Introduction
Aortic dissection (AD) is a deadly condition in
which the mouth of the aortic wall intima is torn, allowing
pulsatile blood flow through the tear, thereby causing the layers
of the aorta to separate (1). A
dissection may significantly reduce vascular wall strength, finally
resulting in vascular expansion or rupture (2). This is associated with the high
mortality rate identified in recent years for this condition: The
global fatality rate from AD and aortic aneurysms combined has
increased from 2.49 per 100,000 in 1999 to 2.78 per 100,000
inhabitants in 2010, representing an increasing global health
burden (3). Investigators have
recently begun to develop methods for the diagnosis and treatment
of thoracic AD (TAD) at the gene and protein levels. One of the
characteristic features of aortic aneurysm and dissection is the
disruption and degradation of structural extracellular matrix (ECM)
proteins by the matrix metalloproteinases (MMPs), particularly MMP2
and MMP9. Therefore, ECM-degrading proteins within the MMP family
may serve as promising biomarkers for the diagnosis and treatment
of AD (4-6).
DNA methylation has a key role in embryonic development and in the
pathogenesis of numerous diseases by regulating gene transcription,
imprinting and immune defense against the invasion of cells by
exogenous genetic material. It was reported that DNA methylation
involves heritable but reversible methylation or demethylation of
nucleotides and has an important role in gene and RNA
transcriptional regulation in cardiovascular diseases (7-9).
In the past 5 years of methylome research, enhancer
regions in particular have been identified as critical sites for
differential methylation in projects including ENCODE and FANTOM5
(10,11). Epigenome-wide association studies
using DNA methylation arrays have become an important tool in
clinical research for discovering differentially methylated regions
(DMRs) associated with phenotypes of interest. For instance,
Illumina's Infinium Beadchips provide unparalleled genome-wide
coverage, allowing the detection of DMRs of the DNA that are likely
to influence transcriptional gene activity and, thus, the
regulation of metabolic processes (12,13).
Indeed, by far the most widely used technology in this field is the
Illumina Methylation EPIC 850K BeadChip, which may be used to
compare the methylation levels of 850,000 cytosines in the context
of CpG dinucleotides between patients and a control group (14).
Changes in DNA methylation may affect the expression
of numerous genes (15,16). In particular, methylation frequently
occurs at the 5'position of the cytosine ring in CpG dinucleotides,
where it has a role in regulating gene expression (17). In a previous study, differences in
the methylation level of promoters of the MMP gene family between
healthy controls and patients with acute AD were identified.
Therefore, it was hypothesized that DNA methylation may have an
important role in the development and progression of TAD. To date,
however, only few studies have been performed on the epigenetic
mechanisms involved in TAD, and no study has focused on DNA
methylation specifically. Thus, the aim of the present study was to
identify and characterize the DMRs and associated genes in TAD in
comparison with healthy controls. The DNA methylation level of MMP
promoters in the peripheral blood was also quantified to determine
whether altered MMP promoter methylation specifically may be
associated with TAD. The present study may help to identify novel
DNA methylation markers and enhance the current understanding and
treatment of TAD.
Materials and methods
Patients
Subjects treated at the inpatient clinic of Ningbo
Medical Center, Li Huili Hospital (Ningbo, China) between May 2016
and December 2016 were selected. The inclusion criteria for the
clinical diagnosis of AD were as follows: i) AD originating in the
descending thoracic aorta and not involving the ascending aorta
(known as type B); ii) proximal landing zone of at least 2 cm; iii)
distance between entry tear and brachiocephalic trunk of at least
0.5 cm; iv) no signs of cardiac tamponade or severe aortic
regurgitation; and v) no sign of aortic branch ischemia. Patients
with cardiac revascularization from the ascending aorta were
excluded. Clinical information regarding the 25 patients with TAD
and 27 healthy controls, from which samples were taken for
methylation analysis, is provided in Table I. All participants were recruited
from Li Huili Hospital (Ningbo, China).
 | Table IClinical characteristics of 25 cases
with thoracic aortic dissection and 27 healthy controls. |
Table I
Clinical characteristics of 25 cases
with thoracic aortic dissection and 27 healthy controls.
| | Illumina Human
Methylation 850K array (n=8) | MMP2 gene methylation
by pyrosequencing (n=44) |
|---|
| Variable | Controls (n=4) | Cases (n=4) | Controls (n=23) | Cases (n=21) |
|---|
| Age (years) | 60.25±5.62
(59-64) | 61.00±2.16
(53-66) | 59.83±5.96
(47-61) | 63.8±14.72
(37-90) |
| Sex | | | | |
|
Male | 4 | 4 | 16 | 19 |
|
Female | 0 | 0 | 7 | 2 |
| Cigarette
smoking | 2 | 0 | 6 | 15 |
| Hypertension | 0 | 4 | 2 | 21 |
Sample collection
A total of 52 blood samples were collected in EDTA
tubes from patients with TAD (25 samples) and healthy controls (27
samples). The samples were centrifuged for 15 min at 936 x g to
obtain the plasma, which was then stored at -80˚C. The 52 samples
were divided into four groups: i) 4 samples from the patients with
TAD for array analysis, ii) 4 samples from the healthy controls for
array analysis, iii) 21 samples from the patients with TAD for
bisulfite pyrosequencing and iv) 23 samples from the healthy
controls for bisulfite pyrosequencing. All of the blood samples
were collected by the same investigator (Xiuying Zhu).
Infinium HumanMethylationEPIC 850
BeadChip assay
The Infinium MethylationEPIC 850K BeadChip assay
(Illumina) was performed in accordance with Illumina's standard
protocol. Bisulfite-converted DNA was amplified followed by
enzymatic end-point fragmentation, precipitation and resuspension.
Sample labeling, hybridization to chips and image scanning were
performed and all 8 samples were processed on the same chipset to
avoid a batch effect. Principal component analysis was performed
between the TAD cases and the controls through PCA analysis. The
distribution of samples was investigated, the rationality of
experimental design was verified and the homogeneity of biological
duplicate samples was demonstrated by two-dimensional graph. The
closer the two-dimensional spatial distribution of the same group
of samples was, the more representative the selection of these
samples was, as well as the better the biological duplicate.
