Introduction
Multiple myeloma (MM) is one of the most common
types of haematological cancer, which is becoming more common in
the ageing population. It is also the cause of a number of fatal
outcomes (1). As a haematological
cancer that originates from the malignant transformation of plasma
cells, MM maintains pathophysiologic heterogeneity due to its
complex genetic background (2).
Several distinct clinical phases of MM have been identified,
including monoclonal gammopathy of undetermined significance and
smoldering multiple myeloma (3).
With the development and progression of MM, several distinct
patterns of genetic aberration are recognized, including
cytogenetic abnormalities, chromosomal aberration and signaling
pathway disorders (4).
DNA methylation, a form of epigenetic control of
gene transcription, refers to cytosine methylation at position 5 in
the pyrimidine ring, which can result in inappropriate silencing of
genes involved in diverse biological processes, including cell
proliferation, apoptosis, migration and cell cycle arrest (5). In normal cells, unmethylated CpG
islands (a cluster of CpG dinucleotides) are usually observed;
however, human malignancies are characterized by the gain of
methylation at promoter associated CpG islands (6). The role of DNA methylation in the
mediation of multiple tumor suppressor gene and microRNA silencing
has been implicated in the development and progression of MM
(7).
Long non-coding RNAs (lncRNAs) are a class of
non-coding RNA with a length of >200 nucleotides, which possess
little to no capacity for protein synthesis (8). Numerous studies have reported that
lncRNAs are deregulated in various types of cancer and are
implicated in carcinogenesis and antitumor pathways (9). Wang et al (10) reported that protein tyrosine
phosphatase L1 could be epigenetically regulated in MM; a process
which can be reversed by 5-Aza-2'-deoxycytidine (5-Aza-CdR),
suggesting a potential therapeutic agent for MM. The maternally
expressed 3 (MEG3) imprinted gene is located on chromosome 14q32,
which produces a non-coding RNA transcript (11). lncRNA MEG3 has been identified as a
tumor suppressor in various types of cancer, including meningioma
(12), breast cancer (13), bladder cancer (14) and hepatocellular carcinoma (15). A previous study also demonstrated the
anticancer effect of MEG3 in MM (16). The promoter region of MEG3 is rich in
CpG islands, and the specific methylated and unmethylated CpG
islands of differentially methylated regions (DMRs) are located
upstream of the MEG3 gene (IG-DMR and MEG3-DMR) (17,18).
Furthermore, lncRNA MEG3 expression has been reported to be induced
following epigenetic modification of DNA methylation in diverse
malignancies, including gliomas (19), ovarian cancer (20) and leukemia (21,22).
In MM, Benetatos et al (21) observed MEG3 promoter hypermethylation
in both bone marrow and peripheral blood samples. This
hypermethylation was correlated with MM stage and subtype.
Therefore, it was hypothesized that MEG3 expression could be
epigenetically controlled by MEG3 promoter hypermethylation, which
may ultimately influence the biological behavior of MM.
Materials and methods
Study subjects
The present study was approved by the Institutional
Review Board of the First Affiliated Hospital of Nanjing Medical
University. All participants provided written informed consent.
Bone marrow biopsy samples were collected from 39 patients with
newly diagnosed MM who were admitted to the First Affiliated
Hospital of Nanjing Medical University between January 2009 and May
2014. Patient information is listed in Table I. The diagnosis of MM was established
according to the standard morphological and immunophenotypical
criteria (23). The subtype of MM
was classified according to the monoclonal component. The stage of
MM was classified according to the Durie-Salmon staging system and
the International Staging System (ISS) (24).
 | Table IDistribution of variables of patients
with multiple myeloma. |
Table I
Distribution of variables of patients
with multiple myeloma.
| Variable | Number (%) |
|---|
| Median age
(range) | 61 (36-82) |
| Sex | |
|
Male | 26 (66.7) |
|
Female | 13 (33.3) |
| Subtypes | |
|
IgG | 18 (46.2) |
|
IgA | 11 (28.2) |
|
Light
chain | 10 (25.6) |
| Durie-Salmon
stage | |
|
I | 4 (10.3) |
|
II | 5 (12.8) |
|
III | 30 (76.9) |
| International
Staging System stage | |
|
I | 5 (12.8) |
|
II | 14 (35.9) |
|
III | 20 (51.3) |
| Serum creatinine
(µmol/l) | |
|
>176.8 | 22 |
|
≤176.8 | 12 |
| Serum calcium
(mmol/l) | |
|
>2.98 | 31 |
|
≤2.98 | 3 |
|
N/A | 5 |
MM cells were isolated from bone marrow samples
using CD138 microbeads and MS-columns (Miltenyi Biotec; cat. no.
130-051-301) according to the manufacter's protocol. The MM cell
line, ARP1 (American Type Culture Collection), was cultured in RPMI
1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin in an incubator at 37˚C with 5%
CO2.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from ARP1 cells and patient
derived MM cells using TRIzol® reagent (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protcol. Total
RNA was reverse transcribed to cDNA using the Primescipt RT Reagent
kit with gDNA Eraser (Takara Biotechnology Co., Ltd.), according to
the manufacturer's protocol.
qPCR was subsequently performed on a StepOne Plus™
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) using a SYBR Green qRT-PCR assay according to the
manufacturer's protocol (Takara Biotechnology Co., Ltd.). The
following primer pairs, designed by Primer Premier 5 (Premier
Biosoft International), were used for qPCR: MEG3 forward,
5'-GGAGCTGTTGAGCCTTCAGT-3' and reverse, 5'-CAAGCCCTGTGCTTTGGAAC-3';
and β-actin forward, 5'-AGCGAGCATCCCCCAAAGTT-3' and reverse,
5'-GGGCACGAAGGCTCATCATT-3'. The following thermocycling conditions
were used for the qPCR: 40 cycles of denaturation at 95˚C for 5
sec, annealing at 60˚C for 30 sec, followed by a final extension at
72˚C for 5 min. MEG3 mRNA levels were quantified according to the
standard curve of MEG3 and β-actin using the 2-∆∆Cq
method (24). β-actin was used as
the internal reference gene.
DNA isolation and methylation-specific
PCR (MSP)
Genomic (g)DNA was extracted from ARP1 cells and
patient derived MM cells using a TIANamp Genomic DNA kit according
to the manufacturer's protocol (Tiangen Biotech Co., Ltd.).
Subsequently, DNA bisulfite conversion was performed on the gDNA
using the EpiTect Plus Bisulfite kit (Qiagen GmbH), according to
the manufacturer's protocol.
