Introduction
Coronary heart diseases, including myocardial
infarction and ischemic heart disease, are a leading cause of
mortality and morbidity worldwide, causing at least 370,000 deaths
in the USA each year (1). Patients
with coronary heart disease, which is characterized by impaired
blood supply to the heart, present with myocardial ischemic injury
in the clinic. Coronary heart perfusion or reperfusion is a
commonly used and effective therapy in protection of the heart
against ischemic injury. However, perfusion can cause additional
damage to the myocardium, termed ischemia/reperfusion (I/R) injury.
Though reperfusion attenuates myocardial ischemia, it can increase
the incidence of arrhythmia and myocardial stunning, and increase
infarct size (2-4). It
would therefore be beneficial to develop a drug to prevent I/R
injury.
I/R injury can be modeled in vitro using a
hypoxia/reoxygenation (H/R) technique. In this model cells are
placed in an hypoxic environment and then returned to a normoxic
environment. This can be a useful tool to investigate
cardioprotective strategies against myocardial injury (5-7).
Dexmedetomidine (Dex),
(+)-4-(S)-[1-(2,3-dimethylphenyl)ethyl]-1H-imidazole, is a
selective and potent α2-adrenoceptor agonist, that is prescribed as
an anti-anxiety medication, a sedative and an analgesic (8,9). As an
α2-adrenoceptor agonist, Dex has potential applications as a
prophylactic in neuroprotection, which has attracted researchers to
study the role of Dex after I/R injury to the brain and other
organs (10). Studies in animal
models have reported that Dex inhibits hepatic and cerebral I/R by
suppressing the inflammatory response (11,12).
MicroRNAs (miRNAs/miRs) are RNAs with a length of
18-24 bp that can inhibit protein translation by binding to the 3'
untranslated region (UTR) of target mRNAs (13). The miR-17-92 cluster, which is one of
the most studied miRNA clusters, has six members including miR-17,
miR-18a, miR-19a, miR-19b-1, miR-20a and miR-92a (14). The miR-17-92 cluster has been
suggested to promote cardiomyocyte proliferation in post-natal and
adult hearts (14). Numerous studies
have also indicated that miR-17-92 cluster expression is related to
the progression of cancer and physiological disorders, such as
genetic bone, lung and septal defects (15-18).
Additional research suggests that miR-17-3p promoted keratinocyte
cells proliferation and metastasis via activating Notch1/NF-κB
signal pathways in cutaneous wound healing (19).
In the present study it was hypothesized that
regulation of miR-17-3p, a component of the miR-17-92 cluster, may
be the method through which Dex reduces I/R and inflammation caused
by I/R. The aim of the present study was to investigate whether Dex
reduced I/R injury to the myocardium using an H/R model in H9C2
cells and to study the relationship between Dex and miR-17-3p.
Materials and methods
H9C2 cell culture and H/R model
H9C2 cells are myoblasts derived from the rat
myocardium, used in the present study as a model of cardiomyocytes,
and were acquired from American Type Culture Collection. Gibco, a
brand of Thermo Fisher Scientific, Inc., supplied all cell culture
reagents. Under normoxic conditions H9C2 cells were cultured in
high glucose Dulbecco's Modified Eagle Medium (DMEM), 10% fetal
bovine serum (FBS), 1% 10,000 U/ml penicillin and 10,000 µg/ml
streptomycin. Under hypoxic conditions the cells were cultured in
PBS in a hypoxic chamber (50x50x60 cm) filled with 5%
CO2 and 95% N2 at 37˚C for different times
(1, 2, 3 and 4 h). The hypoxic chamber was placed in an aseptic
incubator chamber at 37˚C. Gas filling was performed according to
the method of Li et al (20).
After exposure to hypoxia, the cells were reoxygenated with normal
culture medium in 5% CO2 and 95% air at 37˚C for 3
h.
Transfection and treatment
A concentration of 50 nmol/l of miR-17-3p mimic,
miR-17-3p inhibitor and a miR-negative control (NC) were obtained
from Biomics Biotechnologies (Nantong) Co., Ltd. and were dissolved
in FBS-free DMEM medium, containing Lipofectamine™ (Invitrogen;
Thermo Fisher Scientific, Inc.), following the manufacturer's
protocol. Sequences were 3'-GAUGUUCACGGAAGUGACGUCA-5' (mimics),
3'-CUACAAGUGCCUUCACUGCAGU-5' (inhibitor) and
5'-UUUGUACUACACAAAAGUACUG-3' (NC). A Lipofectamine-medium solution
was added to the cells (2x105 cells/well) in 24-well
plate, and cells were cultured for 2-3 h. The medium was then
replaced with normal culture medium and the cells were cultured for
a further 24 h. TAK-242 (TAK), an inhibitor of TLR-4 was purchased
from MedChemExpress. Dex was purchased from Selleck Chemicals.
Cells were treated with different concentrations of
Dex (0, 0.1, 1, 5 and 10 µmol/l) to assess the impact of Dex on
cell viability and miR-17-3p levels in H9C2 cells. Cells treated
with different concentrations of Dex (0, 0.1, 1, 5 and 10 µmol/l)
were applied for detecting function of Dex on cell viability,
apoptosis, miR-17-3p, TLR4 and galectin-3 levels in H9C2 cells with
H/R (3 h hypoxia/3 h reoxygenation). H9C2 cells treated with
mimics, inhibitor, NC or TAK (10 µg/ml) were named the mimics,
inhibitor, NC or TAK groups respectively. Untreated H9C2 cells were
used as a control. Cells in the inhibitor, NC or TAK groups going
through 3 h hypoxia/3 h reoxygenation were named the H/R +
inhibitor, H/R + NC or H/R + TAK groups. Cells in the inhibitor, NC
or TAK groups, treated with 5 µmol/l Dex, going through 3 h
hypoxia/3 h reoxygenation were named the Dex + H/R + Inhibitor, Dex
+ H/R + NC, or Dex + H/R + TAK groups. Cells that had not received
any treatment were used as controls.
Cell viability
Cell Counting kit-8 (CCK-8; Sigma-Aldrich; Merck
KGaA) was used to determine cell viability, according to the
manufacturer's protocol. The cells (5x103 cells/well)
were seeded in 96-well plates for 24 h. After the cells had been
treated with the appropriate reagents or H/R, the CCK-8 reagent was
diluted with FBS-free DMEM at a ratio of 1:9. A total of 200 µl of
CCK-8 solution was applied to each well. The plate was put into an
incubator (Thermo Fisher Scientific, Inc.) for 1 h. The color
change was detected by a microplate reader (Thermo Fisher
Scientific, Inc.) at a wavelength of 450 nm.
