Introduction
Urea cycle disorders (UCDs) are a set of hereditary
metabolic disorders caused by congenital enzymatic defects that are
characterized by high levels of ammonia in the blood (1). The urea cycle eliminates blood ammonia
by transforming toxic ammonia into non-toxic urea, which can then
be excreted in urine (2). Defects in
any of the enzymes associated with urea metabolism [namely
carbamoyl-phosphate synthetase (CPS1), ornithine
carbamoyltransferase, argininosuccinate synthetase,
argininosuccinate lyase, arginase and N-acetyl glutamate synthase]
may lead to the occurrence of UCDs (3). CPS1 deficiency can present as
life-threatening hyperammonemia, a UCD and a rare autosomal
recessive disorder of ureagenesis (4).
The human CPS1 gene is located at chromosome
2q34-35 and contains 38 exons and 4,500 coding nucleotides
(5-8).
CPS1 deficiency affects neonatal development and the severity of
the symptoms is largely parallel to the degree of enzymatic
defects, with complete enzyme deficiency being the most severe
(4). Neonatal onset CPS deficiency
leads to a poor prognosis, and patients who accept treatment with
long-term protein restriction and ammonia medication may experience
severe sequelae (9). The current
study presents the clinical performance, diagnosis, and treatment
of a case of CPS1 deficiency in a neonate and aims to increase
understanding of the causes and symptoms of CPS1 deficiency and
present new options for genetic screening.
Case report
Ethics. The present study was approved by the
Ethics Committee of Hunan Provincial People's Hospital. This study
complied with the World Medical Association Declaration of Helsinki
(10) regarding the ethical conduct
of research involving human subjects. Informed consent was obtained
from the parents of the patient.
Patient
A 3 day-old female was admitted to the Department of
Neonatology, Hunan Provincial People's Hospital, China in June 2016
due to anorexia and lethargy for 1 day. The patient was born to
healthy non-consanguineous Chinese parents following full-term
gestation, with a birth weight of 3.7 kg. Her mother was gravida 3,
para 2. No abnormality was noted within 48 h of the birth. However,
her parents noted drowsiness and poor responsiveness, without
obvious cause, after 72 h. This condition gradually worsened. As
the patient had a healthy 7 year old sister, abnormality based on
family medical history was discarded as a reason for the
condition.
Patient examination
Upon admission, routine examinations, including
physical examination, urinalysis, blood gas analysis, and blood
examination, were performed as described previously (11,12). In
addition, laboratory tests for C-reactive protein, pro-calcitonin,
toxoplasma, rubella, cytomegalovirus and herpes simplex viruses,
respiratory viruses (influenza virus, respiratory syncytial virus
and adenovirus), biochemistry, routine cerebrospinal fluid
analysis, blood ammonia, blood glucose, lactic acid, liver function
and renal function were performed (12,13).
Bacterial cultures of blood, urine, and cerebrospinal fluid samples
were conducted. Electrolytes, myocardial enzyme and magnetic
resonance imaging (MRI) examinations were performed (11). In addition, lumbar puncture manometry
was conducted. Liquid chromatography with tandem mass
spectrophotometry was performed in order to analyze compounds in
the serum of the patient. Venous blood was collected and dropped on
to a filter paper of ~10 mm in diameter and cooled at room
temperature. Amino, organic and fatty acid detection was then
performed as described previously (14).
Whole exome sequencing (WES)
WES was performed according to a previously reported
method (11). Briefly, genomic DNA
was extracted from 2 ml of peripheral blood from each family member
using a BloodGen Midi kit (Beijing CWBio Co., Ltd.). The DNA was
then sheared, hybridized, and enriched following the manufacturer's
protocol. Size, distribution and concentration of DNA was assessed
using an Agilent Bioanalyzer 2100 (Agilent Technologies Inc.). The
libraries were sequenced on an Illumina Hiseq2500 platform
(Illumina, Inc.).
Bioinformatics analysis
Raw sequencing files were processed using BclToFastq
software (version 2; Illumina, Inc.). Variations were only included
in the study where the quality score was ≥20. The National Center
for Biotechnology Information human reference genome (HG-19) was
employed for the alignment of the sequencing reads. Single
nucleotide polymorphisms and insertion-deletions in the sequences
were analyzed with Genome Analysis Toolkit software (version.3.7;
Broad Institute). Four online programs, including SIFT-SNP
prediction (http://snpeff.sourceforge.net/SnpSift.html), PolyPhen2
(v 2, 2013; http://genetics.bwh.harvard.edu/pph2/) (15), Protein Variation Effect Analyzer
(PROVEAN; 2012, http://provean.jcvi.org/index.php), and MutationTaster
(v2, 2014, http://www.mutationtaster.org), were used to predict
the pathogenicity of the variants. In the SIFT-SNP software, a
score >0.05 indicated that a change of amino acids could be
tolerated (16), while a score
<0.05 indicated that a change of amino acids could not be
tolerated, a lower score indicated a potentially poor tolerance of
an amino acid change. In the PolyPhen2 software, a score close to
0.00 meant that the change of amino acids could be tolerated, while
a score close to 1.00 meant that the change of amino acids could
not be tolerated, and a higher score indicated that great damage
could potentially be caused by amino acid change. In the PROVEAN
software, if the score was equal to or below a predefined threshold
(e.g. -2.5) (17), the protein
variant was predicted to have a ‘deleterious’ effect, while if the
PROVEAN score was above the threshold, the variant was predicted to
have a ‘neutral’ effect (18).
