Introduction
p53 was first discovered in 1979, in a study of
Simian Virus 40 (SV40), a virus oncoprotein (1,2). And in
the same year, another research team reported the identification of
its coding genes (3). Until 1989,
p53 had been considered an oncogene; however, in later years, the
wild-type p53 gene was identified as a tumor suppressor gene that
inhibits tumor formation (4), and
the activation of p53 was shown to be able to lead to cell cycle
arrest and apoptosis (5).
p53 is one of the most important tumor suppressor
genes, and its encoded protein is vital for maintaining the
stability of numerous genes, and regulating cell growth,
differentiation and aging, as well as preventing diseases (6). Therefore, it is also called the ‘Police
Gene’ (7,8). Deletion or mutation of the tumor
protein p53 gene has a direct impact on the functions and
activities of p53 protein, leading to cancer occurrence. More than
50% of cancers, including hematological malignancies and solid
tumors, are caused by mutations (4).
Platelets (PLTs) are components of mammalian blood
derived from the cytoplasmic cleavage of megakaryocytes in the bone
marrow, and possess a biologically active, rather than strict small
cytoplasm (9). Megakaryocytes
originally come from directional differentiation of multifunctional
hematopoietic stem cells in the bone marrow, and further become
mature megakaryocytes (10-12).
The major functions of PLTs include coagulation, hemostasis and
repairing damaged blood vessels; they prevent atherosclerosis by
protecting and repairing vascular endothelium (13).
The knowledge of the underlying mechanisms of p53 in
the development of hematological disease and cancer could have
considerable prognostic and therapeutic values in clinical
applications (14). The p53 gene has
been used as a strong indicator in determining the prognosis of
tumors, since its mutation is associated with cancer development
(15). Previous studies have
reported that p53 plays an important role in megakaryocyte
differentiation and apoptosis (16,17).
Knockdown of p53 increases ploidy of megakaryocytes, as indicated
from in vivo and ex vivo studies in 2012(18). p53 deficiency promotes
polyploidization during megakaryopoiesis, suggesting a direct
association between p53 loss and the development of fully
functional megakaryocytes. However, the signaling pathways involved
in p53 regulation of megakaryopoiesis and blood cell
differentiation still remain poorly understood.
In the present study, the changes of PLT parameters
in p53-/- mice were analyzed to investigate the manner
in which p53 regulates PLT differentiation. The results revealed
that the number of PLTs in p53-/- mice is significantly
lower compared with that in wild-type mice, and that p53 regulates
PLT formation via the PI3K signaling pathway in vivo,
providing useful insight for the investigation of the molecular
mechanisms underlying the differentiation of hematopoietic stem
cells into megakaryocytes.
Materials and methods
Animals
p53 knockout mice(total number, 60; age, 18-20
weeks; weight, 20-25 g; 32 males and 28 females), referred to as
p53-/- mice, and wild-type mice with a BL/6J background,
were donated as gifts by Professor Tiebang Kang from Sun Yat-Sen
University Cancer Center (Guangzhou, China). Littermates identified
as the p53+/+ genotype were used as the wild-type
control. Mice were bred and kept under SPF conditions at the Animal
Center of Guangdong Pharmaceutical University (Guangzhou, China).
Feed treated with 60Co irradiation for sterilization was
purchased from the Guangdong Medical Animals Center. Drinking water
was autoclaved and mice received free access to food and water. The
room temperature was kept at 24±2˚C and humidity was maintained at
between 40 and 60%. Noise level was <60 dB. All 18 to 20
week-old mice were sacrificed via cervical dislocation under ether
anesthesia. The experiments on mice were approved by the Animal
Ethics Committee of the Guangdong Pharmaceutical University.
Genotype identification
The genotypes of p53 mice were identified by PCR.
DNA was extracted from mice via the tail. DreamTaq Green PCR Master
Mix for PCR was purchased from Thermo Fisher Scientific, Inc. (cat.
no. K1081). The wild-type primers used were as follows: Wild-type
primer 1, 5'-CAGCGTGGTGGTACCTTAT-3'; and wild-type primer 2,
5'-CTATCAGGACATAGCGTTGG-3', with a PCR product size of 450 bp. The
mutant primers used were as follows: Mutation primer 3,
5'-TATACTCAGAGCCGGCCT-3'; and mutation primer 4,
5'-CTATCAGGACATAGCGTTGG-3', with a PCR product size of 615 bp. PCR
conditions were as follows: Denaturation at 94˚C for 3 min, 94˚C
for 1 min, 60˚C for 2 min and 72˚C for 2 min, for 30 cycles,
followed by extension at 72˚C for 5 min. Gel electrophoresis was
performed using 1.2% agarose gel. Images were captured with an
automatic gel imaging system (Syngene).
Blood collection and routine blood
tests
Blood was collected from the mice (18-20 weeks old).
Blood cell subtypes were detected at the Guangdong Laboratory
Animal Monitoring Institute using the blood cell counter XT-2000i
(Sysmex Corp). Ether was used for anesthesia.
PLT preparation
PLTs were isolated from the whole blood of the mice
(19). Briefly, 6:1 of
citrate-dextrose solution (Sigma-Aldrich; Merck KGaA) and whole
blood were gently mixed and centrifuged at 280 x g for 15 min at
room temperature. The PLT-rich plasma (PRP) was obtained by another
centrifugation at 400 x g for 5 min at room temperature. To obtain
PLT pellets, PRP was further centrifuged at 1,100 x g for 10 min at
room temperature and the PLT pellets were suspended in PBS for
immediate use. In the indicated experiments, PLTs were stained with
CD41 antibody and visualized under a fluorescence microscope or
analyzed by flow cytometry.