Apoptosis
An Annexin V-FITC/propidium iodide apoptosis kit
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for cell
apoptosis analysis. Cells (5.0x105/ml) were seeded on a
75 mm plate for 24 h. After cells were treated with the reagents or
H/R, medium was collected in a centrifuge tube and the cells were
digested with trypsin (Gibco; Thermo Fisher Scientific, Inc.).
Medium was mixed with the cell suspension, and the suspension was
centrifuged using a Cence centrifuge (Changsha Xiangyi Centrifuge
Instrument Co., Ltd.) at 1,200 x g for 5 min at ˚C. The cell pellet
was then resuspended in PBS. Apoptosis kit reagents were added to
the cells and the fluorescence was detected using a BD FACSCalibur
flow cytometer (BD Biosciences) and BD CellQuest™ Pro Software
version 5.1 (BD Biosciences). Procedures were conducted following
the manufacturer's instructions.
Bioinformatic analysis and dual
luciferase assay
The Targetscan web site (http://www.targetscan.org/vert_72/) was used to
predict the binding site between miR-17-3p and TLR4. The TLR4 3'
UTR or mutant (MUT) TLR4 3' UTR [Sangon Biotech (Shanghai) Co.,
Ltd] was cloned into a psiCHECK-2 vector (Promega Corporation).
Cells were transfected with miR-17-3p mimic, miR-NC or cloned
psi-CHECK-2 vector, using Lipofectamine™ (Invitrogen; Thermo Fisher
Scientific, Inc.), following the manufacturer's protocol. After 24
h, the fluorescence was tested with a microplate reader (Thermo
Fisher Scientific, Inc.) using a Pierce™ Gaussia-Firefly luciferase
dual assay kit (Thermo Fisher Scientific, Inc.), according to
manufacturer's protocol.
Western blotting
After the cells were treated with the reagents or
H/R, the protein was extracted using RIPA lysis buffer (Thermo
Fisher Scientific, Inc.) following the manufacturer's instructions.
The concentration of protein was determined by BCA protein assay
kit (Thermo Fisher Scientific, Inc.) using BSA as a standard. 20 µg
protein and protein ladder (Thermo-Fisher Scientific, Inc.) was
separated by 12% SDS-PAGE and transferred onto PVDF membranes
(Sigma-Aldrich; Merck KGaA). The membranes were blocked with 5% BSA
(Beijing Solarbio Science & Technology Co., Ltd.) in TBS
containing Tween-20% (TBST) at room temperature for 2 h. The
primary antibodies, TLR4 (cat. no. ab22048; Abcam), galectin-3
(cat. no. ab2785; Abcam), phosphorylated (p)-p65 (cat. no. 13346;
Cell Signaling Technology, Inc.), p65 (cat. no. 6956, Cell
Signaling Technology, Inc.) and β-actin (1:5,000, cat. no. 3700;
Cell Signaling Technology, Inc.) were incubated with the protein
membranes overnight at 4˚C. The primary antibodies were diluted
with TBST 1:1,000. The protein membranes were then incubated for 2
h with secondary antibody (horseradish peroxidase-conjugated; cat.
no. 7076, Cell Signaling Technology, Inc.) at 37˚C. Enhanced
chemiluminescence (ECL) detection reagents (Amersham) were used and
detected with an Image-Pro Plus version 6.0 (Media Cybernetics,
Inc.) system.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the H9C2 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). cDNA was reverse transcribed using TaqMan MicroRNA reverse
transcription kit (Fermentas; Thermo Fisher Scientific, Inc.) at
42˚C for 50 min, according to the manufacturer's instructions.
SYBR-Green PCR Master mix (Roche Diagnostics) and the TaqMan miRNA
PCR kit (Applied Biosystems, Thermo Fisher Scientific, Inc.) were
used to perform qPCR assays, using the Opticon RT-PCR detection
system (ABI 7500; Thermo Fisher Scientific, Inc.), The
amplification primers were designed by Sigma-Aldrich; Merck KGaA.
The primers in Table I were used to
perform RT-qPCR under the conditions of 95˚C for 10 min for
pre-denaturation; 30 cycles of 95˚C for 30 sec, 55˚C for 30 sec and
72˚C for 60 sec. U6 and β-actin were used for normalization. The
2-ΔΔCq method was used to determine the relative mRNA
level (21).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| | Primer
sequence |
|---|
| Gene | Forward | Reverse |
|---|
| miR-17-3p |
5'-TGCGTTGACGTCACTCCCG-3' |
5'-GTGCAGGGTCCGAGGT-3' |
| Galectin-3 |
5'-CCGGGATCCATGGCAGACGGCTTCTCACTTAA-3' |
5'-CCGCCCATGGCTATCATTAGATCATGGCGTGGGAAGCG-3' |
| TNF-α |
5'-TGAGCACAGAAAGCATGATC-3' |
5'-CATCTGCTGGTACCACCAGTT-3' |
| IL-6 |
5'-GTGACAACCACGGCCTTCCCTA-3' |
5'-GGTAGCTATGGTACTCCA-3' |
| IL-1β |
5'-GACCTGTTCTTTGAGGCTGAC-3' |
5'-TCCATCTTCTTCTTTGGGTATTGTT-3' |
| TLR4 |
5'-ACCTGTCCCTGAACCCTATG-3' |
5'-CTTCTAAACCAGCCAGACC-3' |
| U6 |
5'-CTCGCTTCGGCAGCACA-3' |
5'-ACGCTTCACGAATTTGCGT-3' |
| β-actin |
5'-GTGGACATCCGCAAAGAC-3' |
5'-GAAAGGGTGTAACGCAACT-3' |
Statistical analysis
Data were analyzed using Graph Pad Prism version 6
(Graph Pad Software, Inc.). Data are presented as the mean ± SD.
One-way ANOVA with Tukey's post-hoc test was used for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of hypoxia on H9C2 cell
miR-17-3p levels
miR-17-3p mRNA expression increased significantly
following 1-4 h of hypoxia when compared with that of control cells
without hypoxia. Though miR-17-3p levels appeared to be increasing
over the first 3 h of hypoxia duration, miR-17-3p expression was
lower after 4 h than at other time-points, though it remained
significantly higher than in control cells (Fig. 1A) This result suggested that
prolonged H9C2 hypoxia could reduce miR-17-3p expression. After
cells were removed from a hypoxic environment they were exposed to
reoxygenation for 3 h and miR-17-3p mRNA expression returned to
levels comparable with control cells (Fig. 1B).