The methylation status of MEG3 was determined by
MSP, using a Veriti96 PCR thermocycler (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with Taq PCR MasterMix (Tiangen Biotech
Co., Ltd.). The following primers obtained from previous studies
(25,26) were used for MSP: Methylated primer
pair forward, 5'-GTTAGTAATCGGGTTTGTCGGC-3' and reverse,
5'-AATCATAACTCCGAACACCCGCG-3'; and unmethylated primer pair
forward, 5'-GAGGATGGTTAGTTATTGGGGT-3' and reverse,
5'-CCACCATAACCAACACCCTATAATCACA-3'. PCR was performed using the
following thermocycling conditions: 94˚C for 3 min; 5 cycles of
94˚C for 30 sec, 70˚C for 30 sec and 72˚C for 30 sec; 5 cycles of
94˚C for 30 sec, 65˚C for 30 sec and 72˚C for 30 sec; 30 cycles of
94˚C for 30 sec, 60˚C for 30 sec and 72˚C for 30 sec; and a final
extension at 72˚C for 7 min. The PCR products were run on a 3%
agarose gel and were subsequently identified by ethidium bromide
staining at room temperature (27).
The CpG island usually localizes to the DMR which is located within
~4 kb of the DMR that contains the promoter of the MEG3 gene
(28,29). A 160 bp product represented the
methylated state and a 120 bp product represented the unmethylated
state of MEG3(26).
Cell transfection and 5-Aza-CdR
treatment
ARP-1 cells were cultured in DMEM medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin in an incubator at 37˚C with 5%
CO2. ARP1 cells (2.0x106/well) were plated
into 6-well plates and transfected with the 4 µg pcDNA3.1-MEG3 or 4
µg pcDNA3.1-empty (provided by Professor Wei De, Nanjing Medical
University) using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. For
MEG3 knockdown, the following small interfering (si)RNA sequences
were used for Lipofectamine® 2000 transfection: si-MEG3,
5'-GCUCAUACUUUGACUCUAUTT-3'; and si-negative control (NC),
5'-UUCUCCGAACGUGUCACGUTT-3'. Both sequences were designed and
synthesized by Shanghai GenePharma Co., Ltd.
ARP1 cells were seeded at 2x104
cells/well in 96-well culture plates and incubated with DMEM (200
µl) containing 0.1, 1, 5, 10, 50 or 100 µg/ml 5-Aza-CdR
(Sigma-Aldrich; Merck KGaA) for 72 h at 37˚C. Control cells were
incubated with DMEM containing PBS (20 µl). The Cell Counting Kit-8
(CCK-8) assay (Selleck Chemicals) was used to analyze cell
proliferation according to the manufacturer's protocol. RT-qPCR and
MSP were performed to assess the expression of MEG3 mRNA and the
methylation status of the MEG3 promoter, respectively.
For restoration experiments, ARP1 cells were treated
with 5-Aza-CdR (50 µg/ml for 48 h at 37˚C), followed by MEG3
knockdown. MEG3 expression was detected by RT-qPCR at 48 h
post-transfection. ARP1 cell proliferation following MEG3 knockdown
was analyzed by the CCK-8 assay at 0, 24, 48 and 72 h
post-transfection.
Western blotting
ARP1 cells and patient derived MM cells were lysed
and total protein was extracted using RIPA buffer (Beyotime
Institute of Biotechnology), according to the manufacturer's
protocol. Total protein was quantified using the Bicinchoninic Acid
Protein Assay kit (Beyotime Institute of Biotechnology), according
to the manufacturer's protocol. Subsequently, 50 µg protein/lane
was separated on 10% SDS-PAGE gels by electrophoresis and then
transferred to PVDF membranes. The membranes were blocked with 5%
skimmed milk for ~2 h at room temperature and then incubated with
anti-p53 (1:1,000; Cell Signaling Technology, Inc. cat. no. 2524)
and anti-GAPDH (1:1,000; Cell Signaling Technology, Inc.; cat. no.
5174) primary antibodies overnight at 4˚C. Membranes were washed
for 1 h with TBST buffer. Following the primary incubation,
membranes were incubated for 1.5 h with appropriate secondary
antibodies (horseradish peroxidase conjugated goat anti-rabbit IgG
H&L; 1:4,000; Abcam; cat. no. ab6721) at room temperature.
Protein bands were visualized using the Chemiluminescence
horseradish peroxidase substrate (cat. no. P90720; EMD Millipore)
and the Molecular Imager ChemiDoc XRS+ chemiluminescence system
(Bio-Rad Laboratories, Inc.). Protein expression was quantified
using Image Lab software version 5.0 (Bio-Rad Laboratories, Inc.)
with GAPDH as the loading control.
Flow cytometry
To analyze the cell cycle, ARP1 cells
(2.0x106/well) were plated in 6-well plates and treated
with a series of concentrations of 5-Aza-CdR (0, 5, 10 and 50
µg/ml). After 48 h incubation at 37˚C with 5% CO2, cells
were washed with PBS and fixed with 75% cold ethanol for 24 h at
-20˚C. Subsequently, the cells were washed with PBS and stained
using the Cell Cycle Detection kit according to the manufacturer's
protocol (Nanjing KeyGen Biotech Co., Ltd.) at room temperature for
30 min to analyze the cell cycle with FACS (Becton, Dickinson and
Company).
To analyze apoptosis, ARP1 cells
(2.0x106/well) were plated in 6-well plates and treated
with a series of concentrations of 5-Aza-CdR (0, 5, 10 and 50
µg/ml). After 48 h treatment at 37˚C with 5% CO2, the
cells were washed with PBS. Subsequently, the cells were harvested
and stained using the Annexin V-FITC Apoptosis Detection kit
(Nanjing KeyGen Biotech Co., Ltd.), according to the manufacturer's
protocol. Cells were stained with Annexin V and PI at 4˚C for 15
min in the dark, and subjected to FACS analysis (Becton, Dickinson
and Company).
For restoration experiments, ARP1 cells were treated
with 5-Aza-CdR (50 µg/ml for 48 h at 37˚C with 5% CO2),
followed by MEG3 knockdown. At 48 h post-transfection, cells were
used for cell cycle and apoptosis analyses.
Statistical analysis
The schematic diagram of CpG islands in the human
MEG3 promoter was performed using MethPrimer software (Version 1.0;
www.urogene.org/methprimer). Statistical
analysis was performed using GraphPad Prism (version 5; GraphPad
Software, Inc.). A chi-squared test was used to compare categorical
variables. Data were presented as the mean ± standard deviation
from at least three repeats. Data were compared using a Student's
t-test or one-way ANOVA followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
MEG3 expression is negatively
associated with ISS stage in patients with newly diagnosed MM
In the present study, MEG3 levels in 39 newly
diagnosed MM bone marrow samples were quantified via RT-qPCR. The
clinical characteristics of the patients with MM are listed in
Table I. Five representative samples
were selected to present the methylation pattern of the MEG3 DMRs
(Fig. 1A). The standard curve used
to detect MEG3 and β-actin expression levels is presented in
Fig. 1B. Of the 39 samples, MEG3
expression was detected in 36 and the remaining 3 exhibited an MEG3
expression below the limit of detection (Fig. 1C). MEG3 expression was also
negatively associated with ISS stage in patients with newly
diagnosed MM (Fig. 1D), indicating
that MEG3 may serve as a tumor suppressor in human MM. The
schematic diagram of CpG islands in the human MEG3 promoter is
presented in Fig. 1E.