Mutation Taster scores were obtained using the tools available at
http://www.mutationtaster.org/info/PhyloP_PhastCons_Test.html
and previously described protocols (19).
Sanger sequencing
Sanger sequencing was conducted to validate the
variants identified in the proband as described previously
(12). The primers used were
5'-TCCCTTTAAGGAATGGTTAGTCAAG-3' and 5'-GTCATGATAAGGAACCATGAGTTG-3'
for c.713G>C (p.Arg238Pro); and 5'-TCCTGTGACCTGTGCCTCTATAC-3',
and 5'-ACACTGTCCATGTGTTGATGGTATC-3' for c.2339G>A (p.Arg780His),
which harbor 540 and 517-bp products, respectively. Genomic DNA,
extracted from a blood sample from each of the three family
members, was extracted with Qiagen FlexiGene DNA kit (cat. no.
512206; Qiagen GmbH) according the protocol provided by the
manufacturer. The DNA polymerase used was EasyTaq®
(TransGen Biotech Co., Ltd.). The reaction conditions were as
follows: Initial denaturation at 95˚C for 5 min; 32 cycles of
denaturation at 95˚C for 45 sec, annealing at 56˚C for 45 sec and
extension at 72˚C for 1 min and a final extension at 72˚C for 5
min. The polymerase chain reaction products were sequenced using an
ABI 3730XL (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
were analyzed with DNASTAR 5.0 software (DNASTAR, Inc.).
Structural modeling
The reported mutational sites of CPS1 were
analyzed using the online server of the Human Gene Mutation
Database (HGMD®; www.hgmd.cf.ac.uk; HGMD professional 2016.1 release).
The crystal structure of CPS1 (apo form) was downloaded from the
Protein Data Bank (PDB) database (https://www.ncbi.nlm.nih.gov/Structure/pdb/5DOU,
released in Sep, 2015) in PDB format (20). PyMol (version 1.8.0.7; www.pymol.org) was used for structure displaying and
structural figure preparation.
Clinical characteristics
Physical examination results were as follows:
Temperature 38˚C; Pulse, 168 times/min; Respiratory Rate, 57
time/min; blood pressure, 60/32 mmHg; weight, 3.29 kg; and head
circumference, 34 cm. The proband was in a light coma and her
anterior fontanelle was 2.5x2.5 cm in size, with high tension. Her
pupils had equal diameters and were insensitive to papillary light
reflex. The liver was palpable 2 cm below the right subcostal arch.
The patient exhibited limb hypertonia.
Auxiliary examination results
For accurate diagnosis, an auxiliary examination was
conducted following admission. Normal range values are based on
accepted values in Hunan Provincial People's Hospital. A blood test
revealed white blood cells, 26.48x109 cells/l (normal
range, 5-25x109 cells/l), neutrophil percentage, 61.5%
(normal range, 50-70%); hemoglobin, 183 g/l (normal range, 145-220
g/l); platelet count, 341x109 cells/l (normal range,