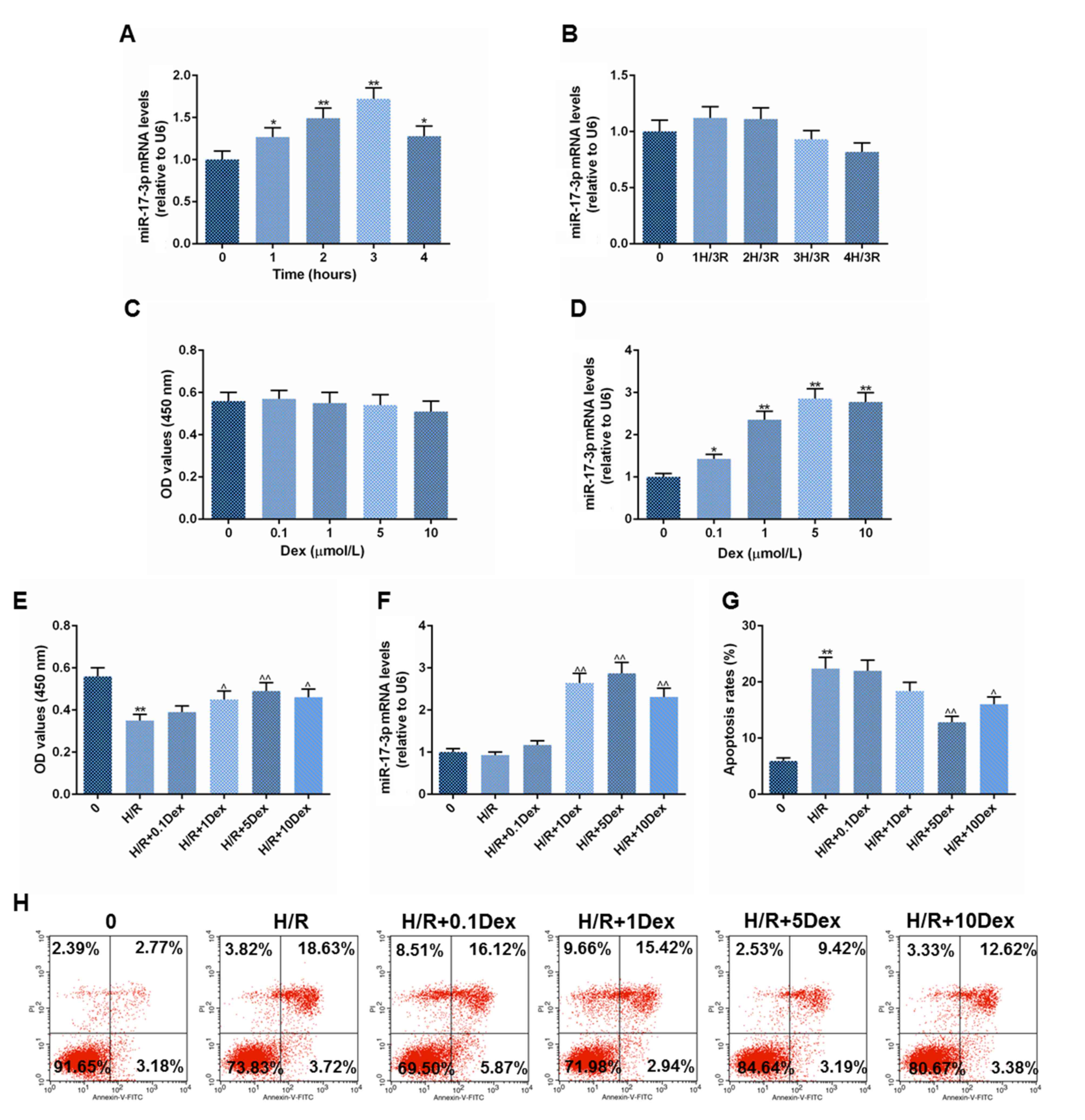 | Figure 1Effects of Dex on H9C2 cell viability
and miR-17-3p levels after H/R. H9C2 cells were incubated under
hypoxic conditions for 1-4 h and miR-17-3p mRNA levels relative to
U6 were determined (A) immediately or (B) after a further 3 h of
reoxygenation. H9C2 cells were additionally treated with a range of
concentrations of Dex for 24 h under normoxic conditions before (C)
cell viability and (D) miR-17-3p expression relative to U6 was
determined. An additional group of H9C2 cells was treated with a
range of concentrations of Dex for 24 h under normoxic conditions
before they were placed in hypoxic conditions for 3 h and then
reoxygenated for 3 h. (E) Cell viability, (F) miR-17-3p mRNA level
relative to U6, and (G) cell apoptosis (early and late stage) level
using (H) flow cytometry were then determined. Data are presented
as the mean ± SD. *P<0.05 and **P<0.01
vs. control group, ^P<0.05 and ^^P<0.01
vs. H/R group. Dex, dexmedetomidine; H/R, hypoxia/reoxygenation;
miR, microRNA, PI, propidium iodide. 0, the control, is cells with
no hypoxia and no reperfusion. 0.1, 1, 5 and 10 Dex are cells
treated with 0.1 µmol/l Dex. |
Effects of Dex on H9C2 cell viability,
cell apoptosis and miR-17-3p levels in H/R
Treatment with 0.1-10 µmol/l Dex had no obvious
effect on the viability of H9C2 cells (Fig. 1C). A dose of 0.1-10 µmol/l Dex
increased the expression of miR-17-3p mRNA compared with that of
the control group (0 µmol/l Dex) in a normoxic culture environment
(Fig. 1D). H/R decreased cell
viability, but Dex (1, 5 and 10 µmol/l) treatment improved H/R
exposed-cell viability significantly compared to H/R only control
cells (Fig. 1E). Dex (1, 5 and 10
µmol/l) also promoted miR-17-3p mRNA expression significantly in
H/R conditions (Fig. 1F). The H/R
model promoted H9C2 cell apoptosis, however, Dex (5 and 10 µmol/l)
decreased the rate of H9C2 cell apoptosis significantly after H/R
(Fig. 1G and H). The results presented in Fig. 1E-H led to the hypothesis that the
protective function of Dex on H9C2 cells in H/R was due to
regulation of miR-17-3p levels.
