the capability to survive
therapy and induce early resistance, which can lead to a later
relapse (3,4). The accumulation of pancreatic CSCs
after gemcitabine treatment has been continually observed (2,5).
Therefore, targeting and eradicating cells with CSC markers
represents a promising strategy to treat cancer.
Aldehyde dehydrogenase (ALDH) is a family of enzymes
involved in the metabolism of intracellular aldehydes to acids by
an NAD(P)+ dependent reaction (6). ALDH activity is regarded as a marker
for both normal and malignant stem cells in the hematological
system and in the solid organ system, including pancreas, lung,
liver, breast, colon and ovary organs (6-13).
As an ALDH isoform, ALDH1 is a detoxifying enzyme responsible for
oxidizing intracellular retinaldehyde to retinoic acid (6,10).
N,N-diethylaminobenzaldehyde (DEAB), utilized as a negative control
in an ALDEFLUOR™ assay, is generally considered as a selective
ALDH1A1 inhibitor (12,14). Further previous studies demonstrated
that DEAB is additionally able to block other ALDH1 isoforms,
including ALDH1A2, ALDH1A3, ALDH1B1, and even ALDH2 and ALDH5A1
(14,15).
In the present study, three pancreatic cancer cell
lines with different ALDH activities were examined and an ALDH
inhibitor (DEAB) was used on these cells to test whether blocking
ALDH expression triggers cancer cell elimination and sensitizes the
cytotoxic effect of gemcitabine.
Materials and methods
Culture of cell lines
MiaPaCa2 and Panc1 cell lines were obtained from The
Cell Center of Zhongda Hospital, and the BxPC3 cell line was
obtained from The Cell Center of Jiangsu Province Hospital. Cells
were cultured in DMEM (Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
at 37˚C and 5% CO2.
Flow cytometry
Cells were stained with ALDEFLUOR™ reagent (Stemcell
Technologies, Inc.) and propidium iodide (PI; BD Biosciences) as
previously described (16).
Activated ALDEFLUOR™ reagent was aliquoted and stored at -20˚C
according to the manufacturer's instructions (Stemcell
Technologies, Inc.). Cells were suspended with ALDEFLUOR™ Assay
Buffer to a concentration of 1x106 cells/ml. In total, 1
ml cell suspension was transferred to a new tube and 5 µl activated
ALDEFLUOR™ reagent per ml was added to the cell suspension. A total
of 0.5 ml ALDEFLUOR™ reagent/cell suspension mixture was
transferred immediately to a new tube with 10 µl DEAB (1.5 mM).
DEAB, used as the negative control, inhibits the enzymatic reaction
of ALDH. All samples were incubated at 37˚C for 30 min. After
incubation, cells were washed in cold ALDEFLUOR™ Assay Buffer,
centrifuged at 250 x g for 5 min at 4˚C to remove supernatant, and
resuspended in 0.5 ml cold ALDEFLUOR™ Assay Buffer on ice. PI
(2/100 µl reaction) was added before measurement. Cells were
recorded and analyzed with a BD LSR II flow cytometer and FlowJo
10.4 (BD Biosciences). A total of three independent analyses were
performed.
Cell cycle analysis was performed as described
previously (17). Cells were first
incubated with Human BD Fc Block (BD Pharmingen; BD Biosciences) at
a final concentration of 1 µg/100 µl for 15 min at 4˚C. Cells were
fixed in 100 µl PBS/1.6% paraformaldehyde (Electron Microscopy
Sciences) for 10 min at room temperature in the dark and then
permeabilized in 300 µl PBS/90% methanol (Carl Roth) for 30 min on
ice. Fixed cells were stained with Ki67-Alexa Fluor® 647
(clone B56; BD Biosciences) for 1 h at room temperature in the dark
and incubated with DAPI (0.5 µg/ml; Sigma Aldrich; Merck KGaA) for
40 min in the dark on ice. Finally, cells were recorded and
analyzed with a BD LSR II flow cytometer and FlowJo 10.4 (BD
Biosciences). In total, four independent analyses were
performed.
Colony-forming assay
Cells were trypsinized and re-plated at 500 cells
per well in 6-well plates in triplicate and incubated for 2 weeks
without medium change. After medium was removed, cells were washed
twice with PBS (HyClone; GE Healthcare Life Sciences) and were
fixed with 2 ml 4% paraformaldehyde (Electron Microscopy Sciences)
for 15 min at room temperature. Then cells were washed three times
with water and stained with 0.05% coomassie blue (Beijing Solarbio
Science & Technology Co., Ltd.) for 5 min at room temperature.
After being washed with water, plates were dried overnight. The
number of colonies with >50 cells was counted under a Nikon
Eclipse E400 polarizing light microscope under 4, 10 and 20X
objective. A total of three independent experiments were
performed.
Cell Counting Kit-8 (CCK-8) assay
The cell viability in the cytotoxicity test was
quantified with CCK-8 (Sigma-Aldrich; Merck KGaA) according to the
manufacturer's protocol. A density of 104 cells per well
was seeded in 96-well plates. Cells were treated with DEAB
(Sigma-Aldrich; Merck KGaA) at concentrations of 0, 50, 100, 150
and 200 µM for 1, 3 and 5 days in quadruplicate. As DEAB was
dissolved in ethanol, the DEAB-untreated well was added to the same
amount of ethanol (Sigma-Aldrich; Merck KGaA) as a control. Cells
in each well were mixed with 10 µl CCK-8 reagent per 100 µl medium
and incubated at 37˚C and 5% CO2 for 2 h in the dark.
The absorbance of each well was measured at 450 nm using an ELISA
plate reader. In total, two independent experiments were
performed.
Cell treatment and apoptosis
analysis
Cells of the three cell lines were cultured at a
density of 5x105 cells per ml medium and treated in the
following ways: i) Ethanol for 3 days; ii) 200 µM DEAB for 3 days;
iii) ethanol for 1 day, then 50 nM gemcitabine (Dailan Meilun
Biology Technology Co., Ltd.) was added for another 2 days; and iv)
200 µM DEAB for 1 day, then 50 nM gemcitabine was added for another
2 days.
Cells were stained with Annexin V-FITC and PI
(Beyotime Institute of Biotechnology) for 15 min at room
temperature in the dark, according to the manufacturer's
instructions. Cells were recorded and analyzed using a BD LSR II
flow cytometer and FlowJo 10.4 (BD Biosciences). Annexin
V+ cells were calculated as apoptotic cells and Annexin
V-PI- cells were calculated as living cells.
