Introduction
The role of uric acid in the causation and
progression of chronic kidney disease (CKD) has been debated;
however, uric acid has been reconsidered as a potential
contributory risk factor for the development and progression of CKD
over the last 15 years (1-2). Excessive uric acid,
usually in the form of monosodium urate (MSU) crystals,
precipitates in synovial cavities and other anatomic location to
induce severe inflammation and debilitating pain (3-6).
In particular, deposits of uric acid in the kidney causes gouty
nephropathy, which is the most serious complication of
hyperuricemia (3-6).
It is considered that the activation of renin-angiotensin,
inflammatory factors, endothelial dysfunction and cyclooxygenase-2
(COX-2) serve important roles in gouty nephropathy (5,6). The
identified underlying mechanisms of gouty nephropathy have revealed
that inflammation has a dominant role in its pathogenesis.
The Nod-like receptor protein 3 (NLRP3)
inflammasome is comprised of NLRP3, apoptosis-associated
speck like protein (ASC) and caspase-1 and is the most extensively
studied inflammasome of recent years. It is involved in certain
human inflammatory and autoimmune diseases, including
cryopyrin-associated periodic syndrome, ischaemia reperfusion
injury and atherosclerosis (7,8).
Following detection of cellular stress, NLRP3
oligomerizes by homotypic interactions between NACHT domains. The
pyrin domains (PYD) of NLRP3 then becomes exposed for
ASC binding. The caspase activation and recruitment domains (CARD)
of ASC in turn recruits pro-caspase-1 through CARD-CARD
interactions (9). Following
NLRP3 inflammasome formation, procaspase-1 is converted
to active caspase-1, which regulates the maturation of
proinflammatory cytokines, including interleukin (IL)-1β and
IL-18(10), further aggravating
renal damage.
MSU crystals cause the formation of inflammasomes
in vivo (11). In addition,
the inflammatory effect of MSU crystals is primarily mediated by
NLRP3 inflammasomes driving the production of IL-1β and
IL-18. IL-1β is likely the main agent that triggers systemic
inflammation (3). Therefore, these
observations prompted the present study to assess the role of the
NLRP3 inflammasome in the mediation of the innate immune
inflammatory response to MSU crystal deposition with regards to
gouty nephropathy. The present study investigated the role of the
NLRP3 inflammasome signalling pathway with the
progression of hyperuricemia and gouty nephropathy, the results of
which may provide a novel theoretical basis and therapeutic target
for the early prevention and treatment of gouty nephropathy.
Materials and methods
Study subjects
A total of 45 male patients (18-70 years old) were
recruited at the People's Hospital of Shenzhen Baoan between July
2016 and December 2017. According to the inclusion and exclusion
criteria, these patients were divided into three groups (n=15): The
control group, the hyperuricaemia group and the gouty nephropathy
group. The present study was approved by the Ethics Committee of
the Affiliated Bao'an Hospital of Shenzhen (approval no.
BYL2016001). Written informed consent was obtained from all
participants.
Inclusion criteria
Patients in the control group received a health
examination. There were no abnormalities in the laboratory
indicators of the selected subjects and patients had no history of
cardiovascular disease or liver disease (including diabetes and
gout). Patients also had no presence of infection or autoimmune
disease. Hyperuricaemia was defined as levels of serum uric acid
>6-7 mg/dl (12). The diagnosis
of gouty nephropathy was based on the diagnosis of primary gout
(13), with one or more of the
following parameters: Urinary protein >150 mg/dl; urine white
blood cells >5/high power field (HPF); urine red blood cells
>3/high power field; serum creatinine >115 µmol/l; blood uric
acid/creatinine ratio >2.5; ultrasound or ureterography
revealing renal calculus and kidney shrinkage. All of the
aforementioned cases excluded urinary tract infections and other
diseases such as cancer.
Exclusion criteria
Exclusion criteria was based on previous literature
(14) and was as follows: female;
<18 years old or >70 years old; patients with secondary
hyperuricaemia or stage 4-5 chronic kidney disease; acute
hyperuricaemia and the presence of acute renal function
deterioration factors; patients with severe cardiovascular disease,
liver and kidney disease, lung disease, fractures, tumors,
infectious and autoimmune disease, and mental illness; diseases
that may affect NLRP3 inflammasome signalling pathways;
patients who had been using uric acid drugs outside the hospital or
had been treated with lipid-lowering drugs or anti-inflammatory and
anti-oxidative drugs during the 4 weeks prior to admission.
Detection of organ function
indicators
Biochemical serum and urine samples were obtained
following 8 h fasting. A total of 15 ml serum sample was collected
from each patient and shipped to the Laboratory Services at the
Affiliated Bao'an Hospital of Shenzhen (Guangdong, China) for
biochemical analysis, which was obtained by centrifugation at 500 x
g for 10 min at 4˚C. Urinary biochemical parameters were measured
from the patients' first morning urine sample. Other standard
parameters were measured including blood and urine analysis. The
parameters tested for blood included white blood cell (WBC), red
blood cell (RBC), hemoglobin (HB), platelets (PLT), total
cholesterol (TC), total triglycerides (TG), low-density lipoprotein
(LDL), high-density lipoprotein (HDL), total protein (TP), albumin
(ALB), alanine aminotransferase (ALT), aspartate aminotransferase
(AST), serum creatinine (Cr), uric acid (UC), blood urea nitrogen
(BUN), prothrombin time (PT), activated partial thromboplastin time
(APTT), thrombin time (TT) and fibrinogen (FIB). Other parameters
tested for urine were urine leukocytes, urine erythrocytes and
urine protein.
Isolation and culture of peripheral
blood mononuclear cells
Centrifugation (speed, 1,600 x g; duration, 10 min;
temperature, 4˚C) was performed on blood samples in an EDTA tube to
obtain the buffy coat. Samples were stored at -80˚C for future use.
Peripheral blood mononuclear cells were isolated with Ficoll-Paque
Plus (GE Healthcare). Monocytes were isolated by magnetic bead
negative selection according to the instructions of
Dynabeads® Untouched™ Human Monocytes kit (Invitrogen;
Thermo Fisher Scientific, Inc.).