Effects of Dex on H9C2 cell expression
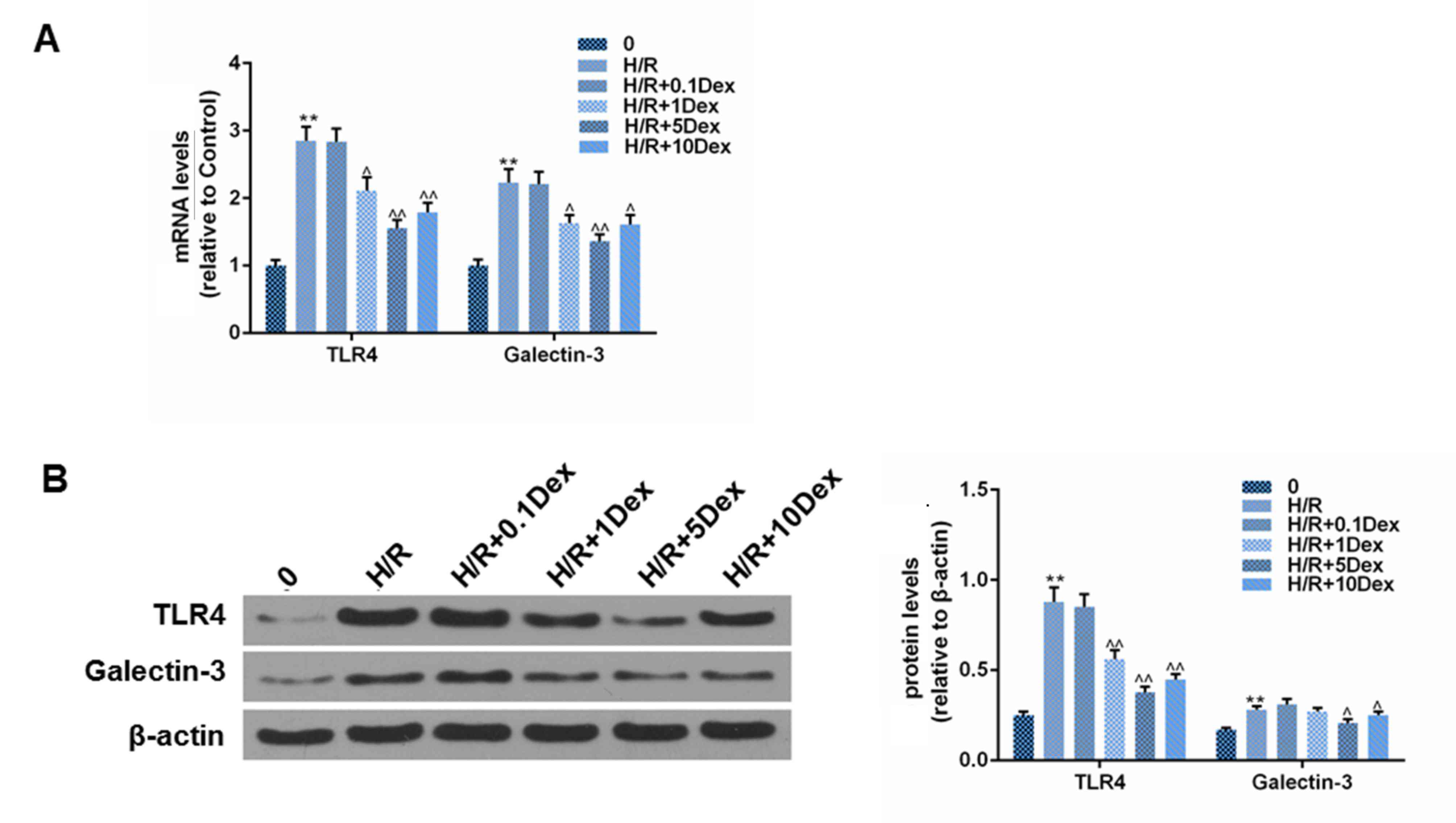
of TLR4 and galectin-3 in H/R
H/R increased TLR4 and galectin-3 expression at the
mRNA and protein level. Treatment with Dex (1, 5, or 10 µmol/l)
reduced the expression of TLR4 and galectin-3 during H/R (Fig. 2).
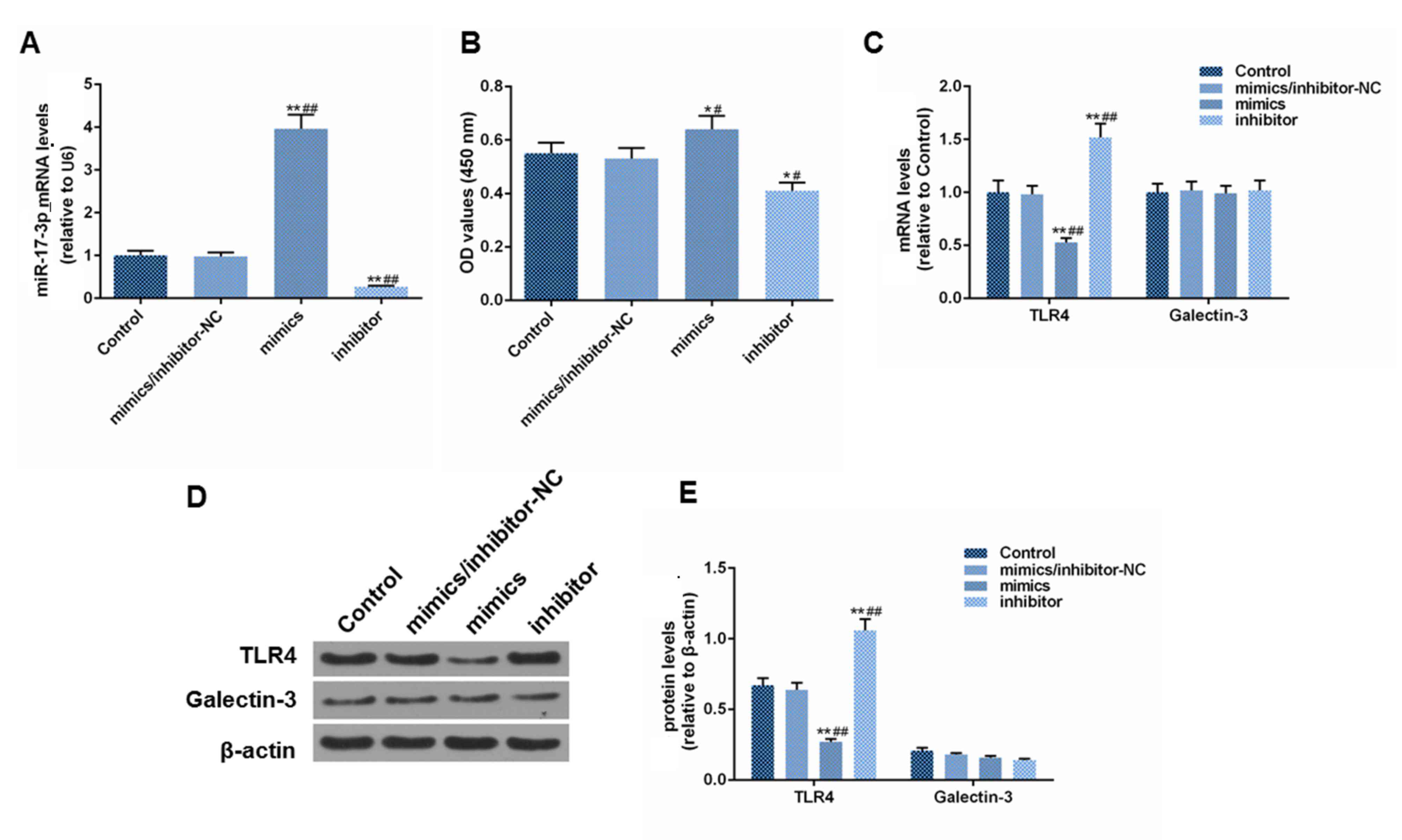
Effects of miR-17-3p inhibitor or
mimic on H9C2 cell viability and the expression of TLR4 and
galectin-3
miR-17-3p mimic increased miR-17-3p expression
(Fig. 3A) and cell viability
(Fig. 3B) and lowered TLR4 protein
and mRNA expression (Fig. 3C-E),
whereas, miR-17-3p inhibitor inhibited miR-17-3p expression and
cell viability but enhanced TLR4 expression (Fig. 3). However, both miR-17-3p mimic and
miR-17-3p inhibitor did not affect galectin-3 expression. It was
hypothesized that there was an association between miR-17-3p and
TLR4 expression.