A total of three independent experiments were performed.
Western blot analysis
Proteins were extracted from cells with RIPA buffer
(Cell Signaling Technology, Inc.), and the protein concentration
was quantified using a BCA Protein Assay kit (Cell Signaling
Technologies, Inc.). Equal amounts of protein (20 µg) from each
sample were loaded to the wells and separated by SDS-PAGE on 10%
gels and then transferred onto a polyvinylidene fluoride membrane
(EMD Millipore). After blocking with 5% skimmed milk for 1 h at
room temperature, the membrane was incubated with primary
antibodies at 4˚C overnight. The primary antibodies of target
proteins were as follows: Anti-B cell lymphoma 2 (Bcl2) associated
X protein (Bax; Cell Signaling Technology, Inc.; cat. no. CST
5023T; 1:1,000), anti-Bcl2 (Cell Signaling Technology, Inc.; cat.
no. CST 2872; 1:1,000) and anti-β-actin (Affinity Biosciences; cat.
no. T0022; 1:1,000). Thereafter, the membrane was incubated with
goat anti-rabbit IgG (H+L) HRP (cat. no. S0001) or goat anti-mouse
IgG (H+L) HRP (cat. no. S0002) secondary antibodies (Affinity
Biosciences; 1:5,000) at room temperature for 1 h. The enhanced
Chemiluminescence Western blot kit (EMD Millipore) was used to
visualize immunoreactive protein bands. Optical density was
measured and relative protein expression was calculated by
normalization to β-actin using ImageJ v1.51 (National Institutes of
Health). A total of four independent experiments were
performed.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) following the manufacturer's instructions. RNA was reverse
transcribed to cDNA using the RevertAid First Strand cDNA Synthesis
kit (Thermo Fisher Scientific, Inc.) at 42˚C for 1 h followed by
inactivation at 70˚C for 5 min. PCR was performed with FastStart
Universal SYBR Green Master (Rox) (Roche Diagnostics) according to
the manufacturer's instructions. PCR was performed with
denaturation at 95˚C for 10 min followed by amplification by 40
cycles of 95˚C for 15 sec, 60˚C for 1 min and 72˚C for 1 min, and a
final extension at 72˚C for 5 min in triplicate. β-actin was used
as an internal control. The relative expression levels of target
genes were calculated using the 2-ΔΔCq method (18). The sequences of primers (Sangon
Biotech Co., Ltd.) were as follows: Bax (forward,
5'-ATGGACGGGTCCGGGGAGCAGCCC-3' and reverse,
5'-GGTGAGCACTCCCGCCACAAAGAT-3'), Bcl2 (forward,
5'-AAGAGCAGACGGATGGAAAAAGG-3' and reverse,
5'-GGGCAAAGAAATGCAAGTGAATG-3') and β-actin (forward,
5'-CTACCTCATGAAGATCCTCACC-3' and reverse,
5'-AGTTGAAGGTAGTTTCGTGGAT-3'). A total of two independent
experiments were performed.
Statistical analysis
Statistical analysis was performed using SPSS
Statistics 19 (IBM Corp.). At least two independent experiments
were performed and all values are presented as the mean ± SEM.
One-way ANOVA was performed to compare multiple groups with Tukey's
as the post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
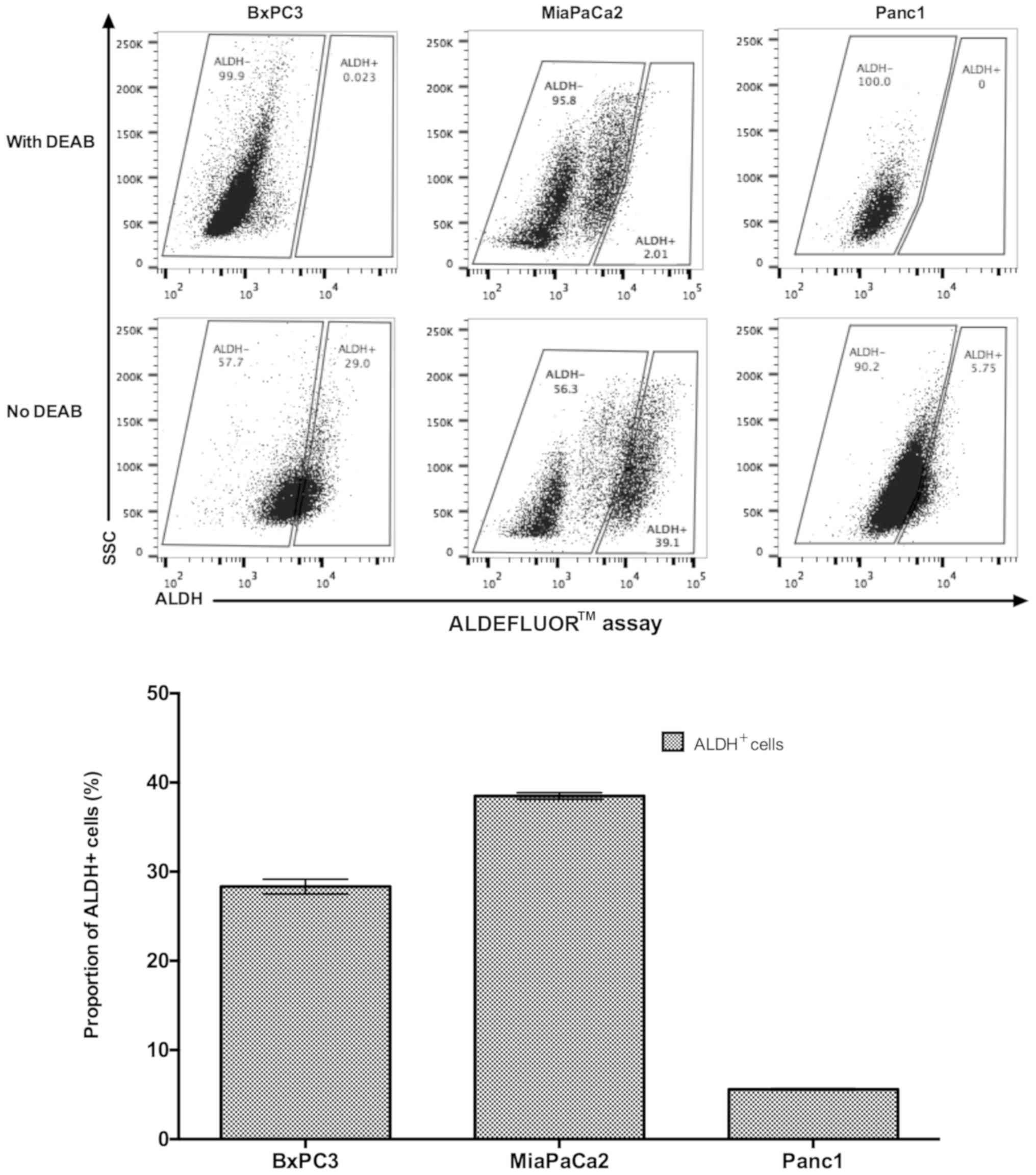
ALDH activities vary among cell lines
and are inhibited under DEAB treatment
The ALDH activities of three pancreatic cell lines,
including BxPC3, MiaPaCa2 and Panc1, were assessed; different
patterns within these three cell lines were identified. On average,
38.5% of MiaPaCa2 cells were ALDH+ followed by BxPC3
cells with 28.33% ALDH+, by contrast in Panc1 cells only
5.59% were ALDH+. BxPC3 and MiaPaCa2 have three
sub-populations with different levels of ALDH expression,
ALDH- includes ALDH-dim and intermediate ALDH, and the
third sub-population is ALDH+. While Panc1 only has
ALDH- and ALDH+ two subpopulations; the
ALDH- sub-population cannot be further divided, unlike
BxPC3 and MiaPaCa2. DEAB demonstrated an ALDH-inhibition effect on
all three cell lines (Fig. 1).