150-600x109 cells/l), C-reactive protein (CRP), <3
mg/dl (normal, ≤3 mg/dl); blood glucose, 4.8 mg/dl (normal range,
2.6-7.0 mg/dl); and blood ammonia, 109.7 µmol/l (normal, ≤40
µmol/l). The index of inflammation was generally normal and the
clinical data did not support infection. Liver and kidney function
together with electrolyte levels were normal. Blood gas results
demonstrated pH, 7.35 (normal range, 7.30-7.40); partial pressure
CO2, 41 mmHg (normal range, 35-45 mmHg); partial
pressure O2, 78 mmHg (normal range, 60-80 mmHg); Base
Excess (BE), -1 mmol/l (normal, -6 mmol/); and lactic acid, 1.3
mmol/l (normal, ≤2 mmol/l). Blood tandem mass spectrometry revealed
that the citrulline level was 3.687 µM (normal range, 4-30 µM) and
that glutamate level was 438.905 µM (normal range, 100-400 µM). The
electrocardiogram (ECG) result was consistent with a normal
neonatal ECG. Fasting blood sugar level was 4.5 mmol/l (normal
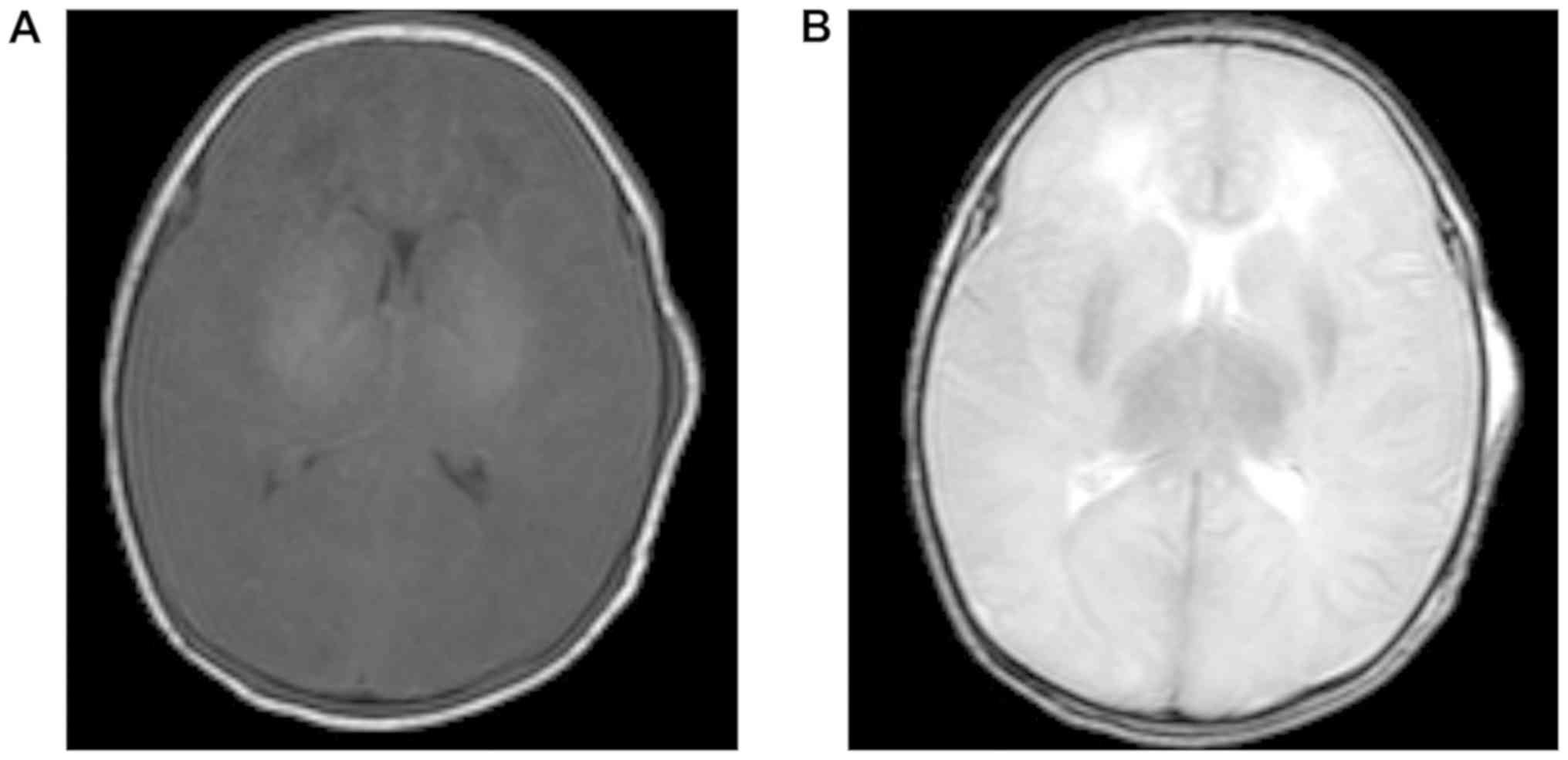
range, 2.6-7.0 mmol/l). MRI of the cerebral hemisphere revealed
diffuse low signal intensity in T1W1 and high signal intensity in
T2W1 (compared with normal neonate) in the cerebral gray matter
(Fig. 1).
Treatment
Head computed tomography excluded intracranial
hemorrhage and the patient was suspected to have an intracranial
infection. Thus, meropenem (20 mg/kg once every 8 h) combined with
vancomycin (10 mg/kg once every 12 h) was administered to treat the
infection, while g-globulin (2.5 g in total), mannitol (0.5 g/kg
once every 6 h), furosemide (0.5 g/kg once every 12 h), and albumin
dehydration (1 g/kg) were used to reduce increased intracranial
pressure and maintain electrolyte stability. On the 2nd
day following admission, increased heart rate, decreased blood
pressure, and mottled skin were noted and the symptoms relieved
following active anti-shock treatment. The neonate lapsed into a
deep coma, with a bulging fontanelle and increased tension. Upon
receiving the results of routine blood analysis, on the
3rd day after admission, tests for the levels of CRP and
procalcitonin were performed. The results of these tests did not
support a bacterial infection. As cerebrospinal fluid may have no
noticeable change from normal at an early stage of infection, the
presence of a virus-induced intracranial infection could not be
ruled out. The neonate was subjected to lumbar puncture manometry
on the 3rd day following admission. Cerebrospinal fluid
routine biochemical and virus examinations showed no evidence of
intracranial infection. Blood ammonia levels were markedly
increased (485.2 µmol/l; normal, ≤150 µmol/l) while blood urea
nitrogen (BUN) was relatively low (1.7 mmol/l; normal, 2.8 mmol/l).