Effects of Dex and miR-17-3p inhibitor
alone or in combination on H9C2 cell viability, apoptosis and
miR-17-3p expression in H/R
miR-17-3p inhibitor reduced cell viability in the
H/R + inhibitor group compared with the H/R + NC group, and reduced
the level of cell viability rescue induced by Dex (5 µmol/l) under
H/R conditions in the Dex + H/R + inhibitor group compared with the
Dex + H/R + NC group (Fig. 4A).
miR-17-3p inhibitor inhibited miR-17-3p mRNA expression, promoted
cell apoptosis in H/R and reduced the protective effect of Dex in
H/R (Fig. 4B-D).
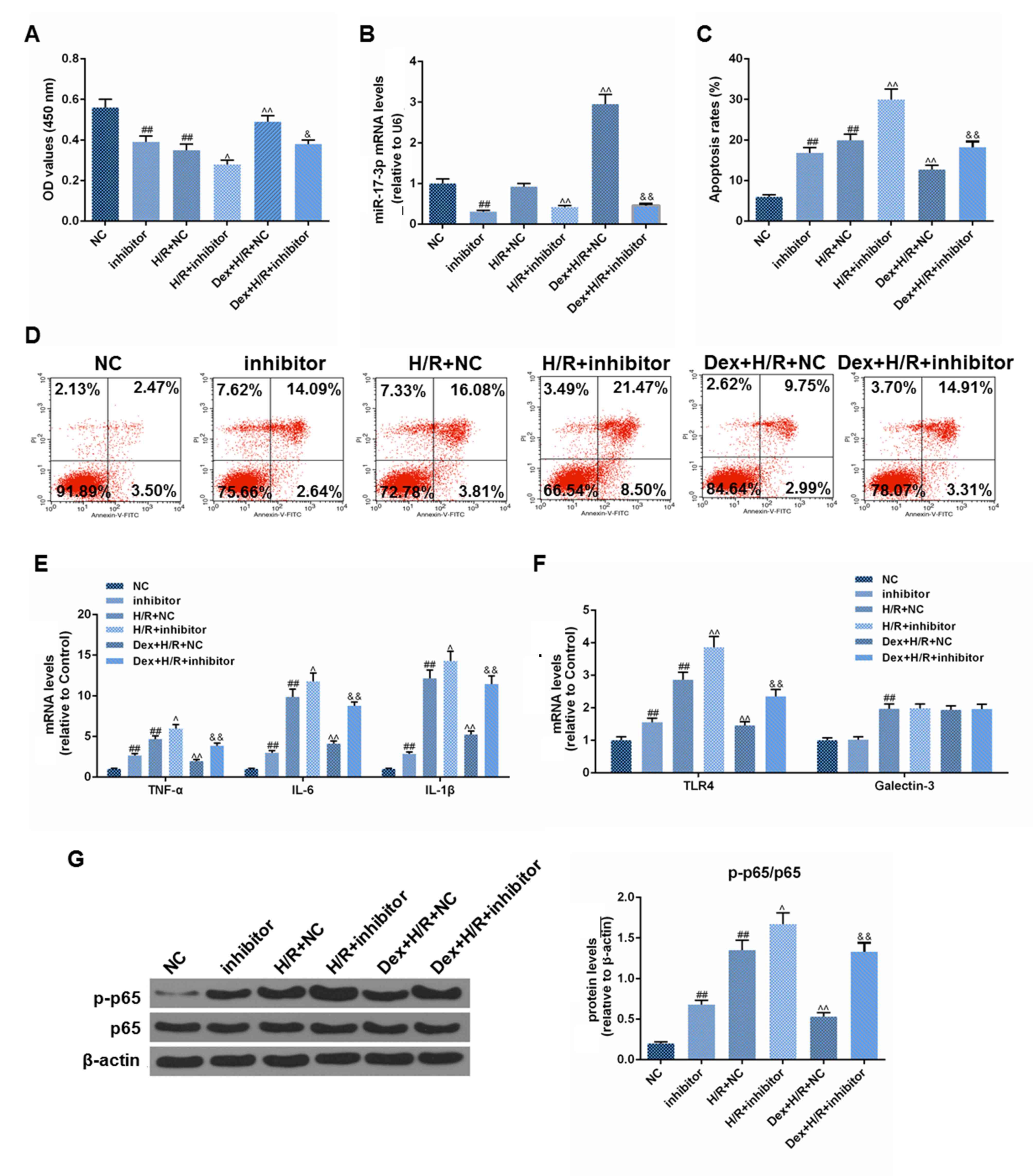 | Figure 4Effects of Dex and miR-17-3p
inhibitor alone or in combination on H9C2 cell viability, apoptosis
and the levels of miR-17-3p, TNF-α, IL-6, IL-1β, p65, p-p65,
galectin-3 and TLR4 during H/R. Dex and miR-17-3p inhibitor alone
or in combination were used to treat H9C2 cells for 24 h. Cells
were then placed in a hypoxic chamber for 3 h and reoxygenated for
3 h. Measurements were taken of (A) cell viability and (B) mRNA
levels of miR-17-3p. (C) Cell apoptosis was determined and (D)
representative plots are presented. mRNA levels of (E) TNF-α, IL-6,
IL-1β and (F) TLR4 and galectin-3 were determined. (G) The protein
levels of p-p65 and p-65 were determined using western blotting.