Malignant proliferation of cancer
cells is weakened under the existence of DEAB
To investigate whether DEAB inhibits proliferation
potential of pancreatic cancer cells in vitro, two adherent
cell lines (BxPC3 and Panc1) were selected for colony-forming
assays. A previous study demonstrated that MiaPaCa2 cells had no
colony- and spheroid-forming potential (19). A colony-forming assay with MiaPaCa2
cells was conducted and it was observed that the numbers of
colonies were extremely low because most were non-adherent cells
(Fig. S1); therefore, MiaPaCa2 was
excluded from this analysis. It was identified that DEAB alone was
able to significantly prevent the colony formation ability of
cancer cells (Fig. 2A).
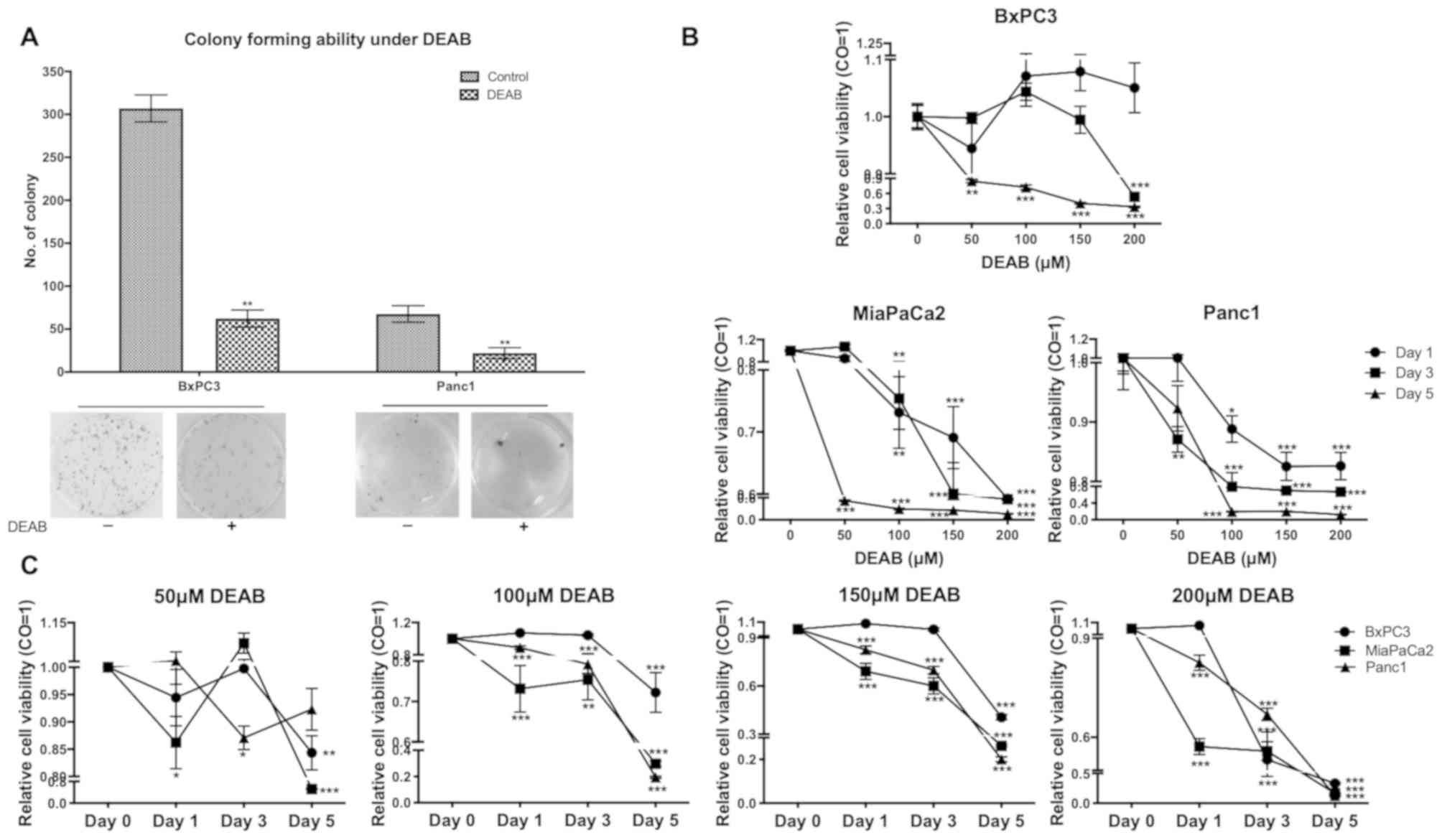 | Figure 2DEAB inhibits colony-forming ability
and cell proliferation. (A) Colony-forming ability of BxPC3 and
Panc1 cells treated or untreated with DEAB was detected by a cell
colony formation assay. Data are representative of three
independent experiments. (B) Proliferation of BxPC3, MiaPaCa2 and
Panc1 cells treated with 0, 50, 100, 150 and 200 µM DEAB were
compared by a CCK-8 assay. (C) Proliferation of BxPC3, MiaPaCa2 and
Panc1 cells at day 0, 1, 3, and 5 were compared using a CCK-8
assay. Cells treated with 0 µM DEAB were used as the control. Data
are representative of two independent experiments. Viability of the
control was set at 1. Data are presented as the mean ± SEM.