In consequence, the patient was diagnosed with encephalopathy
caused by hyperammonemia. Accordingly, albumin was discontinued,
and ammonia-lowering treatment, including high sugar, arginine, and
body fluid supplements, was applied. Blood ammonia was almost
normal (75.5 µmol/l) on the 5th day following admission,
and remained in the normal range during the subsequent tests.
However, the patient was in a state of deep coma, and liver size
was markedly increased. Plasma amino acid examination on the
5th day following admission showed reduced citrulline
levels and increased glutamate levels in comparison with a normal
neonate, supporting the possibility of urea metabolism abnormality.
On the 8th day following hospitalization, as the
condition did not improve, the family discontinued treatment and
the patient succumbed to the disease.
Compound heterozygosity in CPS1 is a
potential candidate pathogenic variant
In order to reveal the genetic causes of the
condition experienced by the patient, WES was performed. Upon
filtration, compound heterozygous variants of c.713G>C,
p.Arg238Pro and c.2339G>A, p.Arg780His in CPS1 (NCBI
reference sequence, NM_001875.4) were regarded as candidate
pathogenic mutations. The American College of Medical Genetics and
Genomics hazard rating (21) for
these variants was PM2 + PM3 + PP3 + PP1, which met the standard of
‘likely pathogenic’. The c.2339G>A, p.Arg780His mutation was
reported to be pathogenic (22). The
p.Arg238Pro variant was expected to ‘affect protein function’ by
SIFT software with a score of 0.00, while it was predicted to be
‘probably damaging’ by PolyPhen2 with a score of 0.976 (using the
HumVar-trained prediction model), ‘deleterious’ by PROVEAN with a
score of -6.33 and ‘disease causing’ by MutationTaster software
with a score of 0.996. The allele frequency of c.713G>C was
explored in four different single nucleotide polymorphism databases
(SIFT, Polyphen2 HVAR, PROVEAN and MutationTaster). The results
suggested that this novel mutation had not been previously
registered (Table I). To validate
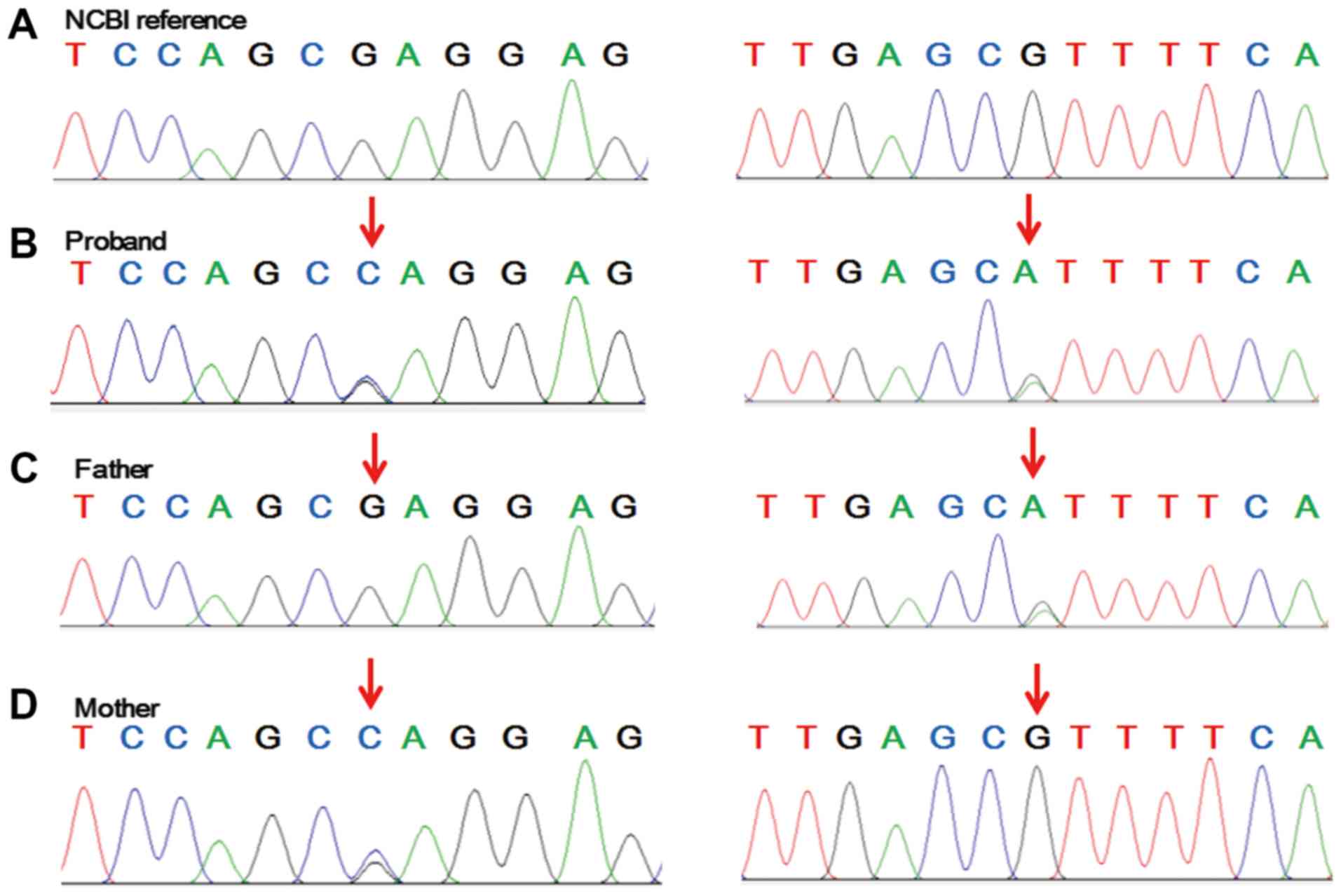
the variants, Sanger sequencing of the family genetic profile was
undertaken. The results illustrated that the mother was
heterozygous for c.713G>C, p.Arg238Pro and the father was
heterozygous for c.2339G>A, p.Arg780His in CPS1, whereas
the proband was heterozygous at both sites (Fig. 2).
 | Table ICharacterization of carbamoyl
phosphate synthetase 1 with the c.713G>C variant found in this
study. |
Table I
Characterization of carbamoyl
phosphate synthetase 1 with the c.713G>C variant found in this
study.
| Amino acid
substitution | SIFT score | Polyphen2 HVAR