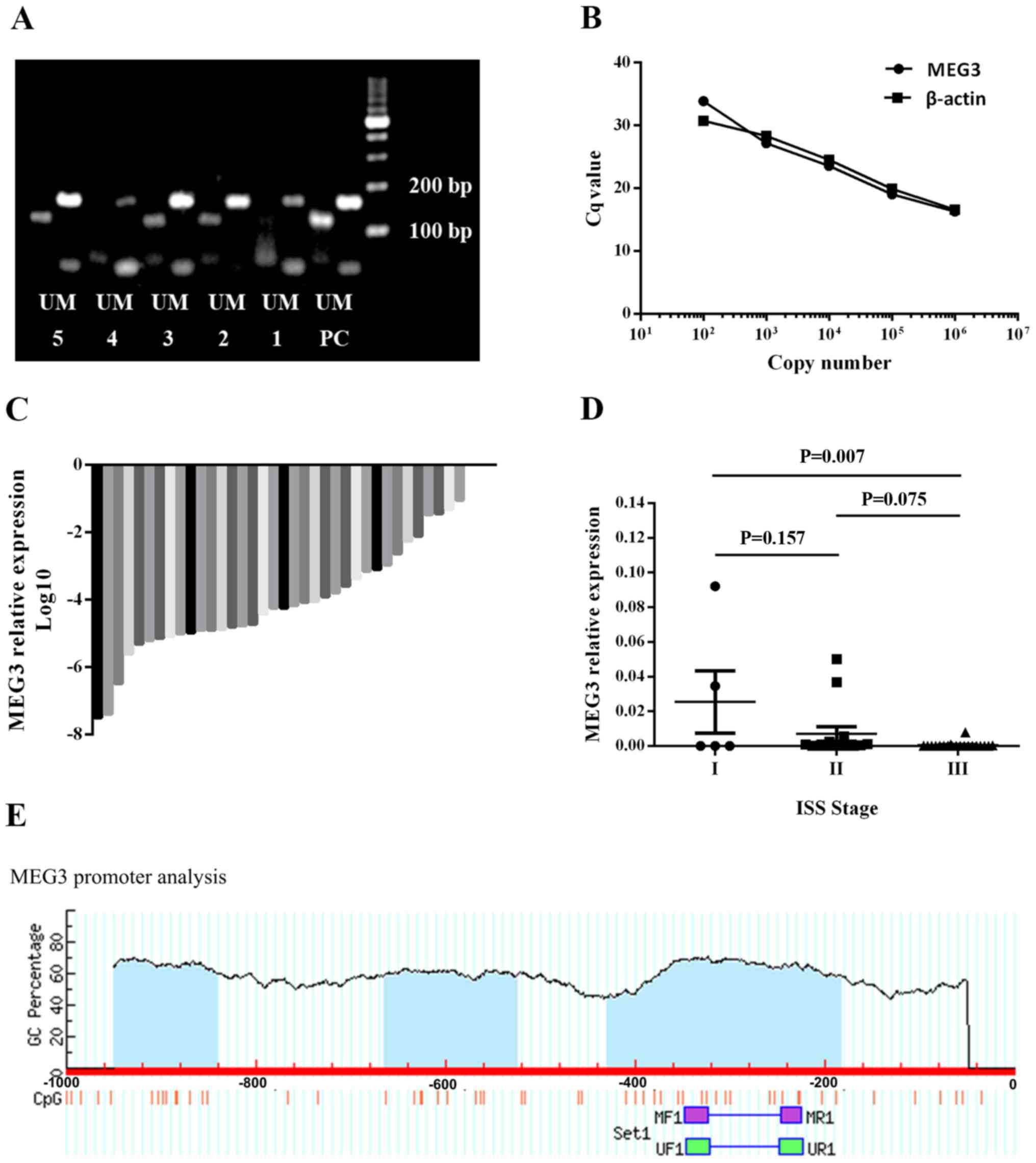 | Figure 1MEG3 methylation status and MEG3
expression in the bone marrow samples of patients with MM. (A) MEG3
methylation status. Samples 1 and 4 exhibited an abnormal
methylation status. Samples 2, 3 and 5 exhibited a normal
methylation status. (B) A standard curve was used to calculate the
expression of MEG3 and β-actin following reverse
transcription-quantitative PCR. (C) Relative expression of MEG3 in
39 bone marrow samples of patients with MM. MEG3 expression was
detected in 36 samples. (D) MEG3 expression was negatively
associated with International Staging System stage. (E) Schematic
diagram of CpG islands (blue area) in the human MEG3 promoter,
identified using MethPrimer software. The position of CpG islands
in relation to the MEG3 transcription starting site and MSP primers
were depicted. MEG3, maternally expressed 3; MM, multiple myeloma;
PC, positive control; M, methylated band; U, unmethylated band;
MF1, methylated forward 1; MR1, methylated reverse 1; UF1,
unmethylated forward 1; UR1, unmethylated reverse 1. |
MEG3 methylation status and expression
is restored after treatment with 5-Aza-CdR
In the 39 newly diagnosed MM samples, an abnormal
methylation pattern of the MEG3 DMRs was identified in eight of the
bone marrow samples. Chi-squared test was used to examine the
association between MEG3 methylation status and ISS stage (Table II); however, a significant
correlation was not observed. ARP1 cells were treated with
different concentrations of 5-Aza-CdR (0, 5, 10 or 50 µg/ml).
Re-expression of MEG3 (Fig. 2B) and
reversed abnormal methylation pattern of the MEG3 promoter
(Fig. 2A) were observed following
treatment with 5-Aza-CdR. These results indicated that CpG
methylation may downregulate MEG3 mRNA expression in MM cells.