*P<0.05, **P<0.01,
***P<0.001 vs. respective control. DEAB,
N,N-diethylaminobenzaldehyde; CCK-8, Cell Counting Kit-8; CO,
control. |
To further confirm these results, a CCK-8 assay was
performed with the three cell lines in the presence of DEAB with a
range of doses from 0 to 200 µM for 1, 3 and 5 days. When a 1
day-treatment on MiaPaCa2 (100, 150 and 200 µM), Panc1 (100, 150
and 200 µM), a 3 day-treatment on BxPC3 (200 µM), MiaPaCa2 (100,
150 and 200 µM), Panc1 (50, 100, 150 and 200 µM), and a 5
day-treatment on BxPC3, MiaPaCa2, Panc1 (100, 150 and 200 µM) were
performed, significant cytotoxicity with increased concentrations
of DEAB was observed (Fig. 2B).
Treatments of cells with 50 µM DEAB at day 1 (MiaPaCa2), day 3
(Panc1) and day 5 (MiaPaCa2 and BxPC3); 100 µM DEAB at day 1
(MiaPaCa2 and Panc1), day 3 (MiaPaCa2 and Panc1) and day 5
(MiaPaCa2, Panc1 and BxPC3); 150 µM DEAB at day 1 (MiaPaCa2 and
Panc1), day 3 (MiaPaCa2 and Panc1) and day 5 (MiaPaCa2, Panc1 and
BxPC3); 200 µM DEAB at day 1 (MiaPaCa2 and Panc1), day 3 (MiaPaCa2,
Panc1 and BxPC3) and day 5 (MiaPaCa2, Panc1 and BxPC3) demonstrated
significant cytotoxicity and the cytotoxicity was more enhanced
with longer incubation time of DEAB (Fig. 2C). The proliferation of cancer cell
lines was significantly inhibited by DEAB, and both dose- and
time-dependent manners were observed in
DEAB-induced-cytotoxicity.
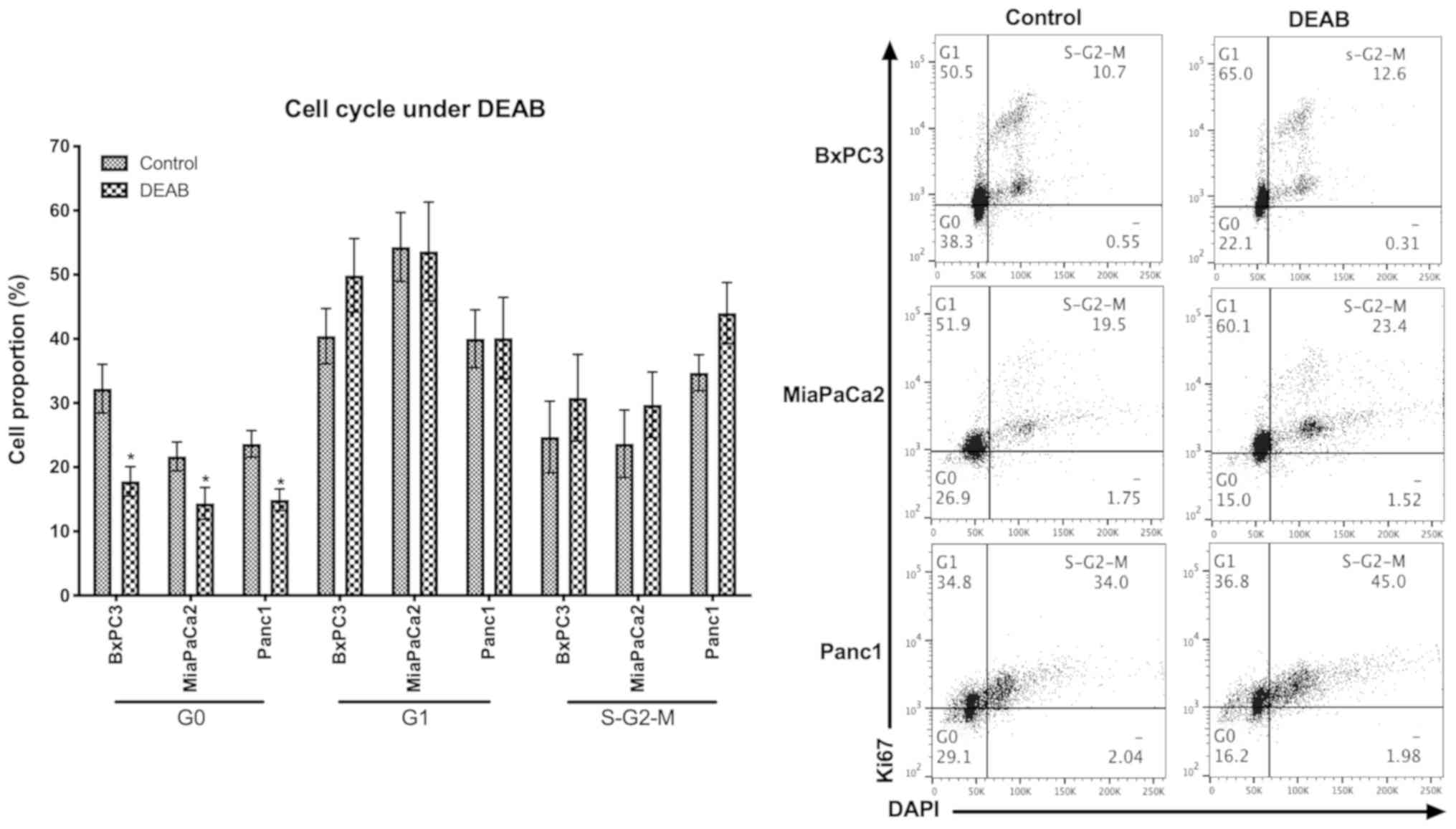
DEAB treatment promotes cell
cycle
As cell quiescence has been suggested to be
associated with chemotherapy resistance (17), the cell cycle of pancreatic cancer
cells under DEAB was analyzed. Cells treated with DEAB contained
significantly decreased proportions of quiescent cells (defined as
the G0 phase) and an accumulation of cells at
S-G2-M phases, compared with the controls (Fig. 3).