Bisulfite treatment
Genomic DNA was extracted from plasma using a QIAamp
Mini kit (Qiagen). Finally, the QIAamp Mini spin column was placed
in a new 1.5-ml microcentrifuge tube, 200 µl Buffer AE or distilled
water was added and the column was incubated at room temperature
for 1 min. Subsequently, the column was centrifuged at 6,654 x g
for 1 min to elute the DNA. Bisulfite treatment was performed with
the EpiTect DNA Bisulfite kit (Qiagen), in accordance with the
manufacturer's protocol, using 500 ng genomic DNA isolated from
each sample.
Pyrosequencing to determine MMP2
methylation levels
PCR amplification of bisulfite-treated DNA was
performed in a total volume of 38 µl of premix, using 50 pmol of
each primer and 100 ng of bisulfite-treated DNA. A total of 40
cycles of PCR were performed using the PCR conditions described in
a previous study by our group (18).
PCR products were visualized on 1.5% agarose gels. Pyrosequencing
was performed in accordance with the manufacturer's protocol on a
PyroMark Q96 ID System (Qiagen). A total of 4 CpG sites in the
promoter of the MMP2 gene were analyzed with the Pyro Q96-CpG
software 2.5.8 (Qiagen). The mean percentage of methylation of
these 11 CpG sites analyzed was calculated. The primers were as
follows: MMP2 sequencing primer, 5'-ACTACCAACTCTTTATCC-3'; forward
primer, 5'-TTTGTTTTTTGGGTTGTTTGTTGA-3' and reverse primer,
5'-CTCACCACTACCAACTCTTTATC-3'.
Quality control of methylation data
and data analysis
DNA quality was controlled using BiQ Analyzer
software. Pathway and gene ontology (GO) analyses were performed.
Statistical analyses were performed using R software (version
2.1.1). The quality of sequencing data was controlled using
GraphPad Prism 8 software (GraphPad Software, Inc.). Values are
expressed as the mean ± standard deviation and P<0.05 was
considered to indicate statistical significance. Student's t-test
was used to compare differences in methylation levels between the
patients with TAD and the controls. Analysis of variance was used
for comparing positions 1-4 in the TAD group.
Results
Participant characteristics
The present study aimed to compare the DMRs in cases
with TAD vs. healthy controls. The clinical and pathological
characteristics of the subjects are provided in Table I, with 27 controls (20 of which were
male) and 25 cases (23 of which were male). A total of 4 male
patients with TAD (mean age, 61±2.16 years) and 4 male healthy
donors (mean age, 60.25±5.62 years) were recruited. A total of 4
patients in each group were used for the Illumina Human Methylation
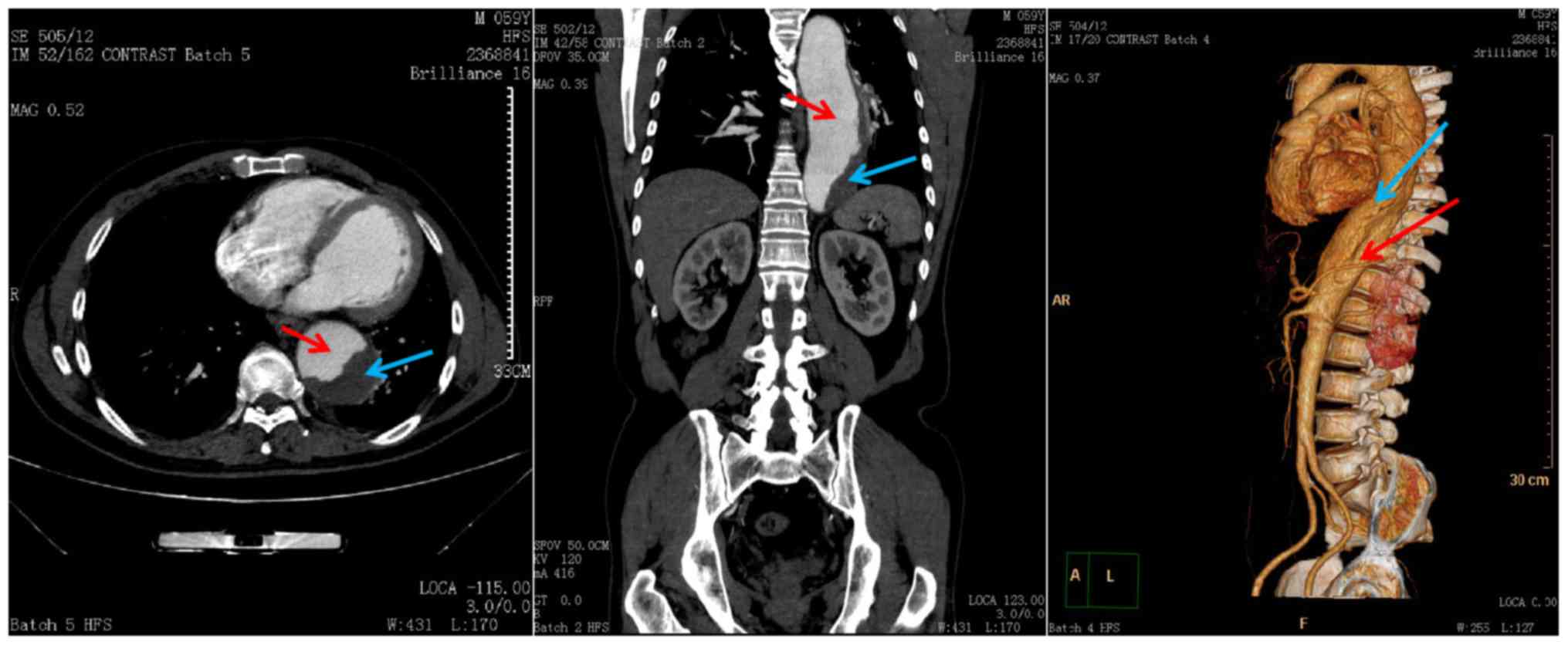
850K array. Pre-operative aortography of one of the patients with
TAD (Fig. 1) revealed that the true
lumen was narrow in the middle of the descending aorta. The
remaining 21 patients with TAD (mean age, 63.8±14.72) and 23
healthy controls (mean age, 59.83±5.96) were used to assess MMP2
gene methylation by pyrosequencing.