Data are presented as the mean ± SD. ##P<0.01 vs. NC
group, ^P<0.05 and ^^P<0.01 vs. H/R +
NC group, &P<0.05 and
&&P<0.01 vs. Dex + H/R + NC group. Dex,
dexmedetomidine; IL, interleukin; miR, microRNA; NC, negative
control; PI, propidium iodide; p, phosphorylated; TNF-α, tumor
necrosis factor-α. |
Effects of Dex and miR-17-3p inhibitor
alone or in combination on H9C2 cell mRNA levels of TNF-α, IL-6,
IL-1β, TLR4, galectin-3, p-p65 and p65 in H/R
Treatment with miR-17-3p inhibitor and exposure to
H/R led to increased mRNA levels of tumor necrosis factor-α
(TNF-α), interleukin (IL)-6, IL-1β, TLR4 and increased levels of
NF-κB phosphorylated (p)-p65/p65 protein in H9C2 cells (Fig. 4E-G). In addition, miR-17-3p inhibitor
treatment increased mRNA levels of TNF-α, IL-6, IL-1β, TLR4 and
increased levels of p-p65/p65 protein during H/R and in H9C2 cells
treated with Dex during H/R. However, Dex attenuated these
H/R-induced changes in TNF-α, IL-6, IL-1β, TLR4 and p-p65/p65
levels (Fig. 4E-G). miR-17-3p
inhibitor had no obvious effect on galectin-3 expression (Fig. 4F).
Effects of TAK-242 and Dex alone or in
combination on H9C2 cell viability and TLR4 expression
The Targetscan web site (TargetScan: http://www.targetscan.org/vert_72/) was used to
predict the binding site between miR-17-3p and TLR4 (Fig. 5A). miR-17-3p reduced luciferase
activity in double fluorescein carrier with TLR4 (Fig. 5B). TAK-242, a TLR4 inhibitor,
improved cell viability in H9C2 cells during H/R or in H9C2 cells
treated with miR-17-3p inhibitor during H/R (Fig. 5C-F). However, TAK-242 had no
significant effect on Dex efficacy in H9C2.
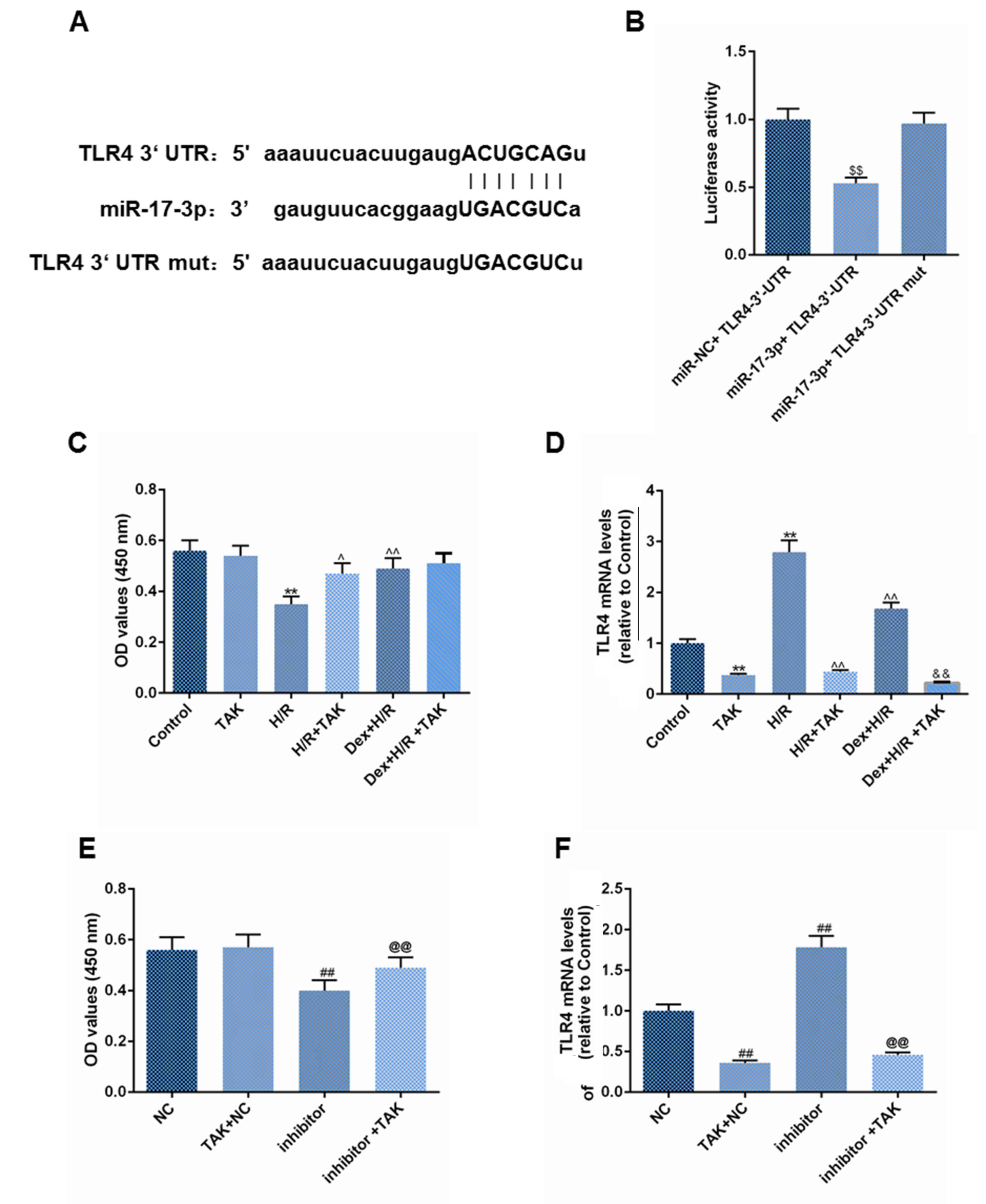 | Figure 5Effects of Dex and TAK-242 alone or
in combination on H9C2 cell viability during H/R. (A) The binding
site between miR-17-3p and TLR4 was predicted. (B) Cells were
transfected with miR-17-3p, miR-NC and TLR4 3'-UTR and cultured for
24 h. Dual luciferase assay was performed to analyze changes in
fluorescence. Cells were treated with 1 µmol/l TAK-242 cells for 2
h, and then with Dex for 24 h. Cells were placed in a hypoxic
chamber for 3 h and reoxygenated for 3 h. (C) Cell viability and
(D) TLR4 mRNA level was then assessed. Cells were treated with1
µmol/l TAK-242 for 2 h, and then transfected with miR-17-3p
inhibitor and cultured for 24 h. (E) Cell viability and (F) TLR4
mRNA level was assessed. Data are presented as the mean ± SD.