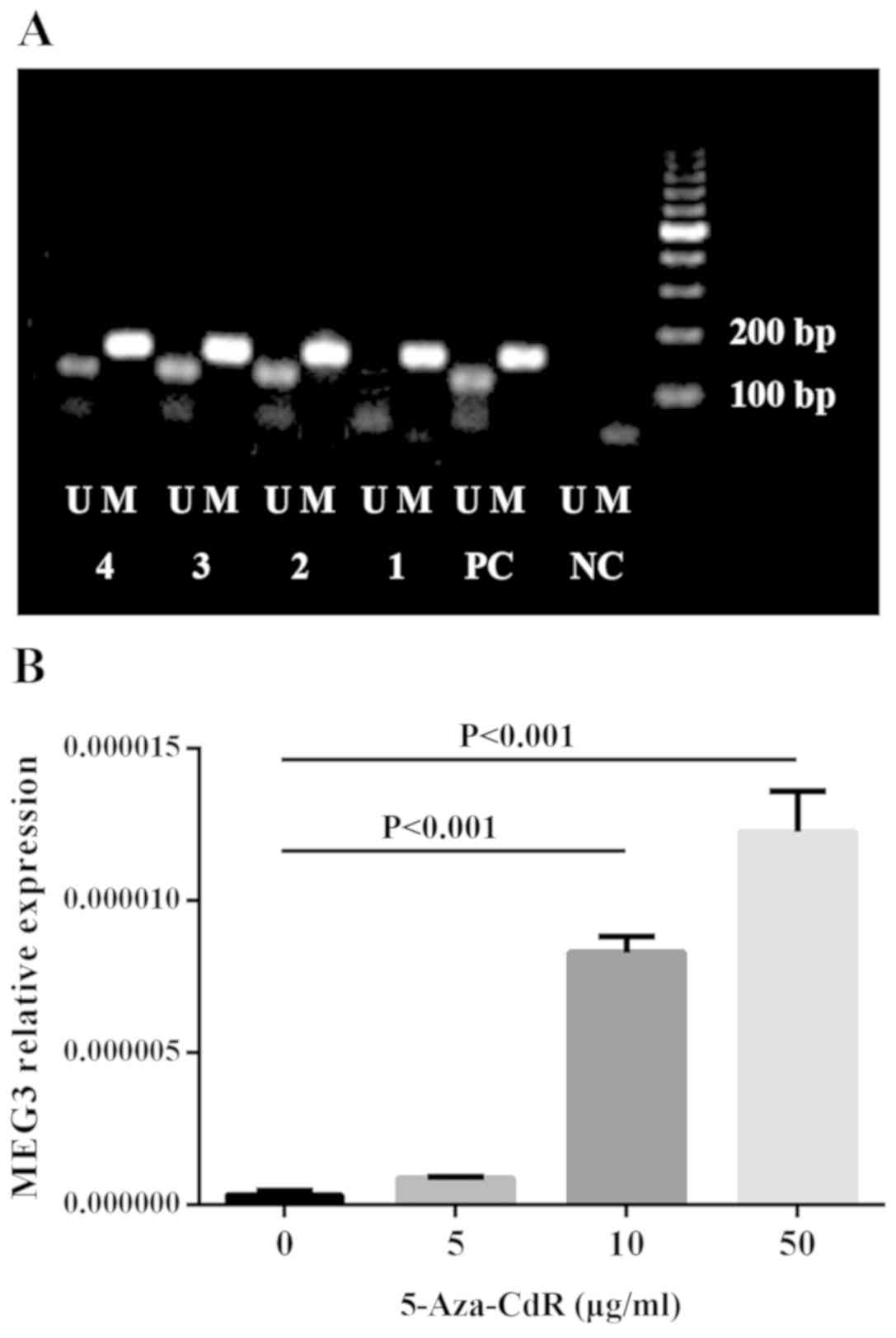 | Figure 25-Aza-CdR restores the normal MEG3
methylation status and MEG3 expression of ARP1 cells. (A) MEG3
methylation status and (B) expression of ARP1 cells. 5-Aza-CdR,
5-Aza-2'-deoxycytidine; MEG3, maternally expressed 3; NC, negative
control; PC, positive control; M, methylated band; U, unmethylated
band; 1, 0 µg/ml 5-Aza-CdR; 2, 5 µg/ml 5-Aza-CdR; 3, 10 µg/ml
5-Aza-CdR; 4, 50 µg/ml 5-Aza-CdR. |
| Table IIDistribution of methylation status
among the International Staging System stages in patients with
multiple myeloma. |
Table II
Distribution of methylation status
among the International Staging System stages in patients with
multiple myeloma.
| | International
staging system stage | |
|---|
| Methylation
status | I | II | III | P-value |
|---|
| Unmethylated | 4 (12.9) | 12 (38.7) | 15 (48.4) | 0.748 |
| Methylated | 1 (12.5) | 2 (25.0) | 5 (62.5) | |
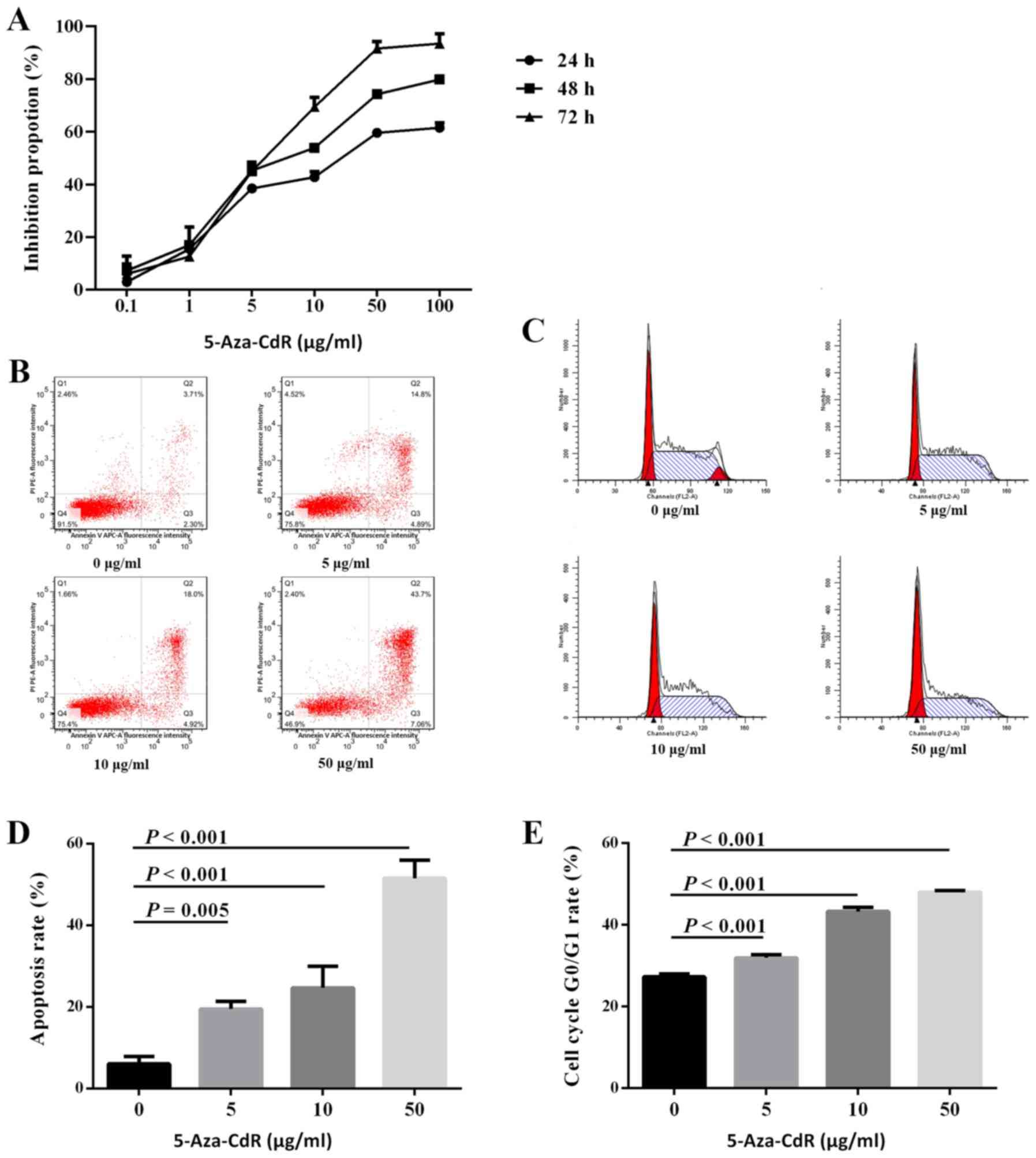
5-Aza-CdR contributes to the
inhibition of MM cells
To investigate the role of 5-Aza-CdR in cell
proliferation, ARP1 cells were treated with different
concentrations of 5-Aza-CdR. The results of the CCK-8 assay
revealed that proliferation was inhibited by 5-Aza-CdR in a
dose-dependent manner (mean inhibition proportion of 0, 5, 10, 50