Isolated monocyte lysates were separated from which
5 µl was extracted for viability verification. Trypan Blue staining
confirmed that the viability of the cells was >95%. Cell
concentration was then adjusted to 5x105 cells/ml using
a hemocytometer using the following formula: Number of cells/ml =
number of cells counted in 100 small grids/100x400x10,000x dilution
factor. The total number of monocytes was counted to be
34.311±19.912x105. A third of which was used for RNA
extraction, whilst the rest were used for western blotting. The
number of monocytes for RNA extraction and western blot analysis
were divided 1:2. A total of 200 µl TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.) was added to monocytes for RNA extraction,
whereas 30-50 µl PBS (Invitrogen; Thermo Fisher Scientific, Inc.)
was added for western blotting experiments.
Reverse transcription-quantitative PCR
(RT-qPCR) to determine
NLRP3, ASC and caspase-1 mRNA expression in
peripheral blood mononuclear cells. Total RNA was extracted from
monocytes using TRIzol solution (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. RNA
concentration and purity were assessed via electrophoresis and a UV
spectrophotometer (Thermo Fisher Scientific, Inc.), respectively.
RNA was reverse transcribed to cDNA following the protocol provided
by the RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.). cDNA was stored at -20˚C prior to qPCR
amplification using the TransStart® Tip Green qPCR
Supermix 2X kit (TransGen Biotech Co., Ltd.), using the SYBR-Green
I fluorophore, according to manufacturer's protocol. All qPCR
reactions were performed in an Applied Biosystems 7500 system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) using the
following thermocycling conditions: Initial denaturation at 95˚C
for 30 sec, followed by 40 cycles of 95˚C for 5 sec and 60˚C for 30
sec. Finally, the melting curve was generated (dissociation, 60˚C
at 30 sec and 95˚C at 15 sec) to determine normality. Gene
expression was calculated using the 2-ΔΔCq
method (15). β-actin was used as an
internal reference for the mRNA levels of NLRP3, ASC and
caspase-1 detected. The primer sequences used for qPCR are
presented in Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Forward primer | Reverse primer |
|---|
| β-actin |
5'-AACCGCGAGAAGATGACCCAGAT-3' |
5'-GGATAGCACAGCCTGGATAGCA-3' |
|
NLRP3 |
5'-ATGGGTTTACTGGAGTACCTTTC-3' |
5'-CTGTCTTCAATGCACTGGAATCTG-3' |
| ASC |
5'-GATGCTCTGTACGGGAAGGTC-3' |
5'-TCCAGTTCCAGGCTGGTGT-3' |
| Caspase-1 |
5'-GGAAGACTCATTGAACATATGCAAG-3' |
5'-CTTGTCAAAGTCACTCTTTCAGTG-3' |
Western blot analysis to
determine
NLRP3, ASC and caspase-1 protein
expression in peripheral blood mononuclear cells. Cells were lysed
using ice-cold RIPA lysis buffer (Beijing Solarbio Science &
Technology Co., Ltd.) and protein concentration was quantified
using Bicinchoninic Acid Assay kit (Thermo Fisher Scientific,
Inc.). Protein samples (10 µg) were separated by 8, 10 or 12%
SDS-PAGE before being transferred to PVDF membranes. The membranes
were then blocked with 5% milk dissolved in TBS supplemented with
1% Tween-20 at room temperature for 1 h, prior to incubation with
primary antibodies against NLRP (1:300; cat. no. sc-134306; Santa
Cruz Biotechnology, Inc.), caspase-1 (1:300; cat. no. sc-622; Santa
Cruz Biotechnology, Inc.), ASC (1:300; cat. no. sc-514414; Santa
Cruz Biotechnology, Inc.) or GAPDH (1:1,000; cat. no. A01020;
Abbkine, Inc.) diluted in PBS supplemented with 0.1% Tween-20
(PBST) and 1% bovine serum albumin [Sangon Biotech (Shanghai) Co.,
Ltd.] at 4˚C overnight. Subsequently, the PVDF membranes were
incubated with horseradish peroxidase-conjugated goat anti-mouse
(1:1,000; cat.no. A0216; Beyotime Institute of Biotechnology) or
goat anti-rabbit (1:10,000; cat. no. A21020-1; Abbkine, Inc.)
secondary antibodies diluted in PBST supplemented with 5% milk at
room temperature for 1 h with agitation. The proteins were then
visualized using Pierce™ ECL Western Blotting Substrate (Thermo
Fisher Scientific, Inc.) and all bands were analysed using Gel Doc™
XR+ Gel Documentation system (Bio-Rad Laboratories, Inc.) to
calculate the densitometric values.
ELISA for the determination of IL-1β
and IL-18 levels in plasma
IL-1β (cat. no. 70-EK101B2) and IL-18 (cat. no.
70-EK1182) ELISA kits [Hangzhou Multi Sciences (Lianke) Biotech
Co., Ltd.] were used to determine the concentration of IL-1β and
IL-18 in 2 ml total plasma, according to manufacturer's
protocol.
Statistical analysis
Each experiment was repeated three times. All data
were analysed using SPSS v.17.0 (SPSS, Inc.) software and expressed
as the mean ± standard deviation. One-way analysis of variance was
performed to compare the mean of multiple groups and a least
significant difference post hoc test was used. P<0.05 was
considered to indicate a statistically significant difference.
Results
Kidney function is damaged in
hyperuricaemia and gouty nephropathy groups
Patient general characteristics are provided in
Table II, Figs. 1 and 2. The expression of uric acid (Fig. 1C), blood urea nitrogen (Fig. 1B) and blood lipids (Fig. 1F) including total cholesterol, total
triglycerides and low-density lipoprotein was higher in the
hyperuricaemia and gouty nephropathy groups compared with the
control group, but the level of high-density lipoprotein exhibited
the opposite trend. Levels of urine leukocytes and erythrocytes
were higher in the gouty nephropathy group compared with both the
control and hyperuricemia groups (Fig. 2E-F). Serum creatinine
(Fig. 1A), uric acid (Fig. 1C), blood urea nitrogen (Fig. 1B) were greater in the gouty
nephropathy group and hyperuricaemia group compared with the
control group. Serum creatinine and urine protein are common
indicators of renal function and used to monitor the progression of
chronic kidney disease (16). These
results demonstrated that the kidney had been further damaged in
the progression from hyperuricaemia to gouty nephropathy.