score | PROVEAN score | MutationTaster
score | gnomAD | ExAC | 1000G | HGMD |
|---|
| c.713G>C | Damaging, 0.001 | Probably damaging,
0.976 | Deleterious,
-6.33 | Disease causing,
0.996 | Not registered | Not registered | Not registered | Not registered |
Structural modeling demonstrates that
the variants may influence the function of CPS1
To further assess the possible pathogenicity of
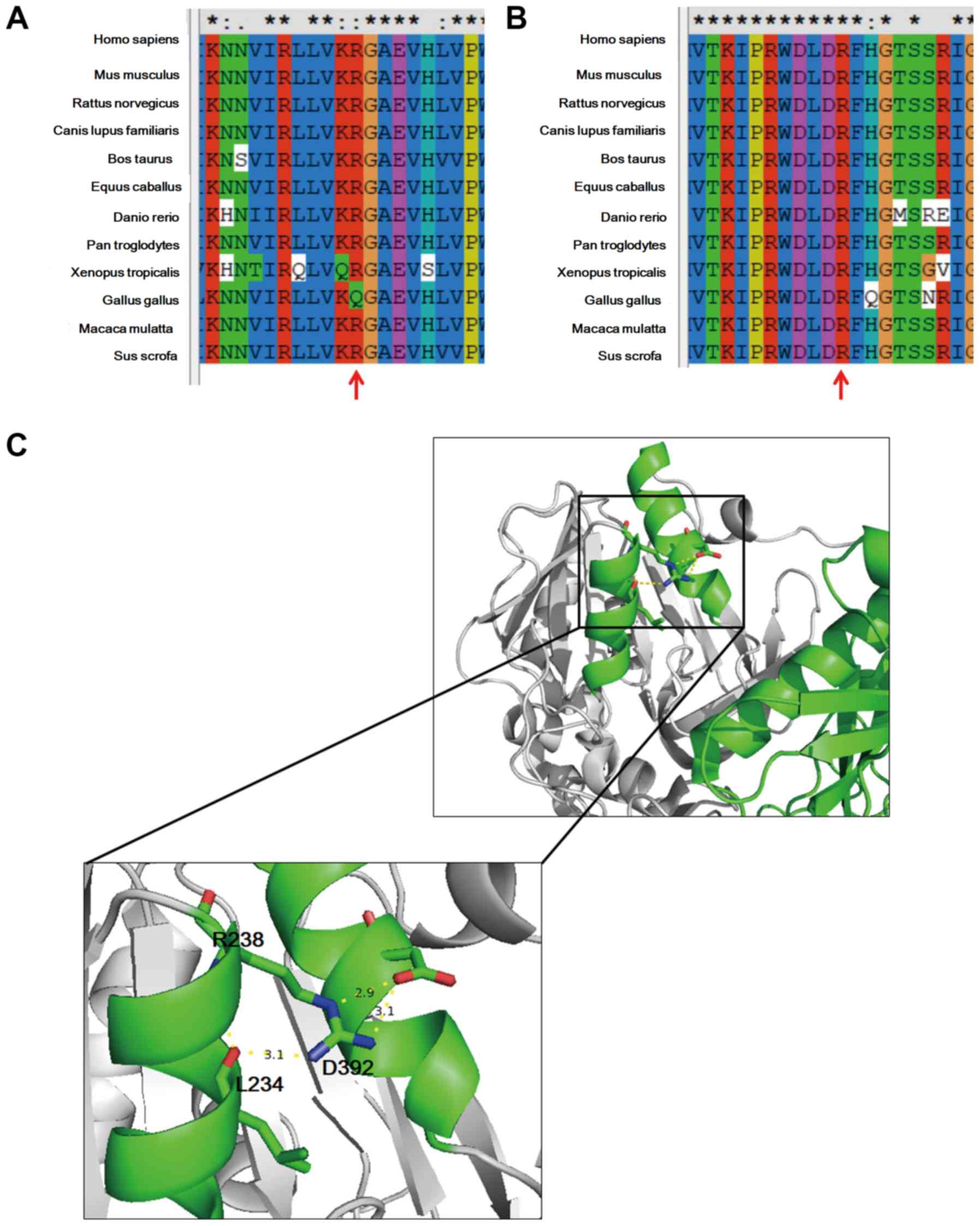
these variants, sequence alignment together with structural
modeling were undertaken. The whole sequence, including the
identified sites of CPS1, is evolutionarily conserved across the
species analyzed (Fig. 3A and
B). In general, proteins with a
highly conserved amino acid sequence are considered to be sensitive
to variants, and hence, the identified sites were likely to be
responsible for the etiology of the patient's condition (23). Based on structural data available
online (http://www.rcsb.org/pdb/gene/) the
apo form of the crystal structure of CPS1 was also studied. As
depicted in Fig. 3C, R238 is located
at the terminal region of an α-helix and forms hydrogen bonds with
D392, which is located on an adjacent α-helix. In addition, R238
forms hydrogen bonds with L234. These interactions serve crucial
roles in the local structure of these two helixes. The R238P
variant changes the charge characteristic of the side chain and may
disrupt the local structure. In light of this, it is possible that
the R238P variant may affect the function of CPS1. The R780H
variant maps in a loop key for the transmission of the allosteric
signal (20), and this mutation has
been previously reported to be pathogenic (22). Accordingly, the variants identified
in the proband may cause CPS1 deficiency.
Discussion
The current study presented the clinical
performance, diagnosis, and treatment of CPS1 deficiency in a
neonate. At the time of admission, the patient exhibited a mild
coma, persistent fever, high fontanelle tension, high muscle
tension and repeated convulsions. Additionally, the patient
temperature at admission was ~40.2˚C. As a result, the following
diseases were initially suspected: Neonatal encephalopathy,
intracranial infection, intracranial hemorrhage, septicemia or
hereditary metabolic disease. The patient was finally diagnosed
with UCD, due to the symptoms of encephalopathy and hyperammonemia.
Abnormal blood ammonia in comparison with the level in a normal
child was not noted at the time of admission. However, ammonia
level had significantly increased by the 3rd day
following admission when compared with the level in a normal child.
A possible cause for this may be that no milk intake occurred
within 20 h; thus, there was no protein substrate intake, masking