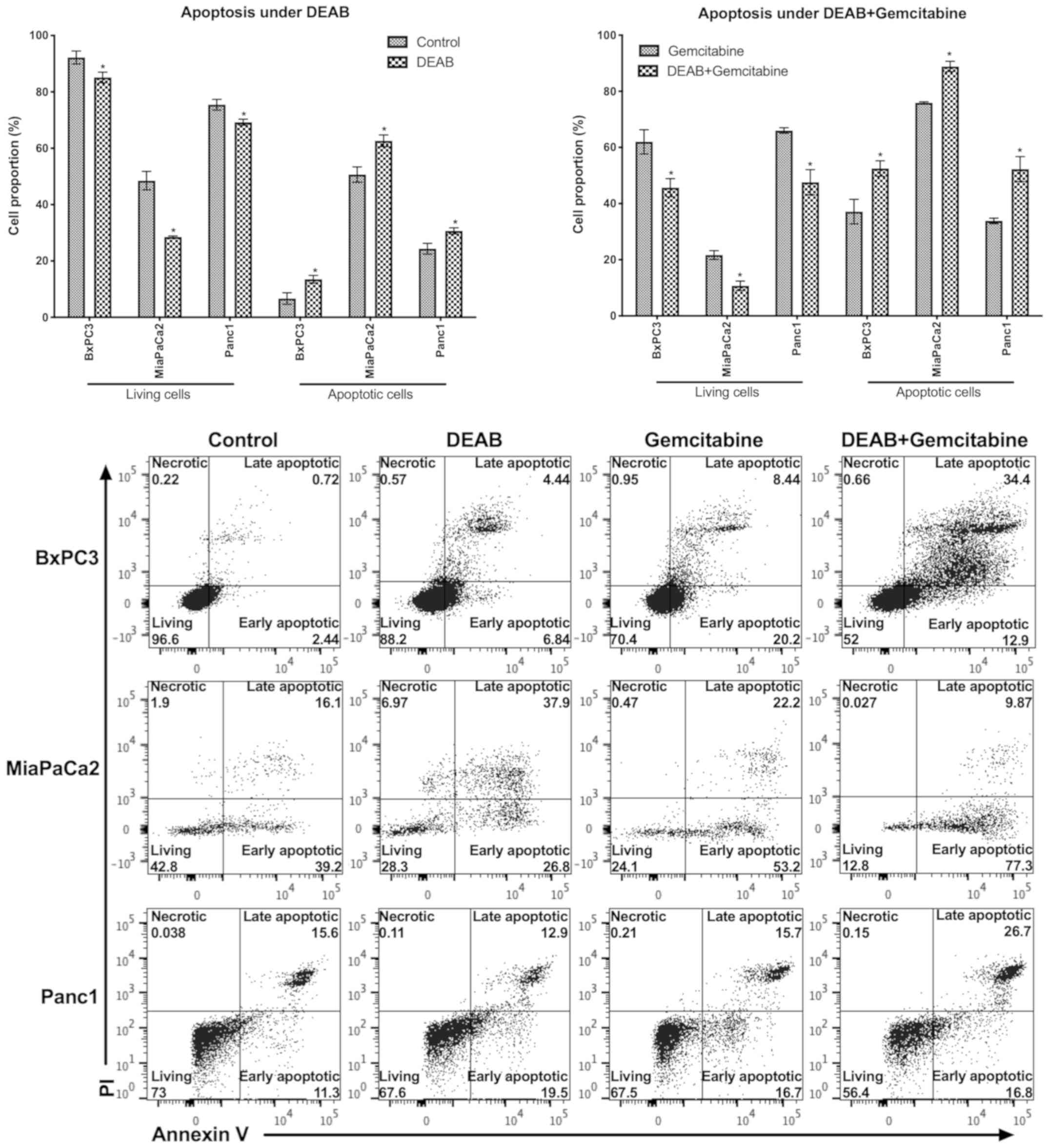
DEAB induces cancer cell apoptosis and
sensitizes the cytotoxic effects of gemcitabine
In order to evaluate the effect of DEAB on
pancreatic cancer cells and gemcitabine-induced cell death, the
cell viability and apoptosis were measured. In all three cell lines
analyzed, it was observed that compared with their respective
controls (ethanol and gemcitabine treatments), both DEAB alone and
DEAB-gemcitabine combination treatments induced a significant
decrease of living cells and a significant increase of apoptotic
cells (Fig. 4).