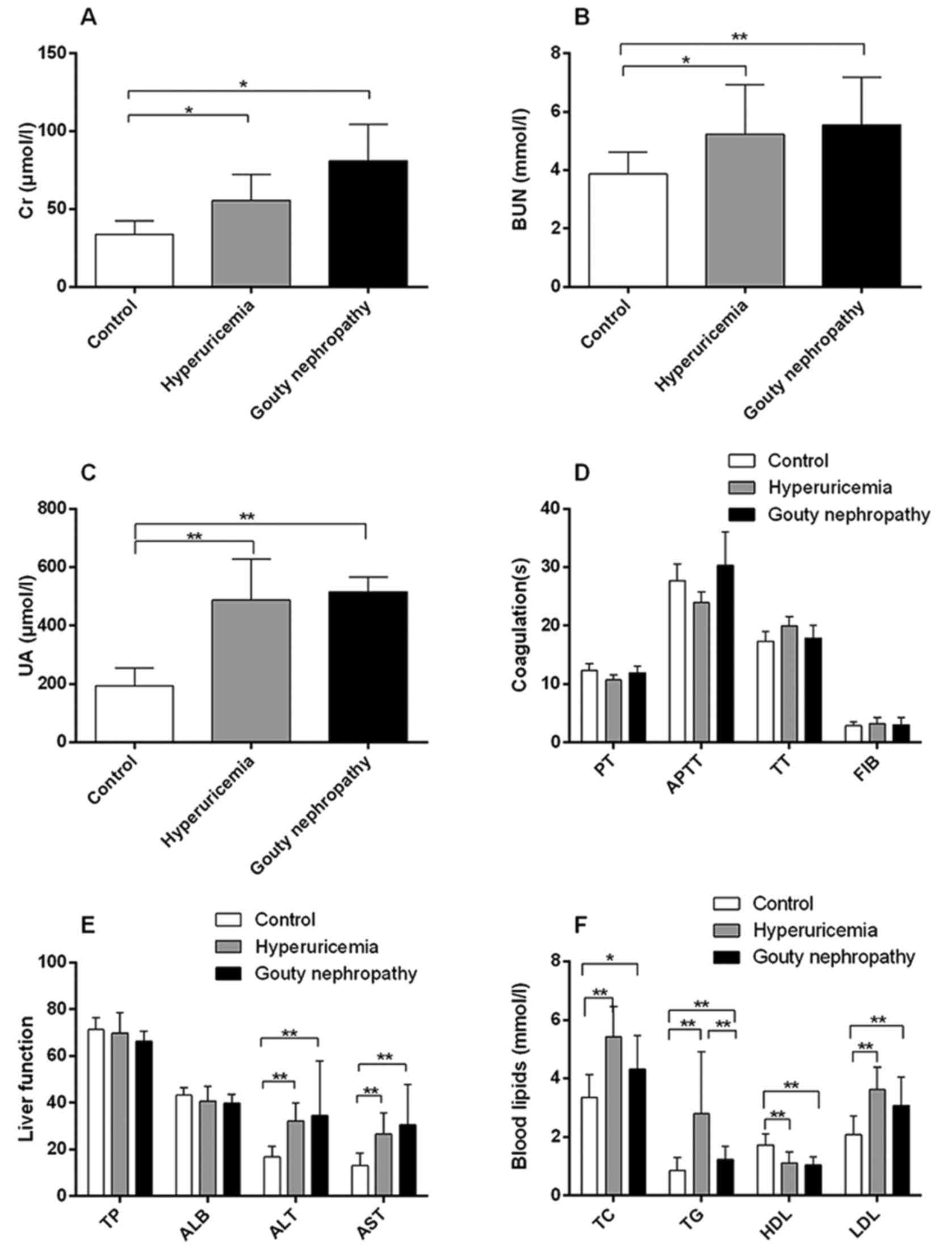 | Figure 1Organ function indicators of enrolled
patients, including renal and liver function, coagulation and blood
lipids. (A) Levels of Cr, (B) BUN, (C) UA, (D) coagulation
functions, (E) liver function and (F) blood lipids of patients in
the control, hyperuricemia and gouty nephropathy groups.
*P<0.05 and **P<0.01, with
comparisons indicated by lines. Cr, creatinine; BUN, blood urea
nitrogen; UA, uric acid; PT, prothrombin time; APTT, activated
partial thromboplastin time; TT, thrombin time; FIB, fibrinogen;
TP, total protein; ALB, albumin; ALT, alanine aminotransferase;
AST, aspartate aminotransferase; TC, total cholesterol; TG, total
triglycerides; HDL, high density lipoprotein; LDL, low density
lipoprotein. |
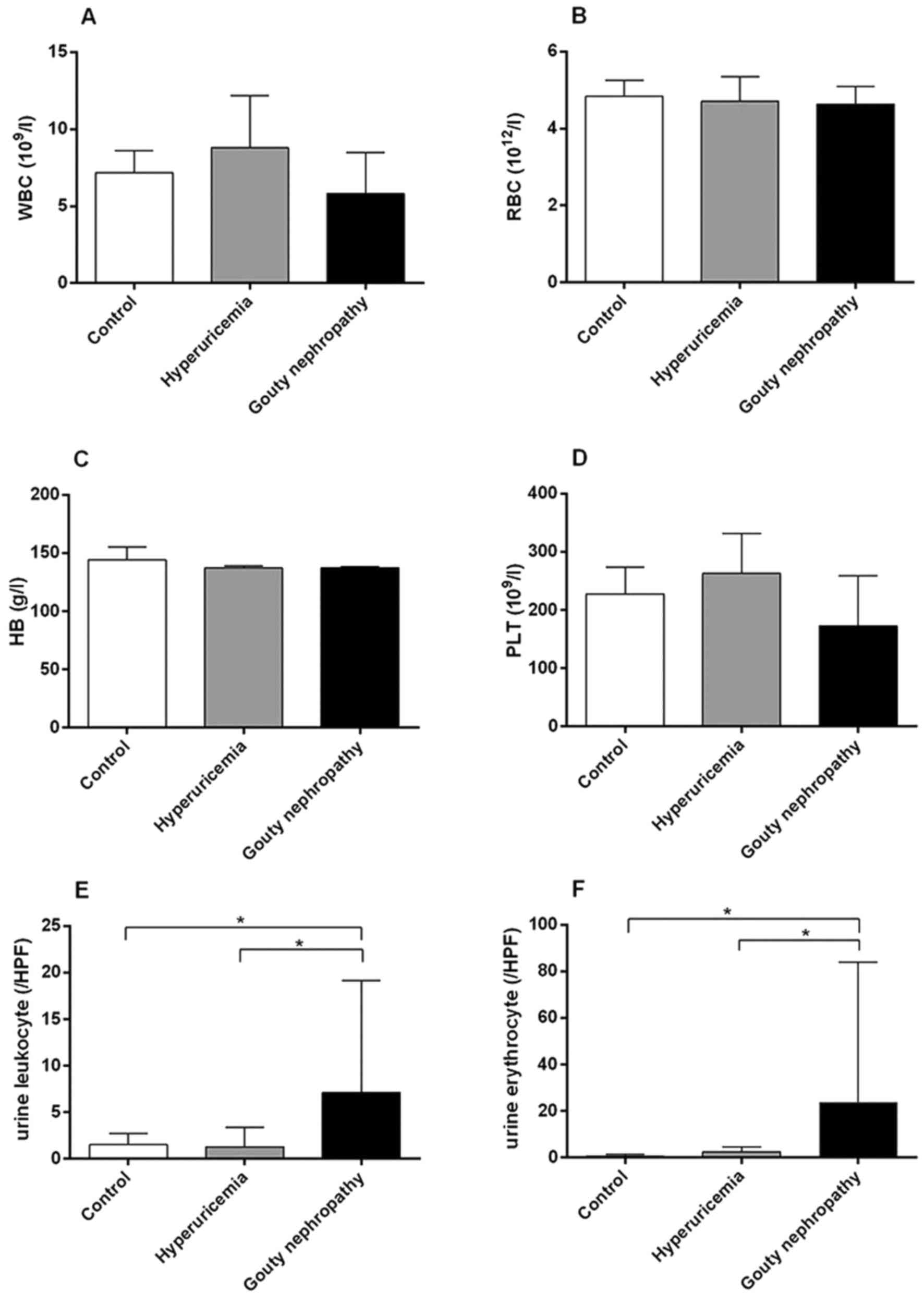 | Figure 2Organ function indicators of enrolled
patients, including blood and urine analysis. (A) WBC, (B) RBC, (C)
HB, (D) PLT, (E) urine leukocytes and (F) urine erythrocytes levels
of patients in the control, hyperuricemia and gouty nephropathy
groups. *P<0.05, with comparisons indicated by lines.