Quality control of the methylation
array data
Genome-wide DNA methylation profiles of the 8
participants were generated using the Illumina Human Methylation
EPIC 850 BeadChip. Methylation at each locus was measured using
β-values, which were generated using the Illumina GenomeStudio
software 1.8 (Illumina) based on the intensity of the methylated
and unmethylated probes. The workflow of the procedure used to
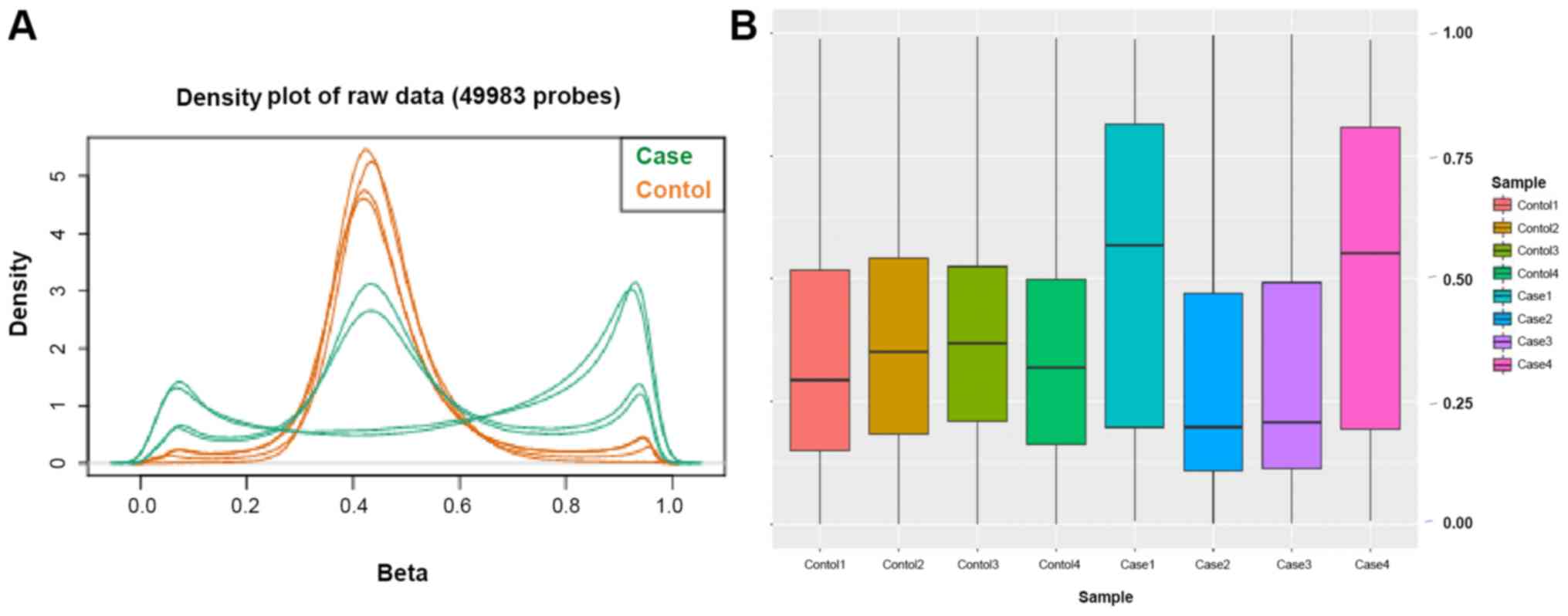
identify the DMRs by array analysis is presented in Fig. S1. The density distribution of the
β-values indicated a typical bimodal distribution (Fig. 2A), in which the first peak
corresponded to low or unmethylated probes with a β-value close to
0, while the second peak corresponded to highly or fully methylated
probes with a β-value close to 1. Furthermore, boxplots of the
β-value distributions of the 8 participants were presented
(Fig. 2B). Using the average
β-values of the two data groups, the overall distribution and
concentration trend of the two groups of data in AD group and
normal group were evaluated in a homogeneous distribution.
Comparison of differential methylation
between patients with TAD and healthy controls
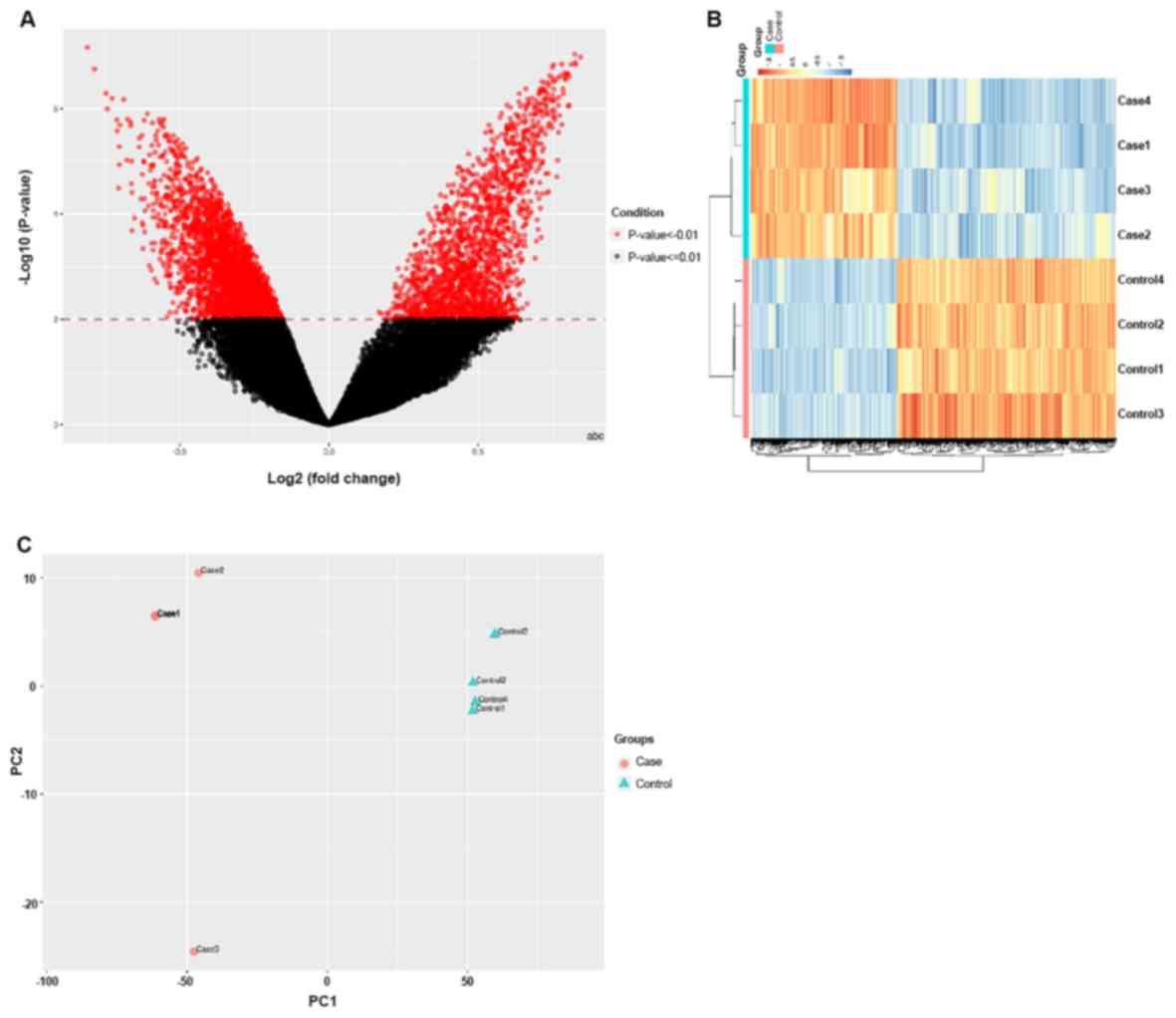
Using the BeadChip, 3,362 DMRs were identified with
a significance of P<0.05, while 1,223 were identified with a
significance of P<0.01. Among these, 2,019 CpG sites were
hypermethylated and 1,343 were hypomethylated in the TAD group
relative to the healthy controls. A volcano plot was used to
graphically represent the distribution of significant CpG sites
from the site-level test sorted by the mean β-differences and
P-values (Fig. 3A). Fig. 3B presents the differentially
methylated CpG sites between the TAD cases and the controls.