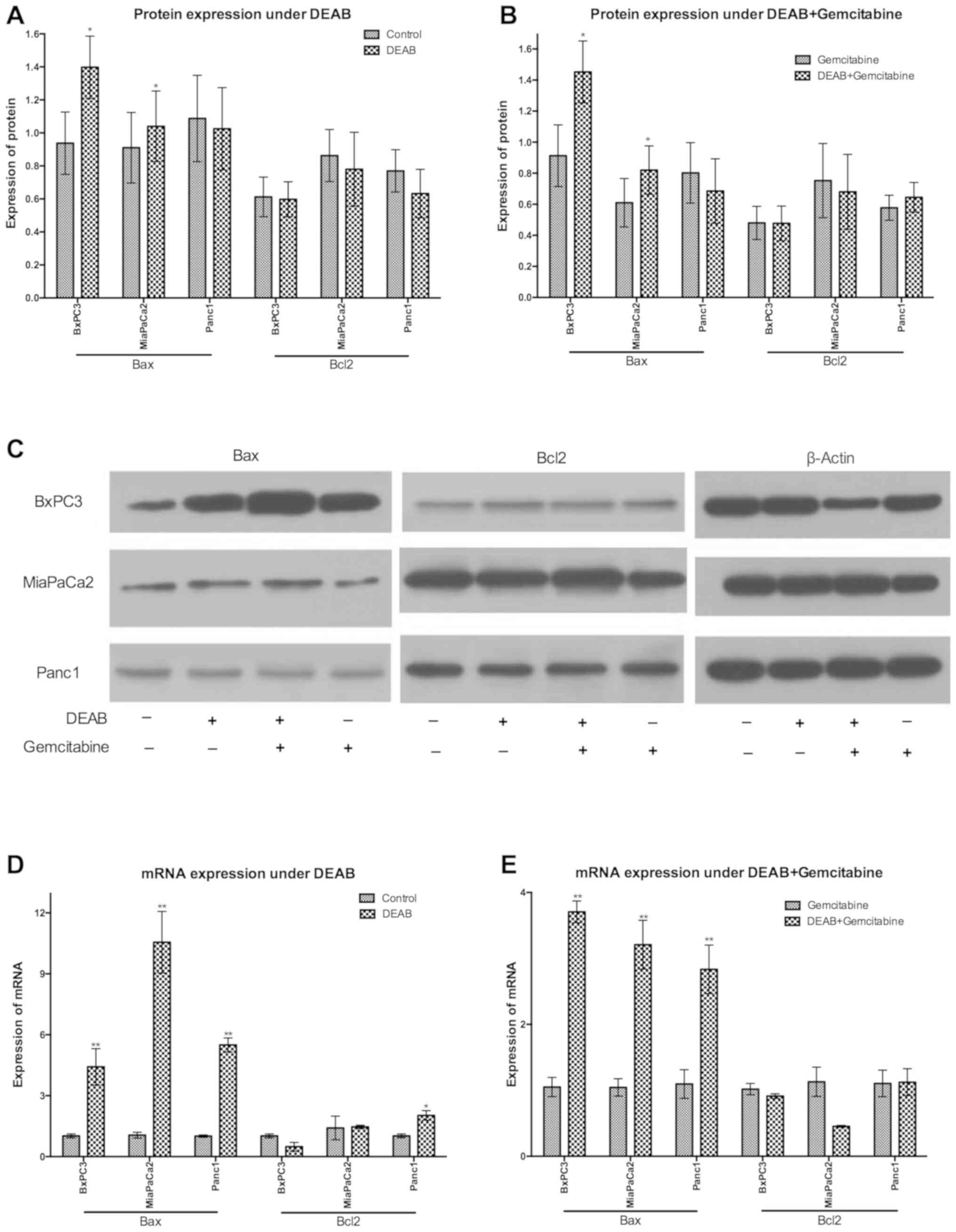
DEAB effect is associated with
upregulation of Bax expression
To understand the underlying mechanisms of the
anti-cancer effects of DEAB, the expressions of Bax and Bcl2 at the
protein level were comparatively analyzed. When compared with the
control, BxPC3 and MiaPaCa2 cells treated with DEAB demonstrated a
significant increase of Bax protein expression and no significant
regulation of Bcl2 protein expression (Fig. 5A and C).
Cells undergoing gemcitabine single treatment and
DEAB-gemcitabine combination treatment were compared, and it was
identified that DEAB exposure significantly upregulated Bax
expression of BxPC3 and MiaPaCa2 cells; however, did not
significantly regulate Bcl2 expression at the protein level
(Fig. 5B and C).
To further examine the mechanisms, the
expressions of Bax and Bcl2 at the mRNA level were analyzed using
RT-qPCR. It was observed that in the presence of DEAB, Bax mRNA was
significantly upregulated in BxPC3 and MiaPaCa2 cells compared with
the controls. Moreover, in Panc1 cells with DEAB treatment, the
present results demonstrated a significant increase of Bax and Bcl2
mRNA (Fig. 5D).
In contrast to cells treated with gemcitabine,
BxPC3, MiaPaCa2 and Panc1 cells treated with DEAB-gemcitabine
demonstrated a significant increase of Bax mRNA (Fig. 5E).
Discussion
In the present study, the role of the ALDH inhibitor
DEAB in anti-cancer efficacy was investigated. To evaluate the
inhibitory ability of DEAB on ALDH activity, three pancreatic
cancer cell lines with inconsistent ALDH activities were utilized.
Similar to the cell lines, the ALDH expressions vary among patients
with pancreatic cancer (20). The
inhibition of ALDH activity induced by DEAB was independent of the
ALDH levels of cells; this finding suggested the possibility of a
universal clinical application of DEAB with unselected patients
with pancreatic cancer.
Gemcitabine preferentially targets cells
characterizing rapid proliferation and differentiation; to some
extent, the quiescence and other stem cell-like characteristics of
ALDH+ cancer cells may explain their gemcitabine
resistance (21,22). The contribution of high ALDH activity
to gemcitabine resistance indicates that inhibiting ALDH may
increase the sensitivity of tumor cells to gemcitabine (2,5,19). Consequently, studies regarding ALDH
inhibition in pancreatic cancer treatment have emerged. Knockdown
of the ALDH gene of MiaPaCa2 cells using small interfering RNAs
reduced cell proliferation and overcame gemcitabine resistance in a
previous study (2). Disulfiram, an
irreversible ALDH inhibitor, inhibited in vitro pancreatic
cancer cell proliferation and in vivo tumor growth combined
with gemcitabine (5). Sulforaphane
enriched in broccoli compound suppressed the enrichment of
ALDH+ cells induced by gemcitabine and enhanced the
cytotoxic effect of gemcitabine (19). The therapeutic potential of ALDH
inhibition was also demonstrated in other solid cancer types. In
cholangiocarcinoma, the reduction of ALDH activity in
gemcitabine-resistant cells by metronidazole resulted in the
enhancement of chemosensitivity (23). In lung cancer, inhibiting ALDH with
DEAB and disulfiram suppressed the viability of cancer cells and
sensitized the cancer cells to chemotherapy (10,11).
Consistent with these data, in the present study, after comparing
untreated cells and cells treated with DEAB, it was observed that
an ALDH inhibitor (DEAB) reduced cell viability, cell quiescence
and furthermore, enhanced gemcitabine-induced cytotoxicity in
vitro. Taken together, the present results established the
status of DEAB as a potential chemotherapeutic reagent or at least
a chemosensitizer to overcome gemcitabine resistance.