**P<0.01 vs. control group, ##P<0.01
vs. NC group, ^P<0.05 and ^^P<0.01 vs.
H/R group, &&P<0.01 vs. Dex + H/R group,
$$P<0.01 vs. miR-NC + TLR4 3'UTR group,
@@P<0.01 vs. inhibitor group. Dex, dexmedetomidine;
miR, microRNA; mut, mutant; NC, negative control; TLR4, toll-like
receptor 4; UTR, untranslated region. |
Discussion
The results of the present study indicated that Dex
(1-10 µmol/l) did not cause severe damage to H9C2 cells, but
reduced H/R-induced injury and increased miR-17-3p expression. In
order to investigate the relationship between miR-17-3p and Dex,
miR-17-3p was used in combination with Dex to prevent H/R-induced
H9C2 injury. The results suggested that the protective effect of
Dex was reduced when miR-17-3p expression was inhibited. The effect
of Dex and miR-17-3p on inflammatory cytokines, TLR4 and galectin-3
expression was also explored.
TLRs are a family of pattern recognition receptors
that play an important role in protection against infection
(22). Studies suggest that TLRs
have an important role in tissue homeostasis through regulation of
inflammation and tissue repair (22). TLR4 signaling has been suggested to
be important in regenerative biology, as it has been shown to have
a pronounced effect on healing in models of injury and inflammatory
disease (23,24). TLR4 mRNA levels were increased by
H/R. TAK-242, which is an exogenous synthetic antagonist for TLR4,
is a small molecule, also known as resatorvid, that binds to TLR4
and inhibits its transduction (25,26).
TAK-242 was used to study whether Dex prevents H/R injury through
action on TLR4. The results of the present study revealed that
inhibition of TLR4 could attenuate H/R injury, but that it had no
obvious effect on the protective effect of Dex. This was unexpected
as Chen et al (27) reported
that Dex relieved retinal I/R injury by acting on TLR4.
Galectin-3, a 32-35 kDa member of galectin family of
β-galactoside-binding lectins, plays multiple roles in cell growth,
differentiation and aggregation (28). Galectin-3 expression is low in normal
rat, murine and human heart; however, galectin-3 expression is
rapidly increased in heart failure and progression (29). H/R caused a high expression of
galectin-3. However, neither Dex nor miR-17-3p inhibitor caused a
marked change in galectin-3 expression after H/R. Hence, Dex might
alleviating I/R injury through other pathways.
The present study also evaluated the levels of
various inflammatory cytokines. TNF-α is an important inflammatory
cytokine, with pleiotropic functions including regulation of
apoptosis and survival (30,31). Human IL-6 is made up of 212 amino
acids including a 28-amino-acid signal peptide (32) and is rapidly produced in response to
infection and tissue injury. Dysregulated synthesis of IL-6 has a
pathological effect on inflammation (32). IL-1β, a member of the IL-1 family,
has pro-inflammatory activity and promotes tissue injury (33,34).
NF-κB was initially discovered in B cells, and is made up of a
family of subunits consisting of p65/RelA, p50, p52, RelB and c-Rel
(35). p65 activation promotes
cardiac fibrosis, inflammation, and apoptosis in a mouse model of
heart failure (36). The results of
the present study suggest that H/R and miR-17-3p inhibitor
increased the inflammatory response; however, Dex decreased this
H/R-induced inflammatory response. Inhibition of miR-17-3p in
Dex-treated H9C2 during H/R promoted inflammation, suggesting that
miR-17-3p plays an important role in the protection induced by
Dex.
In the present study the relationship between
miR-17-3p and TLR4 was explored, and the results suggested that
miR-17-3p binds with TLR4, and the inhibition of miR-17-3p
increased TLR4 expression. Whether TLR4 participated in the
regulation of inflammatory cytokines including IL-6, IL-1β, TNF-α
and p65, in combination with Dex in hypoxia, needs to be further
studied.
In conclusion, the findings of the present study
suggested that Dex reduced H/R-induced injury in H9C2 cells via
inhibition of inflammatory signaling, and that the inflammatory
factors were regulated by miR-17-3p.
Acknowledgements
Not applicable.
Funding
This study was supported by the Characteristic
Innovation Projects of Guangdong Provincial Department of Education
(Natural Science Category; grant no. 2017KTSCX040), the National
Natural Science Foundation (grant no. 81704036) and the Natural
Science Foundation of Guangdong Province (grant no.
2017A030310128).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TY and YuC made substantial contributions to
conception and design of the study. ZY, SX, YaC and WC were
responsible for data acquisition, data analysis and interpretation.
TY drafted the article and critically revised it for important
intellectual content. LW and WL performed the majority of the
experiments. All authors approved the final version of the
manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Writing Group Members. Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Heart disease and stroke
statistics-2016 update: A report from the American heart
association. Circulation. 133:e38–360. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Javadov S, Jang S, Parodi-Rullan R,
Khuchua Z and Kuznetsov AV: Mitochondrial permeability transition
in cardiac ischemia-reperfusion: Whether cyclophilin D is a viable
target for cardioprotection? Cell Mol Life Sci. 74:2795–2813.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jiang B, Liu Y, Liang P, Li Y, Liu Z, Tong
Z, Lv Q, Liu M and Xiao X: MicroRNA-126a-5p enhances myocardial
ischemia-reperfusion injury through suppressing Hspb8 expression.
Oncotarget. 8:94172–94187. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Portal L, Martin V, Assaly R, d'Anglemont
de Tassigny A, Michineau S, Berdeaux A, Ghaleh B and Pons S: A
model of hypoxia-reoxygenation on isolated adult mouse
cardiomyocytes: Characterization, comparison with
ischemia-reperfusion, and application to the cardioprotective
effect of regular treadmill exercise. J Cardiovasc Pharmacol Ther.