Fig. 3C provides the results of the
principal component analysis of the two sets of samples.
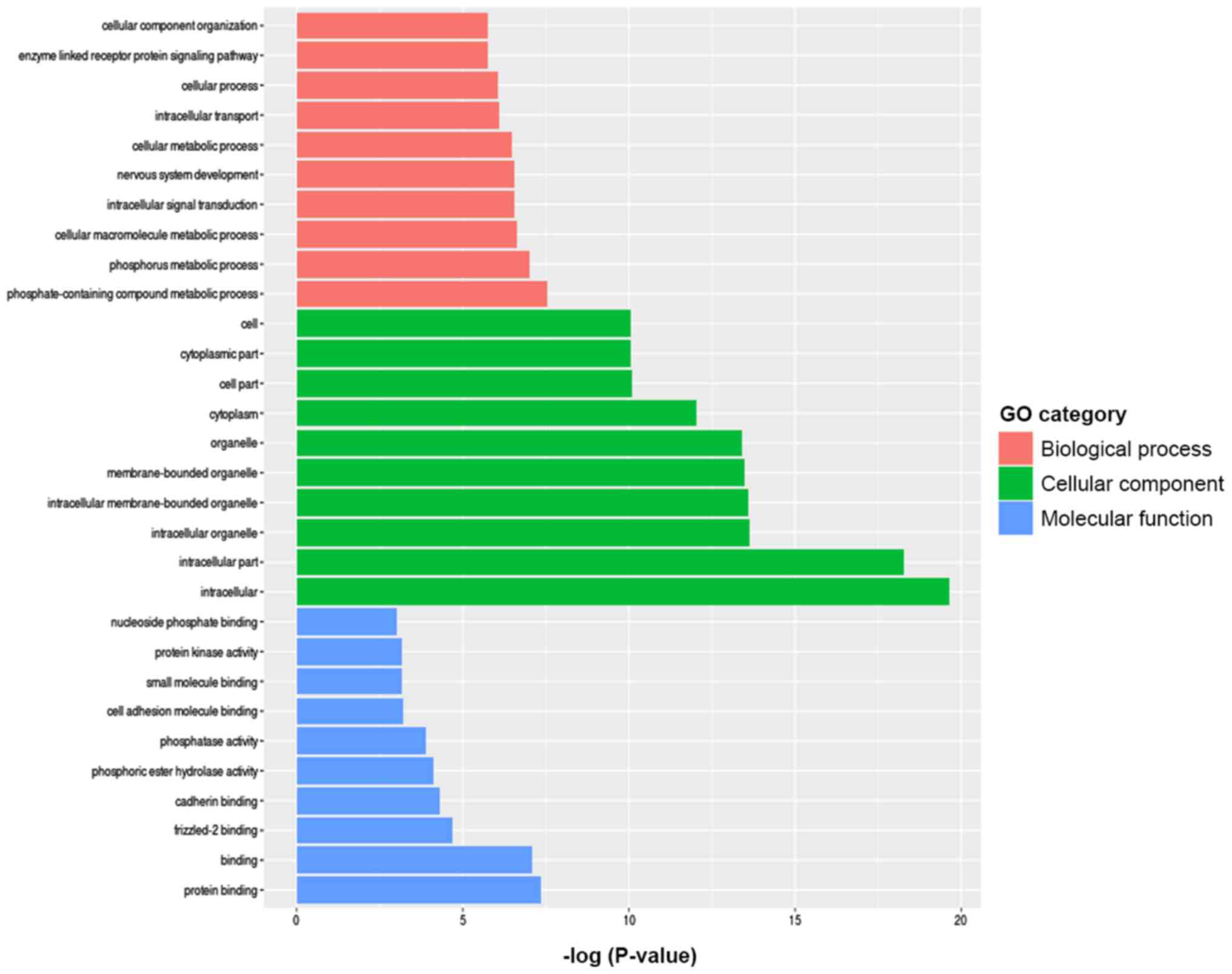
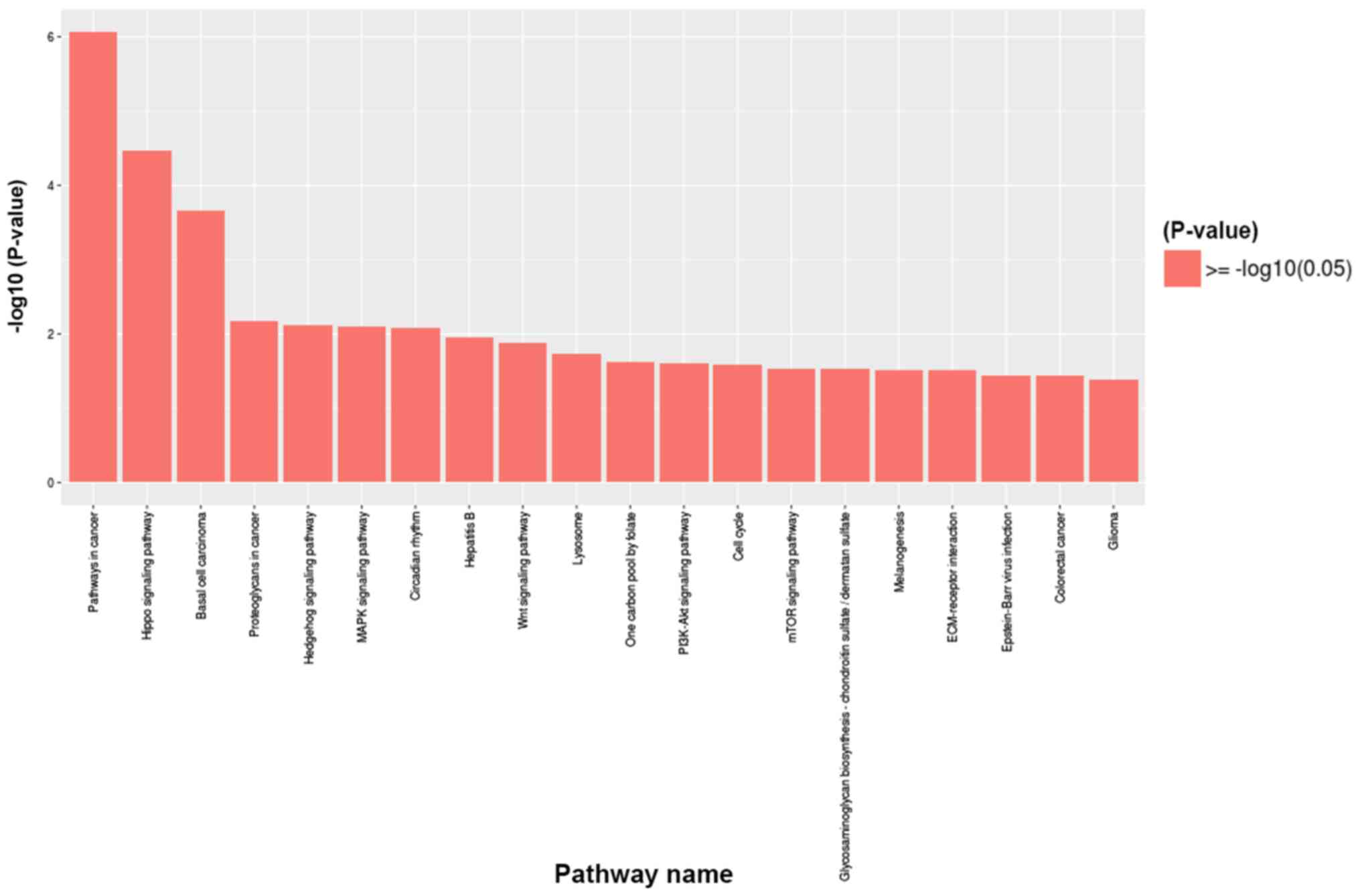
GO and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway analyses
To further investigate the signaling pathways
associated with the differentially methylated genes between the TAD
and control groups, KEGG signaling pathway analysis was used.
According to the criteria of P<0.05 and false discovery rate
<0.05, the 20 most significant major signaling pathways were
identified. GO functional enrichment was also analyzed according to
the differentially methylated sites and the results were annotated
and classified (Fig. 4). Kyoto
Encyclopedia of Genes and Genomes pathway analyses of the
differentially methylated genes are presented in Fig. 5. The key pathways and genes
associated with TAD were identified and also listed in Table II. These results indicate that
changes in the immune system may occur during the pathogenesis of
TAD. The results of the KEGG analysis corresponded well with the
results of the GO analysis.
| Table IIPathway analysis of the differentially
methylated regions and their associated genes in the thoracic
aortic dissection vs. control group based on Kyoto Encyclopedia of
Genes and Genomes pathway analysis. |
Table II
Pathway analysis of the differentially
methylated regions and their associated genes in the thoracic
aortic dissection vs. control group based on Kyoto Encyclopedia of
Genes and Genomes pathway analysis.