and 100 µg/ml for 24 h: 3.0, 38.5, 42.8, 59.6 and 61.5%; 48 h: 7.3,
45.3, 53.9, 74.3 and 79.8%; and 72 h: 6.0, 45.1, 69.5, 91.6 and
93.5%; Fig. 3A). The results of the
apoptosis assay revealed that the number of apoptotic cells
increased by 5-Aza-CdR in a dose-dependent manner (6.02% for 0
µg/ml, 19.45% for 5 µg/ml, 24.58% for 10 µg/ml, and 51.49% for 50
µg/ml; Fig. 3B and D). Similarly, cell cycle analysis
demonstrated that the number of ARP1 cells arrested at the
G0/G1 phase was increased by 5-Aza-CdR in a
dose-dependent manner (27.30±0.74 for 0 µg/ml, 31.93±0.79 for 5
µg/ml, 43.27±1.02 for 10 µg/ml, and 48.00±0.36 for 50 µg/ml;
Fig. 3C and E). The aforementioned results indicated
that 5-Aza-CdR inhibits the proliferation of MM cells.
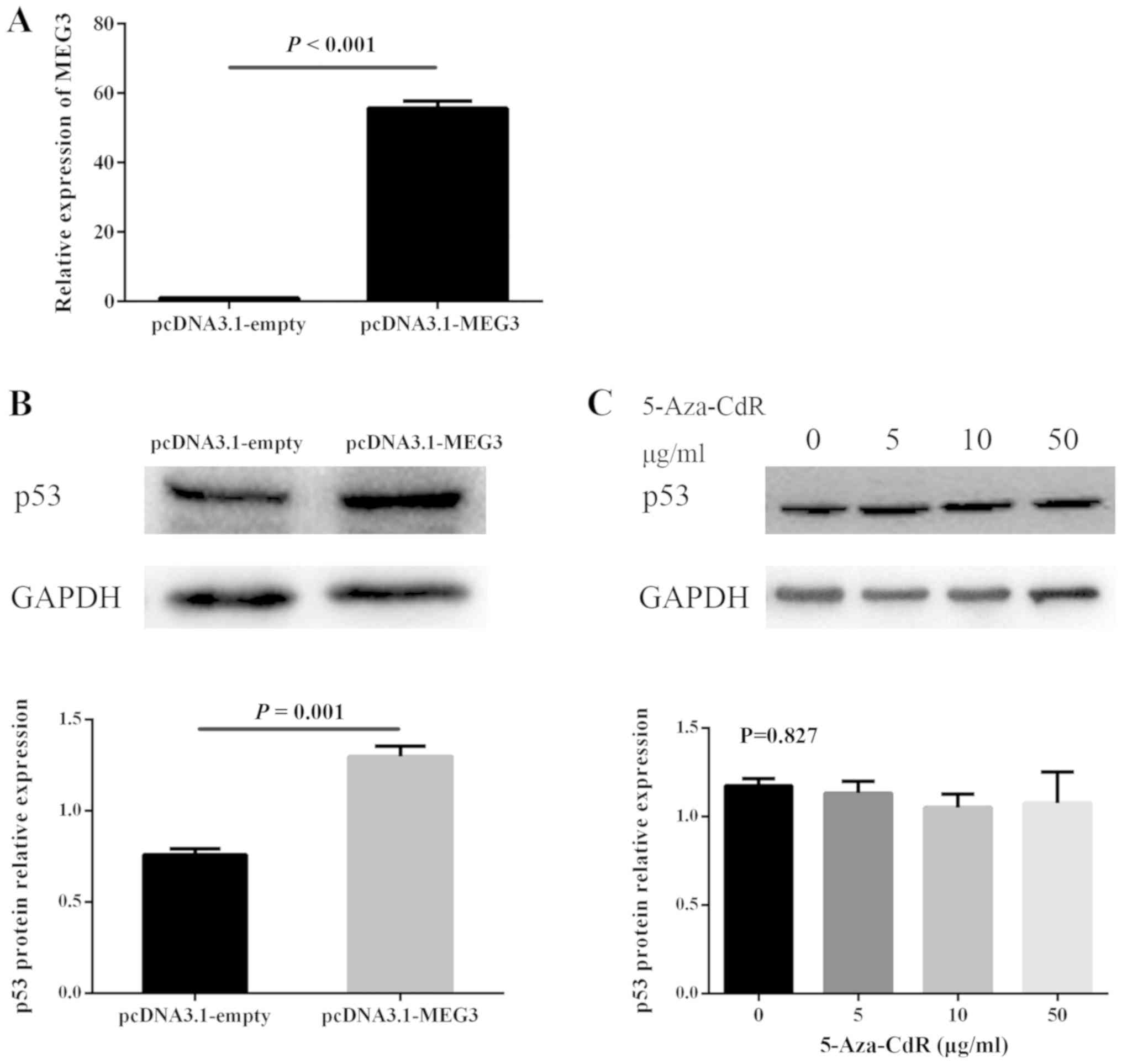
Association between MEG3 and p53
expression
The present study investigated whether MEG3
regulated the expression of p53. ARP1 cells were successfully
transfected with pcDNA3.1-MEG3 or pcDNA3.1-empty (Fig. 4A). The pcDNA3.1-MEG3 group displayed
significantly increased levels of p53 protein (Fig. 4B). Furthermore, whether 5-Aza-CdR
influences the expression of p53 was investigated. The results
revealed that 5-Aza-CdR treatment did not alter the p53 expression
of ARP1 cells (Fig. 4C).