18:367–375. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhang X, Du Q, Yang Y, Wang J, Dou S, Liu
C and Duan J: The protective effect of Luteolin on myocardial
ischemia/reperfusion (I/R) injury through TLR4/NF-kappaB/NLRP3
inflammasome pathway. Biomed Pharmacother. 91:1042–1052.
2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Imarisio C, Alchera E, Bangalore Revanna
C, Valente G, Follenzi A, Trisolini E, Boldorini R and Carini R:
Oxidative and ER stress-dependent ASK1 activation in steatotic
hepatocytes and Kupffer cells sensitizes mice fatty liver to
ischemia/reperfusion injury. Free Radic Biol Med. 112:141–148.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Belleville JP, Ward DS, Bloor BC and Maze
M: Effects of intravenous dexmedetomidine in humans. I. Sedation,
ventilation, and metabolic rate. Anesthesiology. 77:1125–1133.
1992.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Weerink MAS, Struys MMRF, Hannivoort LN,
Barends CRM, Absalom AR and Colin P: Clinical Pharmacokinetics and
Pharmacodynamics of Dexmedetomidine. Clin Pharmacokinet.
56:893–913. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Cai Y, Xu H, Yan J, Zhang L and Lu Y:
Molecular targets and mechanism of action of dexmedetomidine in
treatment of ischemia/reperfusion injury. Mol Med Rep. 9:1542–1550.
2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chen Z, Ding T and Ma CG: Dexmedetomidine
(DEX) protects against hepatic ischemia/reperfusion (I/R) injury by
suppressing inflammation and oxidative stress in NLRC5 deficient
mice. Biochem Biophys Res Commun. 493:1143–1150. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang L, Liu H, Zhang L, Wang G, Zhang M
and Yu Y: Neuroprotection of dexmedetomidine against cerebral
ischemia-reperfusion injury in rats: Involved in inhibition of
NF-κB and inflammation response. Biomol Ther (Seoul). 25:383–389.
2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chen J, Huang ZP, Seok HY, Ding J, Kataoka
M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, et al: mir-17-92 cluster
is required for and sufficient to induce cardiomyocyte
proliferation in postnatal and adult hearts. Circ Res.
112:1557–1566. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. MicroRNAs en route to the clinic: Progress in validating
and targeting microRNAs for cancer therapy. Nat Rev Cancer.
11:849–864. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
de Pontual L, Yao E, Callier P, Faivre L,
Drouin V, Cariou S, Van Haeringen A, Geneviève D, Goldenberg A,
Oufadem M, et al: Germline deletion of the miR-17~92 cluster causes
skeletal and growth defects in humans. Nat Genet. 43:1026–1030.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Mendell JT: miRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–222.
2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261.
2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yan H, Song K and Zhang G: MicroRNA-17-3p
promotes keratinocyte cells growth and metastasis via targeting
MYOT and regulating Notch1/NF-κB pathways. Die Pharmazie.
72:543–549. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li C, Lu J and Zhang B: Development of a
novel chronic intermittent hypoxia chamber. Sleep Breath.
16:177–179. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rakoff-Nahoum S and Medzhitov R: Toll-like
receptors and cancer. Nat Rev Cancer. 9:57–63. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Huebener P and Schwabe RF: Regulation of
wound healing and organ fibrosis by toll-like receptors. Biochim
Biophys Acta. 1832:1005–1017. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lin Q, Li M, Fang D, Fang J and Su SB: The
essential roles of Toll-like receptor signaling pathways in sterile
inflammatory diseases. Int Immunopharmacol. 11:1422–1432.
2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kuno M, Nemoto K, Ninomiya N, Inagaki E,
Kubota M, Matsumoto T and Yokota H: The novel selective toll-like
receptor 4 signal transduction inhibitor tak-242 prevents
endotoxaemia in conscious Guinea-pigs. Clin Exp Pharmacol Physiol.
36:589–593. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sha T, Sunamoto M, Kitazaki T, Sato J, Ii
M and Iizawa Y: Therapeutic effects of TAK-242, a novel selective
Toll-like receptor 4 signal transduction inhibitor, in mouse
endotoxin shock model. Eur J Pharmacol. 571:231–239.
2007.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chen Z, Qiu PY and Ma CG: Dexmedetomidine
preconditioning protects against retinal ischemia/reperfusion
injury and inhibits inflammation response via toll-like receptor 4
(TLR4) pathway. Biomed Pharmacother. 93:1018–1024. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Simon D, Derer A, Andes FT, Lezuo P, Bozec
A, Schett G, Herrmann M and Harre U: Galectin-3 as a novel
regulator of osteoblast-osteoclast interaction and bone
homeostasis. Bone. 105:35–41. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
de Boer RA, Voors AA, Muntendam P, van
Gilst WH and van Veldhuisen DJ: Galectin-3: A novel mediator of
heart failure development and progression. Eur J Heart Fail.
11:811–817. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Marques-Fernandez F, Planells-Ferrer L,
Gozzelino R, Galenkamp KM, Reix S, Llecha-Cano N, Lopez-Soriano J,
Yuste VJ, Moubarak RS and Comella JX: TNFα induces survival through
the FLIP-L-dependent activation of the MAPK/ERK pathway. Cell Death
Dis. 4(e493)2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Crusz SM and Balkwill FR: Inflammation and
cancer: Advances and new agents. Nat Rev Clin Oncol. 12:584–596.
2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Tanaka T, Narazaki M and Kishimoto T: IL-6
in inflammation, immunity, and disease. Cold Spring Harb Perspect
Biol. 6(a016295)2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Striz I: Cytokines of the IL-1 family:
Recognized targets in chronic inflammation underrated in organ
transplantations. Clin Sci (Lond). 131:2241–2256. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wang YX, Ji ML, Jiang CY and Qian ZB:
Upregulation of ICAM-1 and IL-1beta protein expression promotes
lung injury in chronic obstructive pulmonary disease. Genet Mol
Res. 15:2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang YC, Ye H, Zeng Z, Chin YE, Huang YN
and Fu GH: The NF-kappaB p65/miR-23a-27a-24 cluster is a target for
leukemia treatment. Oncotarget. 6:33554–33567. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn
B and Prabhu SD: Cardiomyocyte NF-κB p65 promotes adverse
remodelling, apoptosis, and endoplasmic reticulum stress in heart
failure. Cardiovasc Res. 89:129–138. 2011.PubMed/NCBI View Article : Google Scholar
|