| Pathway_id | Pathway_name | Pathway_class | Genes_in_list |
|---|
| hsa05200 | Pathways in
cancer | Cancers:
Overview | MMP2, WNT1, WNT2B,
IGF1R, PDGFB, BCL2, PGF |
| hsa04151 | PI3K-Akt signaling
pathway | Signal
transduction | FGFR2, IFNAR1,
PPP2R2C, IGF1R, MDM2, TNXB, RPTOR |
| hsa04010 | MAPK signaling
pathway | Signal
transduction | FGFR2, DUSP10, ECSIT,
RASGRF1, GRB2, PDGFB, MAPT, NFATC2 |
| hsa05205 | Proteoglycans in
cancer | Cancers:
Overview | MMP2, WNT1, WNT2B,
IGF1R, MDM2, ITPR3, GRB2, FGF12 |
| hsa04390 | Hippo signaling
pathway | Signal
transduction | WNT1, WNT2B, GLI2,
WNT3A, APC, TCF7L1, PPP2R2B, WNT3, MPP5 |
| hsa05169 | Epstein-Barr virus
infection | Infectious
diseases: Viral | PSMD2, MDM2, PSMD7,
MAP2K6, BCL2, HDAC4, FCER2, CCNA1 |
| hsa05161 | Hepatitis B | Infectious
diseases: Viral | IFNAR1, TICAM1,
NFATC3, GRB2, NFATC2, BCL2,TLR3 |
| hsa04310 | Wnt signaling
pathway | Signal
transduction | WNT1, WNT2B, CTBP2,
NFATC3, FBXW11, NFATC2, SFRP2, RAC1 |
| hsa04142 | Lysosome | Transport and
catabolism | MFSD8, AP3D1,
LAMP1, GBA, NAGPA, CTSA, ARSG, PPT1, CTNS |
| hsa04110 | Cell cycle | Cell growth and
death | MDM2, TFDP2,
MAD1L1, MCM5, CCNA1, E2F2, TGFB1, TFDP1, MCM7 |
| hsa04916 | Melanogenesis | Endocrine
system | WNT1, WNT2B, ADCY7,
ADCY9, PRKCA, PRKCG, WNT5B, WNT5A |
| hsa05217 | Basal cell
carcinoma | Cancers: Specific
types | WNT1, WNT2B, GLI2,
SUFU, APC2, WNT5B, WNT5A, FZD3, WNT3A |
| hsa04512 | ECM-receptor
interaction | Signaling molecules
and interaction | TNXB, COL4A1,
ITGB5, LAMA3, COL4A2, ITGB1, CD44, COL1A |
| hsa04340 | Hedgehog signaling
pathway | Signal
transduction | WNT1, WNT2B, GLI2,
SUFU, FBXW11, WNT5B, WNT5A, WNT3A |
| hsa04150 | mTOR signaling
pathway | Signal
transduction | RPTOR, PRKCA,
PRKCG, RPS6KA2, IKBKB, AKT1S1, MTOR, TSC2 |
| hsa05210 | Colorectal
cancer | Cancers: Specific
types | MLH1, BCL2, RAC1,
APC2, TGFB1, BIRC5, APC, TCF7L1, AKT1 |