Recently, ALDH-targeting based treatment has
attracted increasing attention; however, at present, the mechanisms
involved are still undetermined. The decrease of lung cancer cell
viability induced by disulfiram (through ALDH inhibition) was
attributed to cell cycle arrest in the G2/M phase
(11). ALDH1A1-knockdown stimulated
taxane-resistant ovarian cancer cells to enter the S and
G2 cell cycle phases (12). In pancreatic cancer, it was observed
that the proportion of G0 cells was decreased by DEAB
and more cells entered S-G2-M phases; a previous study
demonstrated that cells with ALDH1A1 knockdown were enriched at the
S phase (2). It was hypothesized
that the cell cycling entry of quiescent cancer cells induced by
ALDH inhibition strengthens the cytotoxicity of cell cycle specific
chemotherapeutic drugs, such as gemcitabine, leading to increased
apoptosis. Inhibition of ALDH activity delayed the process of
retinaldehyde to retinoic acid mediated by ALDH to increase the
production of reactive oxygen species (ROS) (10). The induction of ROS promoted
gemcitabine-related cytotoxicity in pancreatic cancer (2). Moreover, ROS-induced DNA damage and p53
activation contributed to increased apoptosis accompanied by the
accumulation of retinaldehyde (10,24).
The induction of cancer cell apoptosis is a critical
hallmark of anti-cancer therapy; therefore, the present focused on
mitochondrial apoptosis (intrinsic pathway) related Bax and Bcl2 to
elucidate the mechanisms of DEAB-induced-apoptosis (25). Although the apoptosis of all cell
lines analyzed was promoted by DEAB, the latent mechanisms were not
completely the same among the tested cell lines.
DEAB-induced-apoptosis in BxPC3 and MiaPaCa2 is associated with the
mitochondrial pathway induced by significantly upregulated
pro-apoptotic Bax at the protein level, and a significant
consistent trend of mRNA alteration reflected the regulation at the
gene level; however, no significant downregulation of
anti-apoptotic Bcl2 was observed (25,26). In
addition, although Bax and Bcl2 mRNA increased in Panc1 under DEAB,
Bax and Bcl2 proteins did not contribute to DEAB-induced-apoptosis
in Panc1. In a previous study, ALDH1A1-knockdown upregulated the
expression of Bax and induced Bax-mediated apoptosis in ovarian
cancer (27). S-methyl
4-amino-4-methylpent-2-ynethioate, a synthetic suicide inhibitor of
ALDH1, stimulated Bcl2-overexpressing cell apoptosis (28). However, in the present study, the
effect of DEAB on Bcl2 protein expression was not observed. Serving
as an indispensable entry point of the mitochondrial apoptosis
pathway, the abnormal suppression of Bax results in therapeutic
resistance in various cancer types; therefore re-activating Bax is
considered as a strategy in the anticancer field (29). In a recent study, adaptor related
protein complex 5 subunit mu 1 failed to induce apoptotic death in
Bax-/- knockout cells (30); and Bax-/- mice exhibited
increase of some cell types including lymphocytes, certain neurons
and immature germ cells (26).
Therefore, it was hypothesized that the enhancement of
mitochondrial apoptosis-related Bax is responsible for
DEAB-mediated apoptosis of tumor cells. Notably, compared with
Panc1 cells, both BxPC3 cells and MiaPaCa2 cells have relatively
higher ALDH activity in the present study; the apoptosis of these
cells induced by DEAB is likely associated with mitochondrial
apoptosis, by contrast, mitochondrial apoptosis-related proteins
measured were not influenced by DEAB in Panc1. Different patterns
of ALDH expression in pancreatic cancer cell lines have been
observed in the present study and in previous studies (5,7). Based
on the findings that ALDH+ cells possess stem/progenitor
properties, it was suggested that the pancreatic cancer cell lines
with high ALDH activity appear to represent a sub-population with
stem cell-like characteristics, including an effect on the
apoptosis-related pathway (3-5).
The present results suggested that the mechanisms of
DEAB-induced-apoptosis might be associated with the individual
characteristics of a cell line, including the ALDH expression
pattern. To test this hypothesis, future studies could measure the
ALDH activities of more pancreatic cancer cell lines, and compare
additional apoptosis-related proteins and genes of grouped cell
lines based on ALDH activity.
To further elucidate the function of DEAB in an
anti-cancer aspect and clarify the underlying mechanisms,
subsequent studies are required. A future study could knock down
Bax to investigate the direct role of Bax in apoptosis and widely
screen cell cycle, proliferation and apoptosis-related proteins and
genes of cancer cells with different ALDH expressions by
microarray.
The present study demonstrated that DEAB may inhibit
ALDH to suppress cell proliferation, promote apoptosis, activate
the cell cycle, and sensitize pancreatic cancer cells to
gemcitabine by activating apoptosis pathway-related proteins and
genes.
Supplementary Material
Colony-forming ability of MiaPaCa2
cells treated or untreated with DEAB as detected by a cell colony
formation assay. Data are representative of three independent
experiments. Data are presented as the mean ± SEM.
***P<0.001 vs. control. DEAB,
N,N-diethylaminobenzaldehyde.
Acknowledgements
Not applicable.
Funding
The present study was supported by Wuxi Health
Commission Foundation (grant no. Q201601), Nanjing Medical
University Foundation (grant no. 2016NJMU133), Science and
Education Project Foundation of Wuxi Health Commission (grant no.
QNRC004).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WW designed the research. WW, SZ and HG performed
experiments. WW, SZ, HH and BRS analyzed the data. WW, HH and BRS
wrote the manuscript with input from all other authors. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Duong HQ, Hwang JS, Kim HJ, Kang HJ, Seong
YS and Bae I: Aldehyde dehydrogenase 1A1 confers intrinsic and
acquired resistance to gemcitabine in human pancreatic
adenocarcinoma MIA PaCa-2 cells. Int J Oncol. 41:855–861.
2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells-perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344.
2006.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ishiwata T, Matsuda Y, Yoshimura H, Sasaki
N, Ishiwata S, Ishikawa N, Takubo K, Arai T and Aida J: Pancreatic
cancer stem cells: Features and detection methods. Pathol Oncol
Res. 24:797–805. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kim SK, Kim H, Lee DH, Kim TS, Kim T,
Chung C, Koh GY, Kim H and Lim DS: Reversing the intractable nature
of pancreatic cancer by selectively targeting ALDH-high,
therapy-resistant cancer cells. PLoS One. 8(e78130)2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Moreb JS: Aldehyde dehydrogenase as a
marker for stem cells. Curr Stem Cell Res Ther. 3:237–246.