WBC, white blood cell; RBC, red blood cell; HB, hemoglobin; PLT,
platelets; HPF, high power field. |
| Table IIGeneral characteristics of enrolled
patients. |
Table II
General characteristics of enrolled
patients.
|
Characteristics | Control (n=15) | Hyperuricemia
(n=15) | Gouty nephropathy
(n=15) |
|---|
| Age (years) | 45.20±10.19 | 48.27±14.91 | 48.3±11.5 |
| Height (m) | 1.72±0.06 | 1.70±0.05 | 1.66±0.05 |
| Body weight
(kg) | 72.25±6.97 | 69.98±12.08 | 61.39±6.83 |
| BMI
(kg/m2) | 24.28±1.53 | 24.38±4.53 | 22.19±2.43 |
| SBP (mmHg) | 116.00±7.61 | 115.93±9.83 | 121.33±10.87 |
| DBP (mmHg) | 79.27±7.98 | 76.47±7.60 | 74.80±8.78 |
| FBG (mmol/l) | 4.73±0.45 | 5.17±0.70 | 4.77±0.39 |
Successful peripheral blood
mononuclear cell isolation
Peripheral blood mononuclear cells were isolated
from the peripheral blood of 45 patients for in vitro
experiments. Trypan blue staining (Fig.
3) confirmed that >95% were viable cells (Table III).
| Table IIIPeripheral blood mononuclear cells
count. |
Table III
Peripheral blood mononuclear cells
count.
| Group | n | Count
(105) |
|---|
| Control | 15 | 33.047±22.755 |
| Hyperuricemia | 15 | 43.627±18.317 |
| Gouty
nephropathy | 15 | 26.567±13.506 |
NLRP3, ASC and caspase-1
mRNA expression are increased in the hyperuricaemia and gouty
nephropathy groups
NLRP3, ASC and caspase-1 mRNA levels were
significantly increased in the hyperuricaemia and gouty nephropathy
groups compared with the control group (Table IV). Compared with the hyperuricaemia
group, the gouty nephropathy group had a significantly higher
expression of ASC and caspase-1 mRNA (Table IV). The mRNA expression in
peripheral blood mononuclear cells demonstrated that the
NLRP3 inflammasome may serve a pivotal role in gouty
nephropathy.
| Table IVExpression of NLRP3, ASC
and caspase-1 mRNA in peripheral blood mononuclear cells. |
Table IV
Expression of NLRP3, ASC
and caspase-1 mRNA in peripheral blood mononuclear cells.
| Group | n |
NLRP3 | ASC | Caspase-1 |
|---|
| Control | 15 | 23.54±1.19 | 25.88±1.18 | 26.99±1.43 |
| Hyperuricemia | 15 |
26.05±1.89a |
27.49±3.56a |
28.05±2.41a |
| Gouty
nephropathy | 15 |
25.43±1.66a |
32.21±3.01a,c |
30.80±2.80a,b |
Expression of NLRP3, ASC,
caspase-1 protein increases in the hyperuricaemia and gouty
nephropathy group
Gouty nephropathy group had a significantly higher
expression of NLRP3, ASC and caspase-1 protein compared
with the control group (Table V).