| hsa05214 | Glioma | Cancers: Specific
types | IGF1R, MDM2, GRB2,
PDGFB, PRKCA, PRKCG, E2F2, MTOR, AKT1 |
| hsa04710 | Circadian
rhythm | Environmental
adaptation | PRKAG2, FBXW11,
NR1D1, BHLHE41, RORA, RORB, PRKAA1, SKP1 |
Comparison of differential methylation
between patients with TAD and healthy controls with mapping to the
MMP2 gene
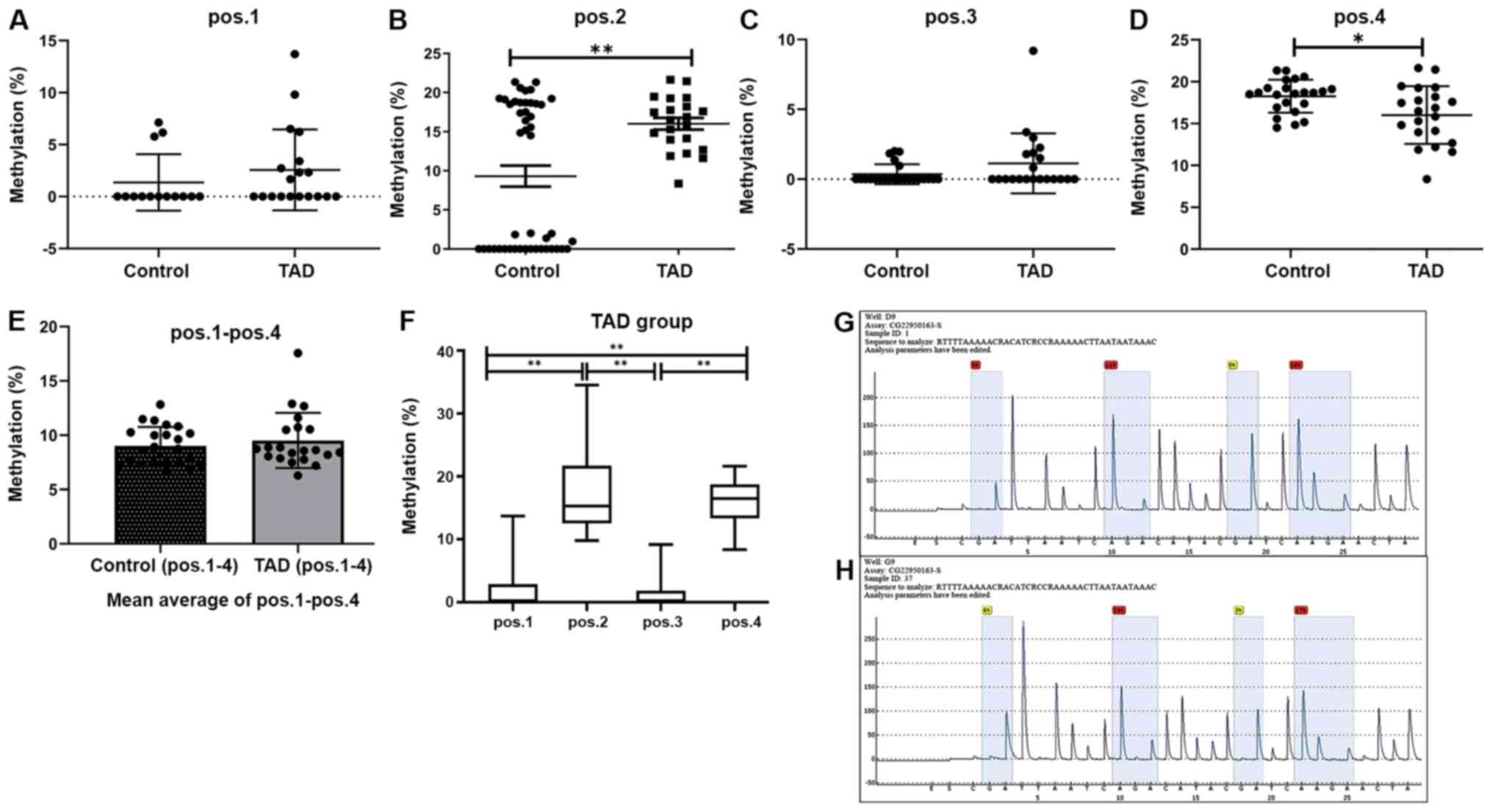
Bisulfite pyrosequencing was then performed for a
fragment of the promoter region of the MMP2 gene and the results
are provided in Fig. 6. This
fragment contained 4 CpG sites that may be measured to evaluate the
methylation levels of the MMP2 promoter (Fig. 6G and H). This analysis revealed significantly
increased methylation levels at position 2 within this fragment in
the TAD cases compared with the controls (P<0.01; Fig. 6B), while decreased methylation levels
were present at position 4 (P<0.05; Fig. 6D). However, no significant
differences in methylation levels were identified at position 1
(Fig. 6A) and position 3 (Fig. 6C). In summary, the mean methylation
level of the 4 CpG sites on the MMP2 gene in the TAD group was
higher than that in the control group (9.51±0.51 vs. 9.00±0.65%;
Fig. 6E). However, this difference
was not significant (P>0.05). Therefore, hypermethylation of the
MMP2 gene may be associated with TAD. The CpG sites that were
hypomethylated and hypermethylated in the TAD group compared with
the healthy controls were subjected to KEGG pathway enrichment
analysis (Table II). The genes
identified to be associated with TAD included MMP2, Wnt family
member 2B (WNT2B) and insulin-like growth factor (IGF), and these
genes are actively involved in carcinogenesis, the PI3K/Akt
signaling pathway, the mitogen-activated protein kinase (MAPK)
signaling pathway, proteoglycans in cancer, ECM-receptor
interactions and the circadian rhythm, which may all have key roles
in the development of cardiopulmonary dysfunction.