2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lin L, Jou D, Wang Y, Ma H, Liu T, Fuchs
J, Li PK, Lü J, Li C and Lin J: STAT3 as a potential therapeutic
target in ALDH+ and CD44+/CD24+ stem cell-like pancreatic cancer
cells. Int J Oncol. 49:2265–2274. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ginestier C, Hur MH, Charafe-Jauffret E,
Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG,
Liu S, et al: ALDH1 is a marker of normal and malignant human
mammary stem cells and a predictor of poor clinical outcome. Cell
Stem Cell. 1:555–567. 2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Huang EH, Hynes MJ, Zhang T, Ginestier C,
Dontu G, Appelman H, Fields JZ, Wicha MS and Boman BM: Aldehyde
dehydrogenase 1 is a marker for normal and malignant human colonic
stem cells (SC) and tracks SC overpopulation during colon
tumorigenesis. Cancer Res. 69:3382–3389. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Park JW, Jung KH, Lee JH, Moon SH, Cho YS
and Lee KH: Inhibition of aldehyde dehydrogenase 1 enhances the
cytotoxic effect of retinaldehyde on A549 cancer cells. Oncotarget.
8:99382–99393. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
MacDonagh L, Gallagher MF, Ffrench B,
Gasch C, Breen E, Gray SG, Nicholson S, Leonard N, Ryan R, Young V,
et al: Targeting the cancer stem cell marker, aldehyde
dehydrogenase 1, to circumvent cisplatin resistance in NSCLC.
Oncotarget. 8:72544–72563. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Landen CN Jr, Goodman B, Katre AA, Steg
AD, Nick AM, Stone RL, Miller LD, Mejia PV, Jennings NB, Gershenson
DM, et al: Targeting aldehyde dehydrogenase cancer stem cells in
ovarian cancer. Mol Cancer Ther. 9:3186–3199. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Fleischman AG: ALDH marks leukemia stem
cell. Blood. 119:3376–3377. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Morgan CA, Parajuli B, Buchman CD, Dria K
and Hurley TD: N,N-diethylaminobenzaldehyde (DEAB) as a substrate
and mechanism-based inhibitor for human ALDH isoenzymes. Chem Biol
Interact. 234:18–28. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Koppaka V, Thompson DC, Chen Y, Ellermann
M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD and
Vasiliou V: Aldehyde dehydrogenase inhibitors: A comprehensive
review of the pharmacology, mechanism of action, substrate
specificity, and clinical application. Pharmacol Rev. 64:520–539.
2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hoang VT, Hoffmann I, Borowski K,
Zepeda-Moreno A, Ran D, Buss EC, Wuchter P, Eckstein V and Ho AD:
Identification and separation of normal hematopoietic stem cells
and leukemia stem cells from patients with acute myeloid leukemia.
Methods Mol Biol. 1035:217–230. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang W, Bochtler T, Wuchter P, Manta L, He
H, Eckstein V, Ho AD and Lutz C: Mesenchymal stromal cells
contribute to quiescence of therapy-resistant leukemic cells in
acute myeloid leukemia. Eur J Haematol. 99:392–398. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kallifatidis G, Labsch S, Rausch V,
Mattern J, Gladkich J, Moldenhauer G, Büchler MW, Salnikov AV and
Herr I: Sulforaphane increases drug-mediated cytotoxicity toward
cancer stem-like cells of pancreas and prostate. Mol Ther.
19:188–195. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kim MP, Fleming JB, Wang H, Abbruzzese JL,
Choi W, Kopetz S, McConkey DJ, Evans DB and Gallick GE: ALDH
activity selectively defines an enhanced tumor-initiating cell
population relative to CD133 expression in human pancreatic
adenocarcinoma. PLoS One. 6(e20636)2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Mueller MT, Hermann PC, Witthauer J,
Rubio-Viqueira B, Leicht SF, Huber S, Ellwart JW, Mustafa M,
Bartenstein P, D'Haese JG, et al: Combined targeted treatment to
eliminate tumorigenic cancer stem cells in human pancreatic cancer.
Gastroenterology. 137:1102–1113. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jimeno A, Feldmann G, Suárez-Gauthier A,
Rasheed Z, Solomon A, Zou GM, Rubio-Viqueira B, García-García E,
López-Ríos F, Matsui W, et al: A direct pancreatic cancer xenograft
model as a platform for cancer stem cell therapeutic development.
Mol Cancer Ther. 8:310–314. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kawamoto M, Umebayashi M, Tanaka H, Koya
N, Nakagawa S, Kawabe K, Onishi H, Nakamura M and Morisaki T:
Combined gemcitabine and metronidazole is a promising therapeutic
strategy for cancer stem-like cholangiocarcinoma. Anticancer Res.
38:2739–2748. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sawada O, Perusek L, Kohno H, Howell SJ,
Maeda A, Matsuyama S and Maeda T: All-trans-retinal induces Bax
activation via DNA damage to mediate retinal cell apoptosis. Exp
Eye Res. 123:27–36. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163.
2005.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326.
1998.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Meng E, Mitra A, Tripathi K, Finan MA,
Scalici J, McClellan S, Madeira da Silva L, Reed E, Shevde LA,
Palle K and Rocconi RP: ALDH1A1 maintains ovarian cancer stem
cell-like properties by altered regulation of cell cycle checkpoint
and DNA repair network signaling. PLoS One.
9(e107142)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Canuto RA, Muzio G, Salvo RA, Maggiora M,
Trombetta A, Chantepie J, Fournet G, Reichert U and Quash G: The
effect of a novel irreversible inhibitor of aldehyde dehydrogenases
1 and 3 on tumour cell growth and death. Chem Biol Interact.
130-132:209–218. 2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Igney FH and Krammer PH: Death and
anti-death: Tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
30
|
Won M, Luo Y, Lee DH, Shin E, Suh DS, Kim
TH, Jin H and Bae J: BAX is an essential key mediator of
AP5M1-induced apoptosis in cervical carcinoma cells. Biochem
Biophys Res Commun. 518:368–373. 2019.PubMed/NCBI View Article : Google Scholar
|