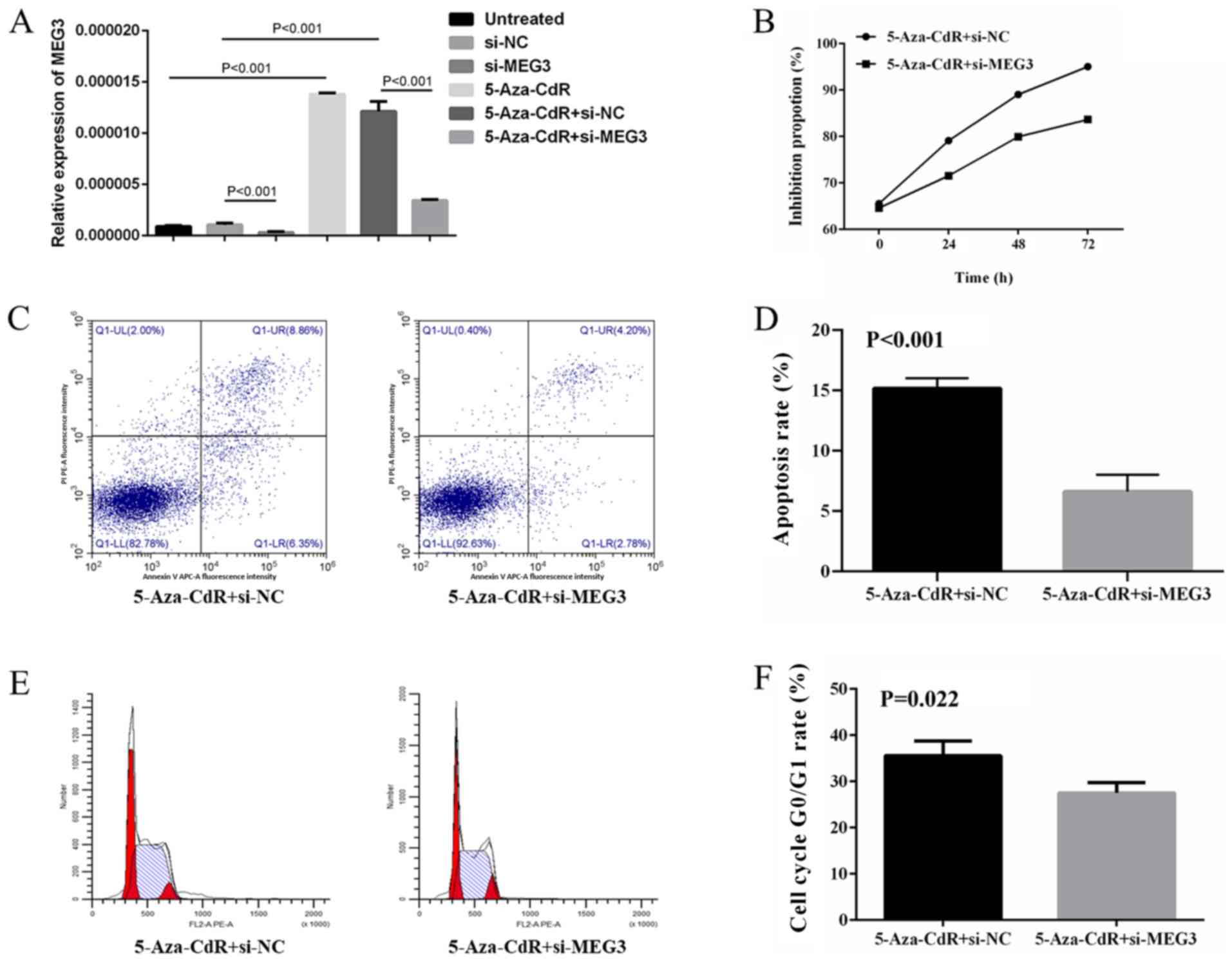
Restoration experiments
ARP1 cells treated with 5-Aza-CdR followed by MEG3
knockdown were used to investigate whether the tumor suppressive
role of 5-Aza-CdR in MM cells may be due to the upregulation of
MEG3. MEG3 expression levels were detected in untreated, si-NC,
si-MEG3, 5-Aza-CdR, 5-Aza-CdR + si-NC and 5-Aza-CdR + si-MEG3
groups, which demonstrated that 5-Aza-CdR could upregulate MEG3
expression in ARP1 cells (Fig. 5A).
In co-treated cells, the increased antiproliferation (Fig. 5B), proapoptotic (Fig. 5C and D) and enhanced cell cycle arrest (Fig. 5E and F) effects induced by 5-Aza-CdR treatment
were partly inhibited by MEG3 knockdown.
Discussion
In the present study, low MEG3 expressions were
associated with higher ISS stages. A previous study reported that
MEG3 overexpression inhibited proliferation, promoted apoptosis and
blocked the cell cycle in ARP1 cells (16). Furthermore, the demethylating agent,
5-Aza-CdR, reversed the hypermethylation status of the MEG3
promoter and increased MEG3 expression in ARP1 cells. Additionally,
5-Aza-CdR produced an antiproliferative effect on ARP1 cells, which
was reversed by MEG3 knockdown. Taken together, the results of the
present study suggested that MEG3 may serve as a tumor suppressor
gene in MM and that MEG3 may be epigenetically modified by the
hypermethylation status of MEG3 promoter.
MEG3 is a maternally expressed imprinted gene, which
encodes an lncRNA (30). Multiple
tumor samples have been tested for MEG3 expression and loss of MEG3
expression has been identified in the majority of tumor types,
including bladder (14), epithelial
ovarian (20) and gallbladder cancer
(31), as well as glioma (19) and nasopharyngeal carcinoma (32). In MM, ISS stage is the most important
prognostic system (23). In the
present study, MEG3 expression was detected in bone marrow samples
derived from patients with MM. Loss of MEG3 expression was
associated with higher ISS stages, indicating that MEG3 may serve
as a prognostic biomarker in MM. The tumor suppressor role of MEG3
in MM identified in the present study is consistent with the
aforementioned role of MEG3 in other malignancies identified in
previous studies.