Discussion
Aortic aneurysm is a severe vascular disease
involving apoptosis of the vascular smooth muscle cells in the
aortic media, endothelial cell injury, and degradation and
pathological remodeling of the ECM (4,19). The
etiology and pathogenesis of aortic aneurysms have remained to be
comprehensively characterized. Characterization of the genetic
landscape of TAD is important to identify potential targets for
personalized therapeutic regimens (20). In the present study, a
high-throughput array platform (Illumina Human Methylation EPIC
850K BeadChip) was used to explore the genome-scale diversity of
DNA methylation in patients with AD in the Han Chinese population.
The incidence of TAD in males is significantly higher than that in
females, and in China, 85% of patients with TAD are male (21). In the present study, 4 samples from
male patients with TAD were selected for analysis with the
Methylation EPIC 850K BeadChip.
Compared with existing tools, DiMmeR may complete
all data analysis steps within only a few minutes, being guided by
an interactive user interface. DiMmeR is the first one-step assay
for the sophisticated identification of statistically robust
differentially methylated CpGs and regions in modern Illumina chip
data. To date, several methylation changes in specific genes have
been reported for TAD and DMRs mainly linked to the
inflammatory/defense response have been detected in aortic aneurysm
(4,22). In the present study, 2,019
hypermethylated and 1,343 hypomethylated CpG sites in the TAD group
compared with the healthy control group were identified.
Furthermore, subsequent GO pathway analysis suggested that, in the
biological process category, the MMP2, WNT2B and IGF genes were
particularly affected. These genes may have key roles in the
development of cardiopulmonary function disorders. Furthermore,
several key signaling pathways were also identified, including the
MAPK signaling pathway. Further comprehensive studies including
whole-genome and detailed epigenetic analyses are warranted to
fully elucidate the role that these genes have in the pathogenesis
of TAD.
Altered DNA methylation in genes including MMP2,
MMP14, WNT1 and BCL2 may mediate the involvement of vascular smooth
muscle cells and inflammatory cells in the pathogenesis and
progression of aortic aneurysm. Therefore, the knowledge obtained
from epigenetic studies of rheumatology may also be applicable to
TAD. For instance, hypermethylation of immune system-associated
genes, including MMP2, WNT1, WNT2B, IGF1 receptor (IGF1R),
platelet-derived growth factor subunit B, BCL2 and placental growth
factor, were identified among the TAD cases. Of these candidate
genes, 4 (MMP2, WNT1, IGF1R and BCL2) are potentially involved in
the pathogenesis of TAD. Aortic aneurysm, which refers to the focal
dilation of the aorta, results from impaired integrity of the
aortic ECM (23,24). MMPs, traditionally known as
ECM-degrading enzymes, have been demonstrated to have an important
role in the myocardial remodeling process; for instance, these
enzymes are active in dilated failing hearts (25). One of the common CpG probes indicated
a mapped gene in the present study (MMP2), which was selected for
further analysis, and genomic comparison of patients with TAD and
healthy controls at this locus suggested that differentially
methylated MMP2 may contribute toward the pathogenesis of TAD. MMP2
has previously been associated with aneurysm in patients and in
animal models (26). It may be
speculated that MMP2 has a pathogenic role during matrix
remodeling, which appears to be important in regulating cardiac
function following stress or injury. Combining the results from the
DNA methylation array and bisulfite pyrosequencing, a significant
difference was observed between patients with TAD and healthy
controls in terms of methylation within the MMP2 promoter, but the
in-depth mechanisms require further study.
In conclusion, a total of 2,019 hypermethylated and
1,343 hypomethylated CpG sites were identified in the TAD group
compared with the healthy controls. Subsequent GO and KEGG pathway
analyses suggested that the MMP2, MMP14, WNT2B and IGF genes in the
biological process category are actively involved in TAD. One of
the genes identified, MMP2, was confirmed to be hypermethylated,
which may be associated with an increased risk of TAD. The
epigenetics of vascular reconstruction and immune-based biomarkers
may not only provide novel insight into the pathogenesis and
progression of TAD but may also assist in identifying novel
therapeutic targets.
Supplementary Material
Workflow of the identification of
differentially methylated regions using array analyses of 4 cases
of thoracic aortic dissection and 4 healthy controls. FDR, false
discovery rate; chr, chromosome; SNP, single nucleotide
polymorphism.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Natural Science Foundation of Ningbo (grant no. 2016A610197), the
Zhejiang Province Medical and Health Project (grant no. 2017RC026),
the Ningbo Public Welfare Project (grant no. 2019C50069), the
Advanced Key Scientific and Technological Programs of Ningbo (grant
no. 2012C5017) and the Ningbo Health Branding Subject Fund (grant
no. PPXK2018-01).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
NL, HL, JG, GS and LS conceived the study,
participated in its design and coordination, and helped draft the
manuscript. NL, HS and DZ performed the experiments. NL, HL, HZ and
GX analyzed the data. JG participated in methylation analysis. XZ
participated in data collection and analysis of clinical samples.
NL, LS and GS wrote the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The Ethics Committee of Li Huili Hospital approved
the present study. Written informed consent was obtained from all
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ohle R: Diagnosing acute aortic
dissection: Both an artery and a science. Acad Emerg Med.
25(1186)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhao H, Wen D, Duan W, An R, Li J and
Zheng M: Identification of CTA-Based predictive findings for
temporary and permanent neurological dysfunction after repair in
acute type a aortic dissection. Sci Rep. 8(9740)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Strzyz P: A sugar rush of DNA methylation.