The expression of ASC and caspase-1 protein was higher in the gouty
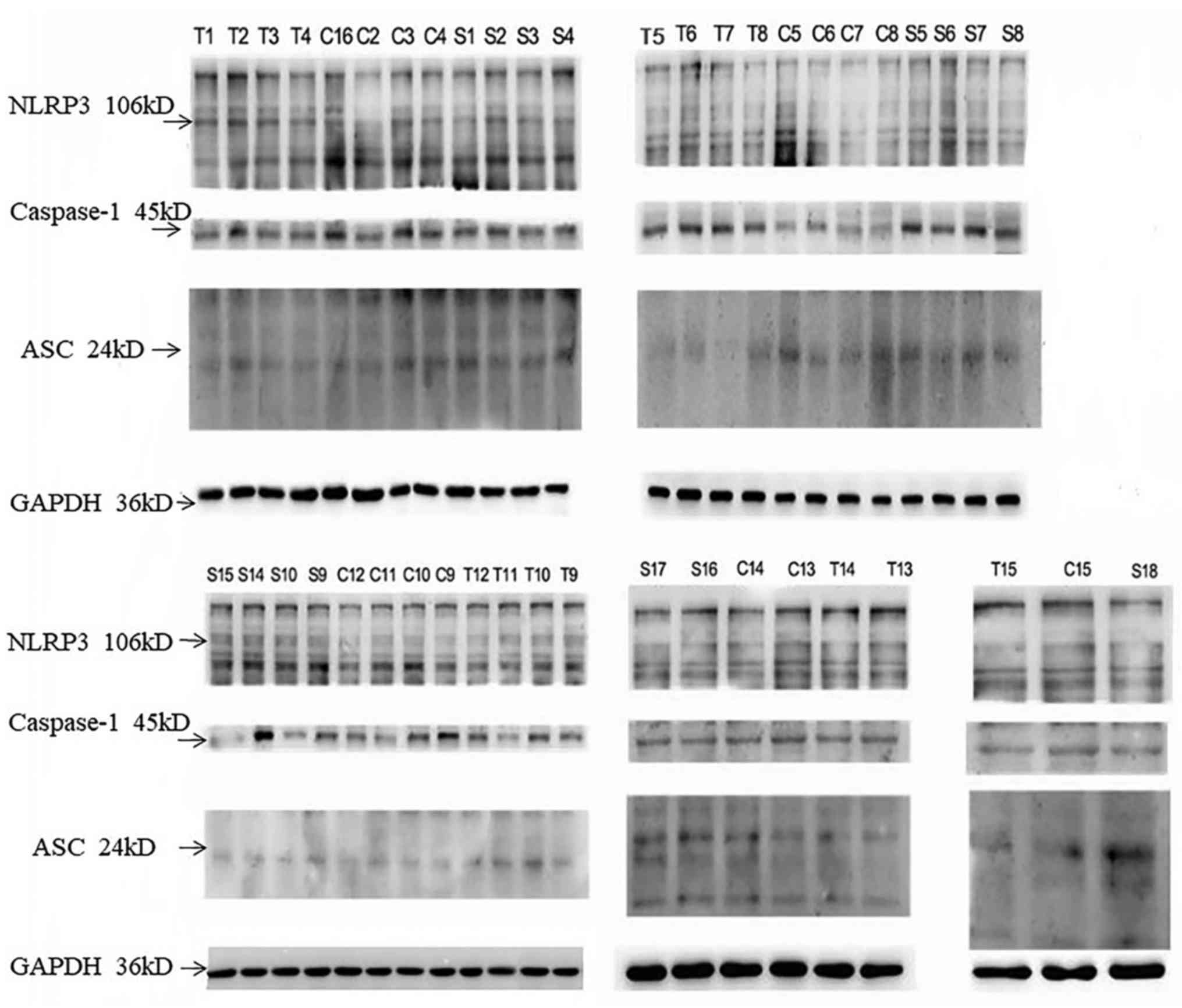
nephropathy group compared with the hyperuricaemia group (Table V; Fig.
4). Peripheral blood mononuclear cell protein expression
(Fig. 4; Table V) also corresponded with mRNA
expression (Table IV), indicating
the important role of the NLRP3 inflammasome in gouty
nephropathy.
| Table VExpression of NLRP3, ASC,
caspase-1 protein in peripheral blood mononuclear cells. |
Table V
Expression of NLRP3, ASC,
caspase-1 protein in peripheral blood mononuclear cells.
| Group | n |
NLRP3/GAPDH | ASC/GAPDH |
Caspase-1/GAPDH |
|---|
| Control | 15 | 0.45±0.18 | 0.22±0.08 | 0.57±0.16 |
| Hyperuricemia | 15 | 0.55±0.12 | 0.26±0.11a | 0.57±0.16 |
| Gouty
nephropathy | 15 |
0.60±0.16b |
0.47±0.19a,c |
0.78±0.15b,c |
Expression of IL-1β and IL-18 is
increased in the hyperuricaemia and gouty nephropathy group
IL-1β and IL-18 levels were significantly higher in
the hyperuricaemia and gouty nephropathy groups compared with the
control group (Table VI). IL-1β
expression was significantly elevated in the gouty nephropathy
group compared with the hyperuricaemia group (Table VI). The increased expression of
inflammatory factors demonstrated that the NLRP3
inflammasome was associated with gouty nephropathy.
| Table VIExpression of IL-1β and IL-18 in
plasma of patients. |
Table VI
Expression of IL-1β and IL-18 in
plasma of patients.
| Group | n | IL-1β (pg/ml) | IL-18 (pg/ml) |
|---|
| Control | 15 | 28.41±14.05 | 323.35±96.16 |
| Hyperuricemia | 15 |
55.19±16.79a |
718.16±211.42a |
| Gouty
nephropathy | 15 |
81.20±17.63a,b |
870.67±371.13a |
Discussion
Uric acid is involved in the pathogenesis of kidney
disease. Increasing evidence supports uric acid as a cause or
exacerbating factor for kidney fibrosis and progressive CKD with an
elevated serum uric acid level independently predicting the
development of CKD (17,18). Elevated uric acid levels induce
oxidative stress and endothelial dysfunction, resulting in the
development of both systemic and glomerular hypertension (19,20). It
is well established that uric acid forms urate crystals in
sufficient concentrations to block renal collecting tubes, which
causes interstitial nephritis, gouty nephropathy and interstitial
fibrosis (21). Furthermore, the
kidney damage caused by uric acid is the result of this sort of
obstruction and crucially, it can be exacerbated by the
inflammatory response that it initiates. Uric acid activates
cytoplasmic phospholipase A2 and inflammatory transcription factor
NF-κB, which increases the production of various systemic
cytokines, including tumour necrosis factor-α, kidney monocyte
chemotactic protein 1 and blood vessel COX-2 (22,23). In
addition, hyperuricaemia aggravates kidney injury by recruiting
IL-1β-secreting macrophages, activating NLRP3
inflammasomes in macrophages and promoting chemokine secretion in
proximal tubular cells (24). Uric
acid, in its soluble form, is responsible for increasing the
production of IL-1β in an NLRP3-dependent manner and is
associated with kidney damage (25).
The inflammatory response is closely associated with
the innate immune system. Intracellular nucleotide-binding
oligomerization domain-like receptors form a group of pattern
recognition receptors that are involved in a wide variety of innate
host immune responses when stimulated by pathogen-associated
molecular patterns dangerous-associated molecular patterns (DAMPs)
(26). One of the most thoroughly
investigated members is the intracellular pattern recognition
receptor NLRP3 inflammasome complex consisting of
NLRP3, ASC and caspase-1(27). Upon activation, NLRP3
oligomerizes via homotypic interactions between NACHT domains and
raises ASC and caspase to form an inflammatory complex, resulting
in activated caspase-1. Activated caspase-1 cleaves intracellular
IL-1β and IL-18 precursors to form mature IL-1β and IL-18, which
are secreted into the extracellular matrix (Fig. 5) (28). The inflammasome is understood to have
a fundamental role in the progression of autoinflammatory diseases
and to have additional roles in infection control, the progression
of immune pathologies and the recognition of tissue damage.