The tumor suppressor function of p53 has long been
recognized and p53 has been reported to be mutated, which may lead
to the loss of wild-type p53 activity in the majority of human
malignancies (33). p53
abnormalities are regarded as independent prognostic markers in MM
(34,35). Previously, a study reported that MEG3
could stimulate p53-dependent transcription (12). p53 exhibits a relative low expression
level due to rapid degradation caused by the ubiquitin-proteasome
pathway (36). The p53 protein is
regulated by MDM2 proto-oncogene (MDM2), which is an E3 ubiquitin
ligase that inhibits the function of p53 and promotes its
degradation (37). MEG3 has been
identified as a tumor suppressor, which exerts its effect by
downregulating MDM2 expression, as well as activating p53(38). Furthermore, MDM2 suppression
contributes to p53 accumulation induced by MEG3(39). Previous studies have demonstrated
that p53 serves as target of MEG3 in multiple types of cancer,
including breast (13), non-small
cell lung (38) and bladder cancer
(40), as well as glioma (19) and hepatoma (41). In the present study, p53 protein
levels were upregulated in ARP1 cells overexpressing MEG3,
indicating that MEG3 may suppress MM cell proliferation by
upregulating p53.
DNA methylation, an epigenetic regulation mechanism,
serves a role in silencing MEG3 gene expression in different types
of cancer (42). The role of the DNA
methylation inhibitor 5-Aza-CdR is associated with the
hypermethylation regulation of various genes in MM (10). Downregulation of MEG3 expression is
associated with the hypermethylation status of the MEG3 promoter in
a number of malignancies, including glioma (19), ovarian cancer (20) and leukemia (21). In MM, Benetatos et al
(21) reported MEG3 promoter
hypermethylation status in 12 out of 21 bone marrow samples and 9
out of 14 peripheral blood samples. In the present study, MEG3
promoter methylation status was identified in the bone marrow
samples of 39 patients with MM and abnormal hypermethylation was
identified in eight samples. The discrepancy between the present
study and previous studies could be attributed to the small sample
size and diverse genetic backgrounds of the patients included in
the current study. Furthermore, no significant association was
identified between MEG3 methylation status and ISS stage, which
could be attributed to the small number of samples and the low
proportion of samples with an abnormal methylation status. The
functional role of 5-Aza-CdR in ARP1 cells was further explored.
The results further demonstrated a demethylation effect of
5-Aza-CdR on MEG3 promoter hypermethylation in ARP1 cells. In
addition, MEG3 levels revealed a dose-dependent relationship with
5-Aza-Cdr concentration. Furthermore, 5-Aza-CdR inhibited
proliferation, promoted apoptosis and induced
G0/G1 cell cycle arrest in ARP1 cells. The
effects of 5-Aza-CdR were reversed by MEG3 knockdown. The results
suggested that the demethylation reagent, 5-Aza-CdR, might restore
MEG3 expression by demethylating the MEG3 promoter and therefore,
may exert an anticancer effect in MM. Furthermore, the effect of
5-Aza-CdR on p53 expression was investigated; however, no
alterations to the levels of p53 protein were observed. This may be
attributed to the different effect of 5-Aza-CdR on different genes.
Therefore, the results indicated that the antitumor effect of
5-Aza-CdR involved MEG3 but was independent of p53.
In conclusion, the present study suggested that MEG3
may serve as a tumor suppressor by upregulating p53 levels in MM.
Furthermore 5-Aza-CdR inhibited MM cell proliferation by
upregulating MEG3 expression. However, this was independent of p53
expression. Further investigation into how 5-Aza-CdR affects MEG3
and why p53 expression is not altered in MM is required.
Additionally, further investigation into the mechanisms of MEG3 may
provide novel therapeutic targets for MM.
Acknowledgements
The authors would like to thank Professor Wei De
(Nanjing Medical University) for providing the pcDNA3.1-MEG3 and
pcDNA3.1-empty plasmid.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81800200 and
81670199) and the Jiangsu Province's Medical Elite Program (grant
no. ZDRCA2016015).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WY and QS assisted with all experiments and wrote
the manuscript. CW and XS were responsible for the flow cytometry
experiments and statistical analysis. JX and LC designed the
current study and critically reviewed the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the First Affiliated Hospital of Nanjing Medical
University. All participants provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kazandjian D: Multiple myeloma
epidemiology and survival: A unique malignancy. Semin Oncol.
43:676–681. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Morgan GJ, Walker BA and Davies FE: The
genetic architecture of multiple myeloma. Nat Rev Cancer.
12:335–348. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Mateos MV and Landgren O: MGUS and
smoldering multiple myeloma: Diagnosis and epidemiology. Cancer
Treat Res. 169:3–12. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Glavey SV, Manier S, Sacco A, Salem K,
Kawano Y, Bouyssou J, Ghobrial IM and Roccaro AM: Epigenetics in
multiple myeloma. Cancer Treat Res. 169:35–49. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wilkins JF: Genomic imprinting and
methylation: Epigenetic canalization and conflict. Trends Genet.
21:356–365. 2005.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Liyanage VR, Jarmasz JS, Murugeshan N, Del
Bigio MR, Rastegar M and Davie JR: DNA modifications: Function and
applications in normal and disease States. Biology (Basel).
3:670–723. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wong KY and Chim CS: DNA methylation of
tumor suppressor protein-coding and non-coding genes in multiple
myeloma. Epigenomics. 7:985–1001. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Schmitt AM and Chang HY: Long noncoding
RNAs in cancer pathways. Cancer Cell. 29:452–463. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang W, Wang J, Chen M, Liang Y, LI Z,
Zhang Z and Jing H: 5-Azacitidine remolds the methylation status
and inhibits growth in multiple myeloma. Blood. 126(4817)2015.
|
|
11
|
Miyoshi N, Wagatsuma H, Wakana S,
Shiroishi T, Nomura M, Aisaka K, Kohda T, Surani MA, Kaneko-Ishino
T and Ishino F: Identification of an imprinted gene, Meg3/Gtl2 and
its human homologue MEG3, first mapped on mouse distal chromosome
12 and human chromosome 14q. Genes Cells. 5:211–220.
2000.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is associated
with meningioma pathogenesis and progression. Cancer Res.
70:2350–2358. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sun L, Li Y and Yang B: Downregulated long
non-coding RNA MEG3 in breast cancer regulates proliferation,
migration and invasion by depending on p53's transcriptional
activity. Biochem Biophys Res Commun. 478:323–329. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ying L, Huang Y, Chen H, Wang Y, Xia L,
Chen Y, Liu Y and Qiu F: Downregulated MEG3 activates autophagy and
increases cell proliferation in bladder cancer. Mol Biosyst.