Nat Rev Mol Cell Biol. 19(617)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Pan S, Lai H, Shen Y, Breeze C, Beck S,
Hong T, Wang C and Teschendorff AE: DNA methylome analysis reveals
distinct epigenetic patterns of ascending aortic dissection and
bicuspid aortic valve. Cardiovasc Res. 113:692–704. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hannum G, Guinney J, Zhao L, Zhang L,
Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al:
Genome-wide methylation profiles reveal quantitative views of human
aging rates. Mol Cell. 49:359–367. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Longo GM, Xiong W, Greiner TC, Zhao Y,
Fiotti N and Baxter BT: Matrix metalloproteinases 2 and 9 work in
concert to produce aortic aneurysms. J Clin Invest. 110:625–632.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Moran S, Arribas C and Esteller M:
Validation of a DNA methylation microarray for 850,000 CpG sites of
the human genome enriched in enhancer sequences. Epigenomics.
8:389–399. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nasu T, Satoh M, Ohmomo H, Shiwa Y, Komaki
S, Ono K, Shimizu A, Taguchi S, Takahashi Y, Osaki T, et al:
Epigenome-wide association study identifies a novel DNA methylation
in patients with severe aortic valve stenosis. Circ Genom Precis
Med. 13(e002649)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Xu H, Du S, Fang B, Li C, Jia X, Zheng S,
Wang S, Li Q, Su W, Wang N, et al: VSMC-specific EP4 deletion
exacerbates angiotensin II-induced aortic dissection by increasing
vascular inflammation and blood pressure. Proc Natl Acad Sci USA.
116(8457)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Xu L, Zheng D, Wang L, Jiang D, Liu H, Xu
L, Liao Q, Zhang L, Liu P, Shi X, et al: GCK gene-body
hypomethylation is associated with the risk of coronary heart
disease. Biomed Res Int. 2014(151723)2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pidsley R, Zotenko E, Peters TJ, Lawrence
MG, Risbridger GP, Molloy P, Van Djik S, Muhlhausler B, Stirzaker C
and Clark SJ: Critical evaluation of the Illumina MethylationEPIC
BeadChip microarray for whole-genome DNA methylation profiling.
Genome Biol. 17(208)2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kirby MK, Ramaker RC, Roberts BS,
Lasseigne BN, Gunther DS, Burwell TC, Davis NS, Gulzar ZG, Absher
DM, Cooper SJ, et al: Genome-wide DNA methylation measurements in
prostate tissues uncovers novel prostate cancer diagnostic
biomarkers and transcription factor binding patterns. BMC Cancer.
17(273)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Silva-Martinez GA, Zaina S and Lund G:
Array probe density and pathobiological relevant CpG calling bias
in human disease and physiological DNA methylation profiling. Brief
Funct Genomics. 17:42–48. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Taylor JY, Wright ML, Crusto CA and Sun
YV: The intergenerational impact of genetic and psychological
factors on blood pressure (InterGEN) study: Design and methods for
complex DNA analysis. Biol Res Nurs. 18:521–530. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zheng D, Chen X, Li N, Sun L, Zhou Q, Shi
H, Xu G, Liu J, Xu L, Duan S and Shao G: Differentially methylated
regions in patients with rheumatic heart disease and secondary
pulmonary arterial hypertension. Exp Ther Med. 14:1367–1372.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Nagata H, Kozaki KI, Muramatsu T, Hiramoto
H, Tanimoto K, Fujiwara N, Imoto S, Ichikawa D, Otsuji E, Miyano S,
et al: Genome-wide screening of DNA methylation associated with
lymph node metastasis in esophageal squamous cell carcinoma.
Oncotarget. 8:37740–37750. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chen X, Shen LH, Gui LX, Yang F, Li J, Cao
SZ, Zuo ZC, Ma XP, Deng JL, Ren ZH, et al: Genome-wide DNA
methylation profile of prepubertal porcine testis. Reprod Fertil
Dev. 30:349–358. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Liu O, Xie W, Qin Y, Jia L, Zhang J, Xin
Y, Guan X, Li H, Gong M, Liu Y, et al: MMP-2 gene polymorphisms are
associated with type A aortic dissection and aortic diameters in
patients. Medicine (Baltimore). 95(e5175)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Shalata A, Mahroom M, Milewicz DM, Limin
G, Kassum F, Badarna K, Tarabeih N, Assy N, Fell R, Cohen H, et al:
Fatal thoracic aortic aneurysm and dissection in a large family
with a novel MYLK gene mutation: Delineation of the clinical
phenotype. Orphanet J Rare Dis. 13(41)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tang W, Yao L, Hoogeveen RC, Alonso A,
Couper DJ, Lutsey PL, Steenson CC, Guan W, Hunter DW, Lederle FA
and Folsom AR: The association of biomarkers of inflammation and
extracellular matrix degradation with the risk of abdominal aortic
aneurysm: The ARIC Study. Angiology. 70:130–140. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Li H, Chan YC and Cheng SW: Contemporary
endovascular treatment of type B aortic dissection in China. Asian
Cardiovasc Thorac Ann. 24:739–749. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bhandari R, Aatre RD and Kanthi Y:
Diagnostic approach and management of genetic aortopathies. Vasc
Med. 25:63–77. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liao M, Zou S, Bao Y, Jin J, Yang J, Liu
Y, Green M, Yang F and Qu L: Matrix metalloproteinases are
regulated by MicroRNA 320 in macrophages and are associated with
aortic dissection. Exp Cell Res. 370:98–102. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li T, Lv Z, Jing JJ, Yang J and Yuan Y:
Matrix metalloproteinase family polymorphisms and the risk of
aortic aneurysmal diseases: A systematic review and meta-analysis.
Clin Genet. 93:15–32. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lin X, Tan JYL, The AL, Lim IY, Liew SJ,
MacIsaac JL, Chong YS, Gluckman PD, Kobor MS, Cheong CY and Karnani
N: Cell type-specific DNA methylation in neonatal cord tissue and
cord blood: A 850K-reference panel and comparison of cell-types.
Epigenetics. 13:941–958. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li YH, Li XM, Lu MS, LV MF and JIN X: The
expression of the BRM and MMP2 genes in thoracic aortic aneurysm
and aortic dissection. Eur Rev Med Pharmacol Sci. 21:2743–2748.
2017.PubMed/NCBI
|