NLRP3 inflammasome is activated in response to a variety
of infectious stimuli or by cellular stress caused by various
sterile danger signals, including high concentrations of
extracellular ATP, a decrease in extracellular osmolality or pH,
crystals of MSU or cholesterol and the degradation of extracellular
matrix components (27).
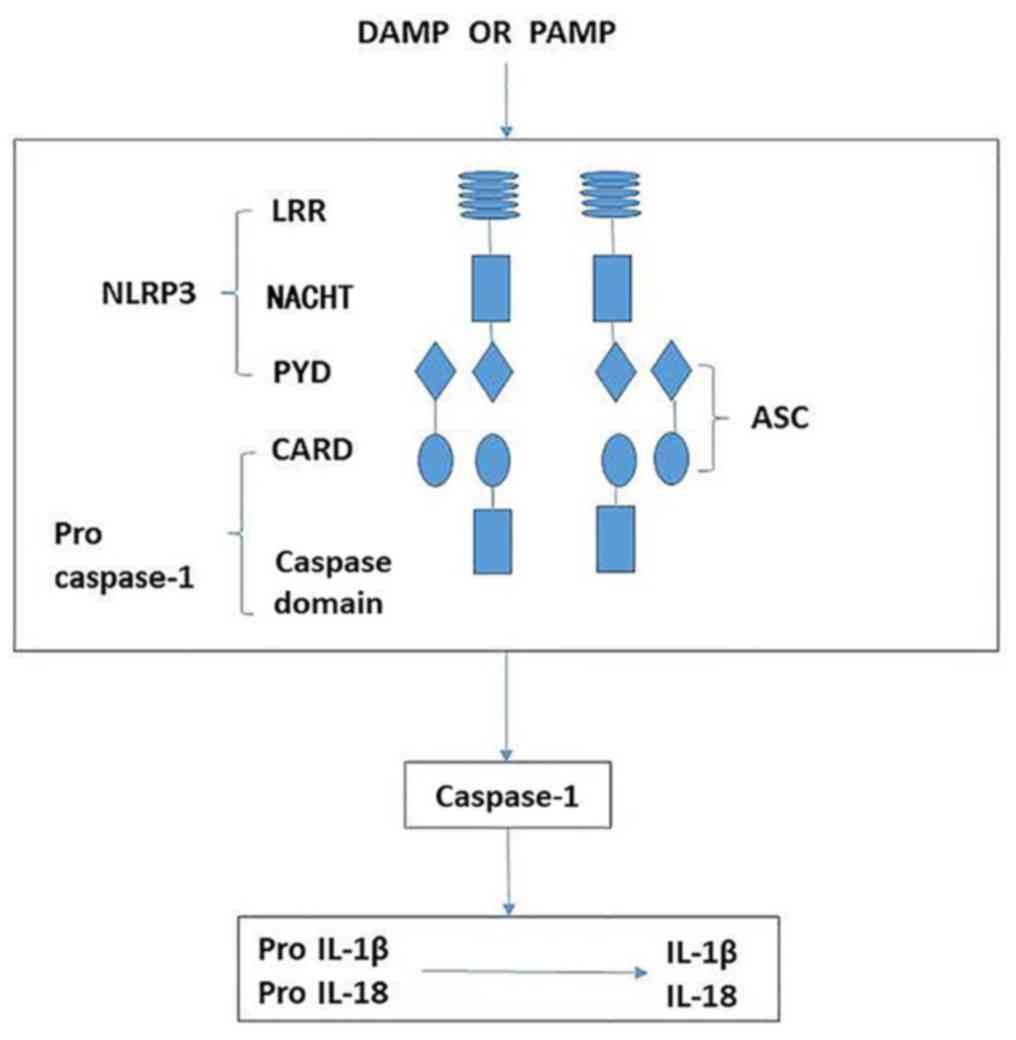 | Figure 5Schematic demonstrating the
activation of NLRP3 leading to release of
proinflammatory factors. The NLRP3 inflammasome is
comprised of NLRP3, ASC and caspase-1. When sensing
danger signals, including PAMP and DAMP, NLRP3
oligomerizes via homotypic interactions between NACHT domains. The
PYD of NLRP3 is then exposed for interaction with the
PYD of ASC. The CARD of ASC in turn recruits pro-caspase-1 through
CARD-CARD interactions, resulting in activated caspase-1. Activated
caspase-1 then cleaves intracellular IL-1β and IL-18 precursors to
form mature IL-1β and IL-18, which are secreted into the
extracellular matrix. NLRP3, nucleotide-binding domain
and leucine-rich repeat protein 3; ASC, apoptosis-associated speck
like protein; LRR, leucine-rich repeat; NACHT, nucleotide-binding
and oligomerization domain; PYD, pyrin domain; CARD, caspase
recruitment domain; IL-1β, interleukin-1β; IL-18, interleukin-18;
PAMP, pattern-associated molecular pattern; DAMP, danger-associated
molecular pattern. |
In the present study, the concentration of uric acid
and creatinine as well as the expression of mRNA and protein in
NLRP3 inflammasomes including NLRP3 mRNA, ASC
mRNA, caspase-1 mRNA and ASC protein levels increased in the
hyperuricaemia and gouty nephropathy groups compared with the
control group. Importantly, the elevated levels of IL-1β and IL-18
indicated that NLRP3 inflammasomes were activated in
hyperuricaemia and gouty nephropathy. The expression of ASC and
caspase-1 mRNA and protein was higher in the gouty nephropathy
group than in the hyperuricaemia group with the level of IL-1β also
being significantly increased. The results indicated that the
NLRP3 inflammasome was activated and induced the
polymerization of the adaptor molecule ASC, leading to the
production of IL-1β. Taken together, these results indirectly
indicated that the NLRP3 inflammasome has a pivotal role
in the progression of gouty nephropathy.
Although the specific activation mechanism of the
NLRP3 inflammasome is unclear, recent studies have
proposed three hypotheses regarding the activation of
NLRP3 inflammasomes: Firstly, all bacteria, viruses,
particles or crystals can stimulate cells to produce reactive
oxygen species, which can activate the NLRP3
inflammasome (29). In the course of
oxidative stress, thioredoxin, following its own oxidation,
releases thioredoxin-interacting protein, activating the
NLRP3 inflammasome (30).