the potential diagnosis (9). Upon
admission, although amino acids were not supplemented, albumin
dehydration and craniofacial pressure treatment were administered.
In addition, protein substrate was added and the blood ammonia
increased significantly. In line with previously published
guidelines, protein intake should be avoided before UCD has been
excluded (24).
CPS1 deficiency presenting as anorexia, irregular
breathing, fever, convulsions, poor mental response, and coma
occurred on the 3rd day after birth. The symptoms were
similar to the presentation of a newborn reported by Yang et
al (25). CPS1 deficiency can
easily be misdiagnosed as septicemia and intracranial infection
(26). The findings of the present
study suggest that CPS1 deficiency should be considered when
intracranial hemorrhage, hypoglycemia, and electrolyte disorders
are suspected. For children with unexplained vomiting, anorexia,
poor mental reaction and convulsions, repeated blood ammonia
monitoring should be performed. Citrulline and orotic acid levels
are helpful in identifying the subtypes of UCD (27). Children with CPS1 deficiency
typically exhibit plasma glutamic acid increase, citrulline
decrease, and normal urinary orotic acid levels (28). Genetic testing such as WES can
further validate a diagnosis and provide a reliable basis for
prenatal eugenics (12). However,
WES is a lengthy process from test to the final diagnosis, and
therefore, it should be performed as soon as possible in patients
who are difficult to give diagnose clinically. The patient in the
present study was identified to have compound heterozygous variants
of c.713G>C, Arg238Pro and c.2339G>A, Arg780His in the
CPS1 gene. The c.2339G>A, Arg780His variant is a known
pathogenic mutation (22) while
c.713G>C,Arg238Pro is, to the best of our knowledge, a novel
heterozygous mutation not included in any database. CPS1
c.2339G>A has been included in the Genome Aggregation Database
(gnomAD) and the Exome Aggregation Consortium (ExAC) with very rare
frequencies (1.629x10-05 and 3.304x10-05,
respectively). This variant has been included in the ClinVar and
HGMD databases and could be classified as likely pathogenic. CPS1
c.713G>C, p.Arg238Pro has not been reported in gnomAD, ExAC,
1000G or HGMD. However, for this amino acid site, p.R238Ter has
been included in HGMD. Therefore, the clinical interpretation of
this variant could be classified as likely pathogenic. Protein
functional prediction and structural modeling suggested that the
mutation was harmful. In line with this, it is possible that the
proband exhibited CPS1 deficiency. Thus, the present report focused
on the genetic spectrum of this disease.