9:407–411. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhuo H, Tang J, Lin Z, Jiang R, Zhang X,
Ji J, Wang P and Sun B: The aberrant expression of MEG3 regulated
by UHRF1 predicts the prognosis of hepatocellular carcinoma. Mol
Carcinog. 55:209–219. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Shen X, Bai H, Zhu H, Yan Q, Yang Y, Yu W,
Shi Q, Wang J, Li J and Chen L: Long non-coding RNA MEG3 functions
as a competing endogenous RNA to regulate HOXA11 expression by
sponging miR-181a in multiple myeloma. Cell Physiol Biochem.
49:87–100. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Astuti D, Latif F, Wagner K, Gentle D,
Cooper WN, Catchpoole D, Grundy R, Ferguson-Smith AC and Maher ER:
Epigenetic alteration at the DLK1-GTL2 imprinted domain in human
neoplasia: Analysis of neuroblastoma, phaeochromocytoma and Wilms'
tumour. Br J Cancer. 92:1574–1580. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kagami M, O'Sullivan MJ, Green AJ, Watabe
Y, Arisaka O, Masawa N, Matsuoka K, Fukami M, Matsubara K, Kato F,
et al: The IG-DMR and the MEG3-DMR at human chromosome 14q32.2:
Hierarchical interaction and distinct functional properties as
imprinting control centers. PLoS Genet. 6(e1000992)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li J, Bian EB, He XJ, Ma CC, Zong G, Wang
HL and Zhao B: Epigenetic repression of long non-coding RNA MEG3
mediated by DNMT1 represses the p53 pathway in gliomas. Int J
Oncol. 48:723–733. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sheng X and Li J, Yang L, Chen Z, Zhao Q,
Tan L, Zhou Y and Li J: Promoter hypermethylation influences the
suppressive role of maternally expressed 3, a long non-coding RNA,
in the development of epithelial ovarian cancer. Oncol Rep.
32:277–285. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Benetatos L, Hatzimichael E, Dasoula A,
Dranitsaris G, Tsiara S, Syrrou M, Georgiou I and Bourantas KL: CpG
methylation analysis of the MEG3 and SNRPN imprinted genes in acute
myeloid leukemia and myelodysplastic syndromes. Leuk Res.
34:148–153. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li ZY, Yang L, Liu XJ, Wang XZ, Pan YX and
Luo JM: The long noncoding RNA MEG3 and its target miR-147 regulate
JAK/STAT pathway in advanced chronic myeloid leukemia.
EBioMedicine. 34:61–75. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Greipp PR, San Miguel J, Durie BG, Crowley
JJ, Barlogie B, Bladé J, Boccadoro M, Child JA, Avet-Loiseau H,
Kyle RA, et al: International staging system for multiple myeloma.
J Clin Oncol. 23:3412–3420. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Murphy SK, Wylie AA, Coveler KJ, Cotter
PD, Papenhausen PR, Sutton VR, Shaffer LG and Jirtle RL: Epigenetic
detection of human chromosome 14 uniparental disomy. Hum Mutat.
22:92–97. 2003.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Benetatos L, Dasoula A, Hatzimichael E,
Georgiou I, Syrrou M and Bourantas KL: Promoter hypermethylation of
the MEG3 (DLK1/MEG3) imprinted gene in multiple myeloma. Clin
Lymphoma Myeloma. 8:171–175. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sigmon J and Larcom LL: The effect of
ethidium bromide on mobility of DNA fragments in agarose gel
electrophoresis. Electrophoresis. 17:1524–1527. 1996.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Schmidt M, Dehne S and Feierabend J:
Post-transcriptional mechanisms control catalase synthesis during
its light-induced turnover in rye leaves through the availability
of the hemin cofactor and reversible changes of the translation
efficiency of mRNA. Plant J. 31:601–613. 2002.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang X, Rice K, Wang Y, Chen W, Zhong Y,
Nakayama Y, Zhou Y and Klibanski A: Maternally expressed gene 3
(MEG3) noncoding ribonucleic acid: Isoform structure, expression,
and functions. Endocrinology. 151:939–947. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jin L, Cai Q, Wang S, Wang S, Mondal T,
Wang J and Quan Z: Long noncoding RNA MEG3 regulates LATS2 by
promoting the ubiquitination of EZH2 and inhibits proliferation and
invasion in gallbladder cancer. Cell Death Dis.
9(1017)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chak WP, Lung RW, Tong JH, Chan SY, Lun
SW, Tsao SW, Lo KW and To KF: Downregulation of long non-coding RNA
MEG3 in nasopharyngeal carcinoma. Mol Carcinog. 56:1041–1054.
2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chng WJ, Price-Troska T, Gonzalez-Paz N,
Van Wier S, Jacobus S, Blood E, Henderson K, Oken M, Van Ness B,
Greipp P, et al: Clinical significance of TP53 mutation in myeloma.
Leukemia. 21:582–584. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Drach J, Ackermann J, Fritz E, Krömer E,
Schuster R, Gisslinger H, DeSantis M, Zojer N, Fiegl M, Roka S, et
al: Presence of a p53 gene deletion in patients with multiple
myeloma predicts for short survival after conventional-dose
chemotherapy. Blood. 92:802–809. 1998.PubMed/NCBI
|
|
36
|
Brooks CL and Gu W: p53 regulation by
ubiquitin. FEBS Lett. 585:2803–2809. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Harris SL and Levine AJ: The p53 pathway:
Positive and negative feedback loops. Oncogene. 24:2899–2908.
2005.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu
WQ, Xie WP and Hou YY: Long non-coding RNA MEG3 inhibits NSCLC
cells proliferation and induces apoptosis by affecting p53
expression. BMC Cancer. 13(461)2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista
DL, Gejman R, Ansell PJ, Zhao J, Weng C and Klibanski A: Activation
of p53 by MEG3 non-coding RNA. J Biol Chem. 282:24731–24742.
2007.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Feng SQ, Zhang XY, Fan HT, Sun QJ and
Zhang M: Upregulation of LncRNA MEG3 inhibits cell migration and
invasion and enhances cisplatin chemosensitivity in bladder cancer
cells. Neoplasma. 65:925–932. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhu J, Liu S, Ye F, Shen Y, Tie Y, Zhu J,
Wei L, Jin Y, Fu H, Wu Y and Zheng X: Long noncoding RNA MEG3
interacts with p53 protein and regulates partial p53 target genes
in hepatoma cells. PLoS One. 10(e0139790)2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhou Y, Zhang X and Klibanski A: MEG3
noncoding RNA: A tumor suppressor. J Mol Endocrinol. 48:R45–53.
2012.PubMed/NCBI View Article : Google Scholar
|