Secondly, crystals or particles, such as MSU, silicon dioxide,
amyloid and cholesterol crystals, are phagocytosed by macrophages
into lysosomes. This accumulation causes lysosomal rupture and the
release of cathepsin B and other proteases, inducing the activation
of the NLRP3 inflammasome (31,32)
Finally, stimulation induces cells to release ATP on the cell
surface of the P2X7 purine receptor, triggering the potassium ion
outflow mechanism and calcium wave transport mechanism leading to
mitochondrial damage. This releases mitochondrial DNA (mtDNA) and
activates the NLRP3 inflammasome (33,34).
Previous studies have demonstrated that NLRP3 can
identify uric acid crystals and other dangerous signals (3,35). Uric
acid crystals trigger IL-1β-mediated inflammation by activating the
NLRP3 inflammasome via the aforementioned three
mechanisms (36).
The present study hypothesized that MSU acts as an
endogenous DAMP, contributing to the progression of the
NLRP3 inflammasome in hyperuricaemia and gouty
nephropathy. When high levels of uric acid in the body cause uric
acid crystals to be deposited in tissues, macrophages directly
phagocytose urate crystals, which are too large to be efficiently
cleared and are likely to induce the production of ROS on their way
to becoming lysosomes (9). Following
the phagocytosis of MSU, macrophages form phagocytic bodies and the
subsequent intracellular rupture of these bodies releases cathepsin
B, which activates the NLRP3 inflammasome (37). MSU also activates the
NLRP3 inflammasome by causing intracellular potassium
efflux (38), leading to
mitochondrial damage. Furthermore, by releasing mtDNA, the
maturation and release of IL-1β and IL-18 is stimulated. A previous
study determined that uric acid activates NLRP3
inflammasomes via mitochondrial ROS production and their results
demonstrated the direct role of hyperuricaemia in activating
NLRP3 inflammasomes in macrophages (39). Berberine may influence the
NLRP3 inflammasome and be involved in MSU
crystal-induced innate immune responses, attenuating the expression
of NLRP3, further inhibiting the downstream signalling
of molecular IL-1β and eventually having a role in the treatment of
gouty arthritis (40). The present
study observed that gouty nephropathy is associated with elevated
levels of uric acid in serum and high expression of
NLRP3, ASC and caspase-1 mRNA and protein, as well as
IL-1β and IL-18 expression. The present results indicated that uric
acid-induced NLRP3 inflammasomes are associated with
gouty nephropathy with the underlying mechanism potentially
involving one of the aforementioned three hypotheses.
The present study has limitations as the exact
association between NLRP3 inflammasomes and gouty
nephropathy was not elucidated. Further studies are therefore
required to explore the role of the NLRP3 inflammasome
in humans and in vivo. In addition to the need for large
clinical trials, more studies are required to better understand the
biology of uric acid. To the best of our knowledge, clinical study
into the regulation mechanism of the NLRP3 inflammasome
signalling pathway with regards to gouty nephropathy has not been
performed.
In conclusion, the present study demonstrated that
the NLRP3 inflammasome was associated with the
progression of hyperuricaemia and gouty nephropathy, leading to the
inflammatory response and renal damage. Although the precise
mechanism of the NLRP3 inflammasome signalling pathway
and the means by which MSU triggers the inflammasome remains a
matter of debate, the importance of the NLRP3
inflammasome in gouty nephropathy has been established. The present
study proposes that future therapeutic strategies for gouty
nephropathy should be based on blocking uric acid or inhibiting its
activation of the NLRP3 inflammasome. Further studies
will be required to explore the mechanism of how uric acid
activates the NLRP3 inflammasome to allow for the early
diagnosis of gouty nephropathy.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Shenzhen Science and Technology Research and Development
Fundamental Research Project (grant no. JCYJ20160427191440905).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZZ and XLS designed the experiments, analyzed the
data and wrote the manuscript. YPX and YZZ performed the
experiments. FJG and ASZ analyzed and interpreted the data. JHC
contributed to the design of the experimental methods, wrote the
manuscript and approved the version to be published and agreed to
be accountable for all aspects of the work. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Affiliated Bao'an Hospital of Shenzhen (approval
no. BYL2016001) and written informed consent was obtained from all
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ryu ES, Kim MJ, Shin HS, Jang YH, Choi HS,
Jo I, Johnson RJ and Kang DH: Uric acid-induced phenotypic
transition of renal tubular cells as a novel mechanism of chronic
kidney disease. Am J Physiol Renal Physiol.
304(F471-F480)2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Siu YP, Leung KT, Tong MK and Kwan TH: Use
of allopurinol in slowing the progression of renal disease through
its ability to lower serum uric acid level. Am J Kidney Dis.
47:51–59. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ghaemi-Oskouie F and Shi Y: The role of
uric acid as an endogenous danger signal in immunity and
inflammation. Curr Rheumatol Rep. 13:160–166. 2011.[J]. PubMed/NCBI View Article : Google Scholar
|
|
4
|
Roddy E, Zhang W and Doherty M: The
changing epidemiology of gout. Nat Clin Pract Rheumatol. 3:443–449.
2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kim IY, Lee DW, Lee SB and Kwak IS: The
role of uric acid in kidney fibrosis: Experimental evidences for
the causal relationship. BioMed Res Int. 2014(638732)2014.
View Article : Google Scholar
|
|
6
|
Saito I, Saruta T, Kondo K, Nakamura R,
Oguro T, Yamagami K, Ozawa Y and Kato E: Serum uric acid and the
renin-angiotensin system in hypertension. J Am Geriatr Soc.
26:241–247. 1978.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gao L, Shen F and Lii YS: Effects of
NLRP3 inflammasome on cerebral ischemia reperfusion
injury. J Shanghai Jiaotong University Medical Science.
35:1896–1899. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Heneka MT, Kummer MP, Stutz A, Delekate A,
Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et
al: NLRP3 is activated in Alzheimer's disease and
contributes to pathology in APP/PS1 mice. Nature. 493:674–678.
2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tschopp J and Schroder K: NLRP3
inflammasome activation: The convergence of multiple signalling
pathways on ROS production? Nat Rev Immunol. 10:210–215.
2010.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Zheng SC, Zhu XX, Xue Y, Zhang LH, Zou HJ,
Qiu JH and Liu Q: Role of the NLRP3 inflammasome in the
transient release of IL-1β induced by monosodium urate crystals in
human fibroblast-like synoviocytes. J Inflamm (Lond).