Treatment for patients with CPS1 deficiency and
clinical symptoms of hyperammonia includes limiting protein intake,
using drugs to reduce serum ammonia levels (L-arginine, sodium
benzoate, and sodium phenylacetate), and supplementing with glucose
(20). For severe ammonia levels,
blood filtration should be considered (24). The patient in the present study had
early onset disease with rapid progress and a poor prognosis. If
limited protein intake prior to coma had been undertaken, excessive
blood ammonia may have been avoided and the prognosis may have been
improved. Reports indicate that the onset of CPS1 deficiency in the
neonatal period is often fatal, and the severity of clinical
manifestations depends on the time of onset and the level of
residual enzymatic activity (29,30).
Certain patients who receive long-term protein restriction and
ammonia removal therapy may also have sequelae (31). A previous study reported a patient
who had remission following liver transplantation (30).
Collectively, hereditary metabolic diseases in the
neonatal period often lead patients to deteriorate rapidly.
Therefore, early recognition of the disease by clinicians is of
great importance (13). The findings
of the present study suggest that blood ammonia monitoring should
be carried out in cases where seemingly healthy newborns display
uncommon symptoms several days after milk intake, in order to
exclude UCD. This monitoring may facilitate early diagnosis and
treatment and improve the quality of life of these patients.
Acknowledgements
Not applicable.
Funding
This study was supported by Changsha Science and
Technology Bureau Project (grant no. KQ1602027).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JX and AZ designed the study. JX collected the
clinical data, performed the experiments and drafted the
manuscript. FH participated in clinical data collection. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Hunan Provincial People's Hospital (Changsha, China)
and informed consent was obtained from parents of the proband.
Patient consent for publication
Consent for publication was obtained from the
parents of the proband.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Soria LR, Allegri G, Melck D, Pastore N,
Annunziata P, Paris D, Polishchuk E, Nusco E, Thöny B, Motta A, et
al: Enhancement of hepatic autophagy increases ureagenesis and
protects against hyperammonemia. Proc Natl Acad Sci U S A.
115:391–396. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ballantyne LL, Sin YY, St Amand T, Si J,
Goossens S, Haenebalcke L, Haigh JJ, Kyriakopoulou L, Schulze A and
Funk CD: Strategies to rescue the consequences of inducible
arginase-1 deficiency in mice. PLoS One.
10(e0125967)2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Braissant O: Current concepts in the
pathogenesis of urea cycle disorders. Mol Genet Metab. 100 (Suppl
1):S3–S12. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Unsinn C, Das A, Valayannopoulos V, Thimm
E, Beblo S, Burlina A, Konstantopoulou V, Mayorandan S, de Lonlay
P, Rennecke J, et al: Clinical course of 63 patients with neonatal
onset urea cycle disorders in the years 2001-2013. Orphanet J Rare
Dis. 11(116)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Summar ML, Dasouki MJ, Schofield PJ,
Krishnamani MR, Vnencak-Jones C, Tuchman M, Mao J and Phillips JA
3rd: Physical and linkage mapping of human carbamyl phosphate
synthetase I (CPS1) and reassignment from 2p to 2q35. Cytogenet
Cell Genet. 71:266–267. 1995.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Haberle J, Schmidt E, Pauli S, Rapp B,
Christensen E, Wermuth B and Koch HG: Gene structure of human
carbamylphosphate synthetase 1 and novel mutations in patients with
neonatal onset. Hum Mutat. 21(444)2003.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Funghini S, Donati MA, Pasquini E,
Zammarchi E and Morrone A: Structural organization of the human
carbamyl phosphate synthetase I gene (CPS1) and identification of
two novel genetic lesions. Hum Mutat. 22:340–341. 2003.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Summar ML, Hall LD, Eeds AM, Hutcheson HB,
Kuo AN, Willis AS, Rubio V, Arvin MK, Schofield JP and Dawson EP:
Characterization of genomic structure and polymorphisms in the
human carbamyl phosphate synthetase I gene. Gene. 311:51–57.
2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Singh RH: Nutritional management of
patients with urea cycle disorders. J Inherit Metab Dis.
30:880–887. 2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
World Medical Association. World medical
association declaration of helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194.
2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jin D, Yu T, Zhang L, Wang T, Hu J, Wang Y
and Yang XA: Novel NFU1 variants induced MMDS behaved as special
leukodystrophy in chinese sufferers. J Mol Neurosci. 62:255–261.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Peng F, Zhong L, Zhang B, Zou R, Nie S,
Tian X, Deng S and He X: Successful application of next-generation
sequencing for pre-natal diagnosis in a pedigree with chronic
granulomatosis disease. Exp Ther Med. 17:2931–2936. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen Q, Bao H, Wu H, Zhao S, Huang S and
Zhao F: Diagnosis of cobalamin C deficiency with renal abnormality
from onset in a Chinese child by next generation sequencing: A case
report. Exp Ther Med. 14:3637–3643. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zytkovicz TH, Fitzgerald EF, Marsden D,
Larson CA, Shih VE, Johnson DM, Strauss AW, Comeau AM, Eaton RB and
Grady GF: Tandem mass spectrometric analysis for amino, organic,
and fatty acid disorders in newborn dried blood spots: A two-year
summary from the new England newborn screening program. Clin Chem.
47:1945–1955. 2001.PubMed/NCBI
|
|
15
|
Adzhubei I, Jordan DM and Sunyaev SR:
Predicting functional effect of human missense mutations using
PolyPhen-2. Curr Protoc Hum Genet. 7(20)2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Choi Y, Sims GE, Murphy S, Miller JR and
Chan AP: Predicting the functional effect of amino acid
substitutions and indels. PLoS One. 7(e46688)2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
de Cima S, Polo LM, Diez-Fernandez C,
Martínez AI, Cervera J, Fita I and Rubio V: Structure of human
carbamoyl phosphate synthetase: Deciphering the on/off switch of
human ureagenesis. Sci Rep. 5(16950)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kurokawa K, Yorifuji T, Kawai M, Momoi T,
Nagasaka H, Takayanagi M, Kobayashi K, Yoshino M, Kosho T, Adachi
M, et al: Molecular and clinical analyses of Japanese patients with
carbamoylphosphate synthetase 1 (CPS1) deficiency. J Hum Genet.
2:349–354. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu D, Wang Y, Yang XA and Liu D: De novo
mutation of paternal IGF2 gene causing silver-russell syndrome in a
sporadic patient. Front Genet. 8(105)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Haberle J, Boddaert N, Burlina A,
Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS,
Santer R, et al: Suggested guidelines for the diagnosis and
management of urea cycle disorders. Orphanet J Rare Dis.
7(32)2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yang X, Shi J, Lei H, Xia B and Mu D:
Neonatal-onset carbamoyl phosphate synthetase I deficiency: A case
report. Medicine (Baltimore). 96(e7365)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ali EZ, Khalid MK, Yunus ZM, Yakob Y, Chin
CB, Abd Latif K and Hock NL: Carbamoylphosphate synthetase 1 (CPS1)
deficiency: Clinical, biochemical, and molecular characterization
in Malaysian patients. Eur J Pediatr. 175:339–346. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Haberle J, Burlina A, Chakrapani A, Dixon
M, Karall D, Lindner M, Mandel H, Martinelli D, Pintos-Morell G,
Santer R, et al: Suggested guidelines for the diagnosis and
management of urea cycle disorders: First revision. J Inherit Metab
Dis. 42:1192–1230. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ono H, Suto T, Kinoshita Y, Sakano T,
Furue T and Ohta T: A case of carbamoyl phosphate synthetase 1
deficiency presenting symptoms at one month of age. Brain Dev.
31:779–781. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Klaus V, Vermeulen T, Minassian B,
Israelian N, Engel K, Lund AM, Roebrock K, Christensen E and
Häberle J: Highly variable clinical phenotype of carbamylphosphate
synthetase 1 deficiency in one family: An effect of allelic
variation in gene expression? Clin Genet. 76:263–269.
2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Foschi FG, Morelli MC, Savini S,
Dall'Aglio AC, Lanzi A, Cescon M, Ercolani G, Cucchetti A, Pinna AD
and Stefanini GF: Urea cycle disorders: A case report of a
successful treatment with liver transplant and a literature review.
World J Gastroenterol. 21:4063–4068. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kasahara M, Sakamoto S, Shigeta T, Fukuda
A, Kosaki R, Nakazawa A, Uemoto S, Noda M, Naiki Y and Horikawa R:
Living-donor liver transplantation for carbamoyl phosphate
synthetase 1 deficiency. Pediatr Transplant. 14:1036–1040.
2010.PubMed/NCBI View Article : Google Scholar
|