12(30)2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Schiltz C, Lioté F, Prudhommeaux F,
Meunier A, Champy R, Callebert J and Bardin T: Monosodium urate
monohydrate crystal-induced inflammation in vivo: Quantitative
histomorphometric analysis of cellular events. Arthritis Rheum.
46:1643–1650. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Xu X, Hu J, Song N, Chen R, Zhang T and
Ding X: Hyperuricemia increases the risk of acute kidney injury: A
systematic review and meta-analysis. BMC Nephrol. 18:27–41.
2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wallace SL, Robinson H, Masi AT, Decker
JL, McCarty DJ and Yü TF: Preliminary criteria for the
classification of the acute arthritis of primary gout. Arthritis
Rheum. 20:895–900. 1977.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang XX, Sun WF, Hou Y, Liu YW and He Y:
Clinical observation on fufang tufuling keli treating 40 cases of
hyperuricemia: a randomize-controlled study. J Tradit Chin Med.
57:41–45. 2016.
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-ΔΔC(T)) method. Methods. 25:402–408. 2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Levey AS, de Jong PE, Coresh J, El Nahas
M, Astor BC, Matsushita K, Gansevoort RT, Kasiske BL and Eckardt
KU: The definition, classification, and prognosis of chronic kidney
disease: A KDIGO Controversies Conference report. Kidney Int.
80:17–28. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Weiner DE, Tighiouart H, Elsayed EF,
Griffith JL, Salem DN and Levey AS: Uric Acid and incident kidney
disease in thy community. J Am soc Nephriol. 19:1204–1211.
2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Weiner DE, Tighiouart H, Elsayed EF,
Griffith JL, Salem DN and Levey AS: Uric acid and incident kidney
disease in the community. J Am Soc Nephrol. 19:1204–1211.
2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sánchez-Lozada LG, Soto V, Tapia E,
Avila-Casado C, Sautin YY, Nakagawa T, Franco M, Rodríguez-Iturbe B
and Johnson RJ: Role of oxidative stress in the renal abnormalities
induced by experimental hyperuricemia. Am J Physiol Renal Physiol.
295(F1134-F1141)2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kang DH, Nakagawa T, Feng L, Watanabe S,
Han L, Mazzali M, Truong L, Harris R and Johnson RJ: A role for
uric acid in the progression of renal disease. J Am Soc Nephrol.
13:2888–2897. 2002.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu LQ: Pathogenesis of hyperuric acid
nephropathy. J Clin Nephrol. 11:53–55. 2011.
|
|
22
|
Kanellis J, Watanabe S, Li JH, Kang DH, Li
P, Nakagawa T, Wamsley A, Sheikh-Hamad D, Lan HY, Feng L, et al:
Uric acid stimulates monocyte chemoattractant protein-1 production
in vascular smooth muscle cells via mitogen-activated protein
kinase and cyclooxygenase-2. Hypertension. 41:1287–1293. 2003.[J].
PubMed/NCBI View Article : Google Scholar
|
|
23
|
Prasad Sah OS and Qing YX: Associations
between hyperuricemia and chronic kidney disease: A Review.
Nephrourol Mon. 7(e27233)2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gul A and Zager P: Does altered uric acid
metabolism contribute to diabetic kidney disease pathophysiology?
Curr Diab Rep. 18(18)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Braga TT, Forni MF, Correa-Costa M, Ramos
RN, Barbuto JA, Branco P, Castoldi A, Hiyane MI, Davanso MR, Latz
E, et al: Soluble uric acid activates the NLRP3
inflammasome. Sci Rep. 7(39884)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Isaka Y, Takabatake Y, Takahashi A, Saitoh
T and Yoshimori T: Hyperuricemia-induced inflammasome and kidney
diseases. Nephrol Dial Transplant. 31:890–896. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Baroja-Mazo A, Martín-Sánchez F, Gomez AI,
Martínez CM, Amores-Iniesta J, Compan V, Barberà-Cremades M, Yagüe
J, Ruiz-Ortiz E, Antón J, et al: The NLRP3 inflammasome
is released as a particulate danger signal that amplifies the
inflammatory response. Nat Immunol. 15:738–748. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xia M, Boini KM, Abais JM, Xu M, Zhang Y
and Li PL: Endothelial NLRP3 inflammasome activation and
enhanced neointima formation in mice by adipokine visfatin. Am J
Pathol. 184:1617–1628. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140.
2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Martinon F: Signaling by ROS drives
inflammasome activation. Eur J Immunol. 40:616–619. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gasse P, Riteau N, Charron S, Girre S,
Fick L, Pétrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B,
et al: Uric acid is a danger signal activating NALP3 inflammasome
in lung injury inflammation and fibrosis. Am J Respir Crit Care
Med. 179:903–913. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jiang H, Yan YQ, Jiang W and Zhou RB:
NLRP3 inflammasome: Activation, regulation, and role in
diseases. Sci Sin Vitae. 47:125–131. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation.
Nature. 469:221–225. 2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jin-Feng LI, Xie D, Ping-Ping HE, Tang YY,
Yu-Lin TU and Yin K: Research advances of the NLRP3
inflammasome and metabolic disease. Prog Biochem Biophys.
41:425–434. 2014. View Article : Google Scholar
|
|
35
|
Martinon F, Pétrilli V, Mayor A, Tardivel
A and Tschopp J: Gout-associated uric acid crystals activate the
NALP3 inflammasome. Nature. 440:237–241. 2006.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xu LL: NLRP3 inflammasomes and
kidney diseases. J Nephrol Dialy Transpl. 20:554–558.
2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hornung V, Bauernfeind F, Halle A, Samstad
EO, Kono H, Rock KL, Fitzgerald KA and Latz E: Silica crystals and
aluminum salts activate the NALP3 inflammasome through phagosomal
destabilization. Nat Immunol. 9:847–856. 2008.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Stutz A, Golenbock DT and Latz E:
Inflammasomes: Too big to miss. J Clin Invest. 119:3502–3511.
2009.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kim SM, Lee SH, Kim YG, Kim S-Y, Seo JW,
Choi YW, Kim DJ, Jeong KH, Lee TW, Ihm CG, et al:
Hyperuricemia-induced NLRP3 activation of macrophage
contributes to the progression of diabetic nephropathy. Am J
Physiol-renal. 308:993–1003. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liu YF, Wen CY, Chen Z, Wang Y, Huang Y
and Tu SH: Effects of berberine on NLRP3 and IL-1β
expressions in monocytic THP-1 cells with monosodium urate
crystals-induced inflammation. Biomed Res Int. 2016(2503703)2016.
View Article : Google Scholar
|