Flow cytometry
Blood samples of 18-20 weeks old p53-/-
and wild-type mice were subjected to flow cytometry analysis. Fresh
blood was taken from ether anesthesia, and then added into
anticoagulant tubes. A total of 10 µl blood was added into labeled
flow tubes and mixed with 90 µl PBS. The mixture was incubated with
antibody CD41-PE [BG-03512-60-100; mouse IgG1, κ-isotype; BioGems,
Ltd. 1:100 with 1% BSA (Sangon Biotech Co, Ltd. cat. no. A600332)]
at room temperature in the dark for 30 min. Finally, 1 ml 1%
pre-cooled paraformaldehyde was added and the fixed mixture was
preserved at 2-8˚C in the dark (20). The samples were gated to exclude
irrelevant constituents, such as lymphocytes and cell debris before
analysis. The positive rate of the specimens and the intensity of
fluorescence were measured. BD FACSCanto™ II system (BD
Biosciences) was used for analysis. EDTA-K2 anticoagulant was used
in blood collection. After being treated with ACD anticoagulant
(C3821; Sigma Aldrich; Merck KGaA), the blood samples were used to
analyze the PLTs by flowJo 7.6.1 (FlowJo LLC).
Immunofluorescence
For immunofluorescence staining, bone marrow and
blood smearing sections were fixed with 1% paraformaldehyde at 4˚C
for 25 min and stained with the PLT marker CD41-PE for 30 min
(BG-03512-60-100, mouse IgG1, κ-isotype; BioGems, Ltd.
dilution:1:100 with 1% BSA) at room temperature, and then the
sections were counterstained with DAPI (100 ng/ml) for 10 min at
room temperature in the dark and all sections were visualized under
a fluorescent microscope (BX53 Olympus microscope; Olympus,
Corp).
Bleeding time test
According to the method utilized in previous studies
(21,22), a 50 ml glass centrifuge tube filled
with sterile isotonic saline was pre-warmed in water bath at 37˚C
for 30 min. Next, 3 mm of the tail of each anesthetized mouse was
cut with a scalpel and immediately placed in physiological saline.
Bleeding time was recorded as the time from the moment bleeding
started until bleeding had stopped, for a minimum of 10 sec. The
assay was automatically stopped if the total bleeding time exceeded
20 min.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from PLTs in bone marrow
using TRIzol®, and was quantified by the NanoDrop
nucleic acid protein analyzer (NanoDrop Technologies; Thermo Fisher
Scientific, Inc.). Reverse transcription was carried out according
to the following protocol: 42˚C for 2 min, stored at 4˚C; 37˚C for
15 min and 84˚C for 5 sec, storage at 4˚C with the Reverse
PrimeScript RT reagent kit (RR047A, Takara Bio Inc.). All gene
primers purchased from Sangon Biotech Co. Ltd, which are listed in
Table SI. The PCR kit was purchased
from Thermo Fisher Scientific, Inc. The PCR and specificity of the
primers were evaluated based on the standard curve of
amplification. PCR volume was comprised of 10 µl SYBR Green Master
Mix, 1 µl upstream primer and 1 µl downstream primer, 1 µl target
cDNA and 7 µl ddH2O. Reaction conditions were as
follows: Pre-denaturation at 94˚C for 5 min, denaturation at 94˚C
for 30 sec, annealing at 60˚C for 30 sec and extension at 72˚C for
30 sec, for 40 cycles, followed by extension at 72˚C for 10 min.
The results were analyzed based on the dissolution curve. The Cq
value of the threshold cycle was measured to calculate
2-ΔΔCq and determine the expression levels of the target
genes (23).
Administration of PI3K inhibitor
The 18-20 week mice were administered with a PI3 K
inhibitor (LY294002, cat. no S1105; Selleck Chemicals). Starting
from 20 weeks, a short-term assay was used with intraperitoneal
administration of ≥200 µl dose of PI3 K inhibitor (15 µg/g body
weight; stock concentration: 50 mg/ml; needle gauge, 25G) each per
day, for 7 consecutive days.
Statistical analysis
All data were analyzed using GraphPad Prism 5.0
software (GraphPad Software, Inc.) and presented as the mean ±
standard deviation. A two-tailed t-test or one-way analysis of
variance (ANOVA), followed by Dunnett's post-hoc test, was used for
the comparison of the results and the evaluation of the statistical
differences between the groups. *P<0.05 was
considered as statistically significant; **P<0.01 and
***P<0.001.
Results
Identification of genotype and routine
blood tests
p53+/+, p53+/- and
p53-/- mice were obtained by crossbreeding
p53+/- male mice with p53+/- female mice. The
size of the PCR product in the p53-/- mice was 615 bp,
whereas that in the wild-type (p53+/+) mice was 450 bp.
The presence of both bands indicated heterozygous p53+/-
mice. The mouse designated as 1# was a homozygote p53-/-
mouse (Fig. 1A), the mice designated
as 2#, 3# and 6# were heterozygous p53+/- mice, and the
mice designated as 4# and 5# were homozygote p53+/+
mice. These results were representative.
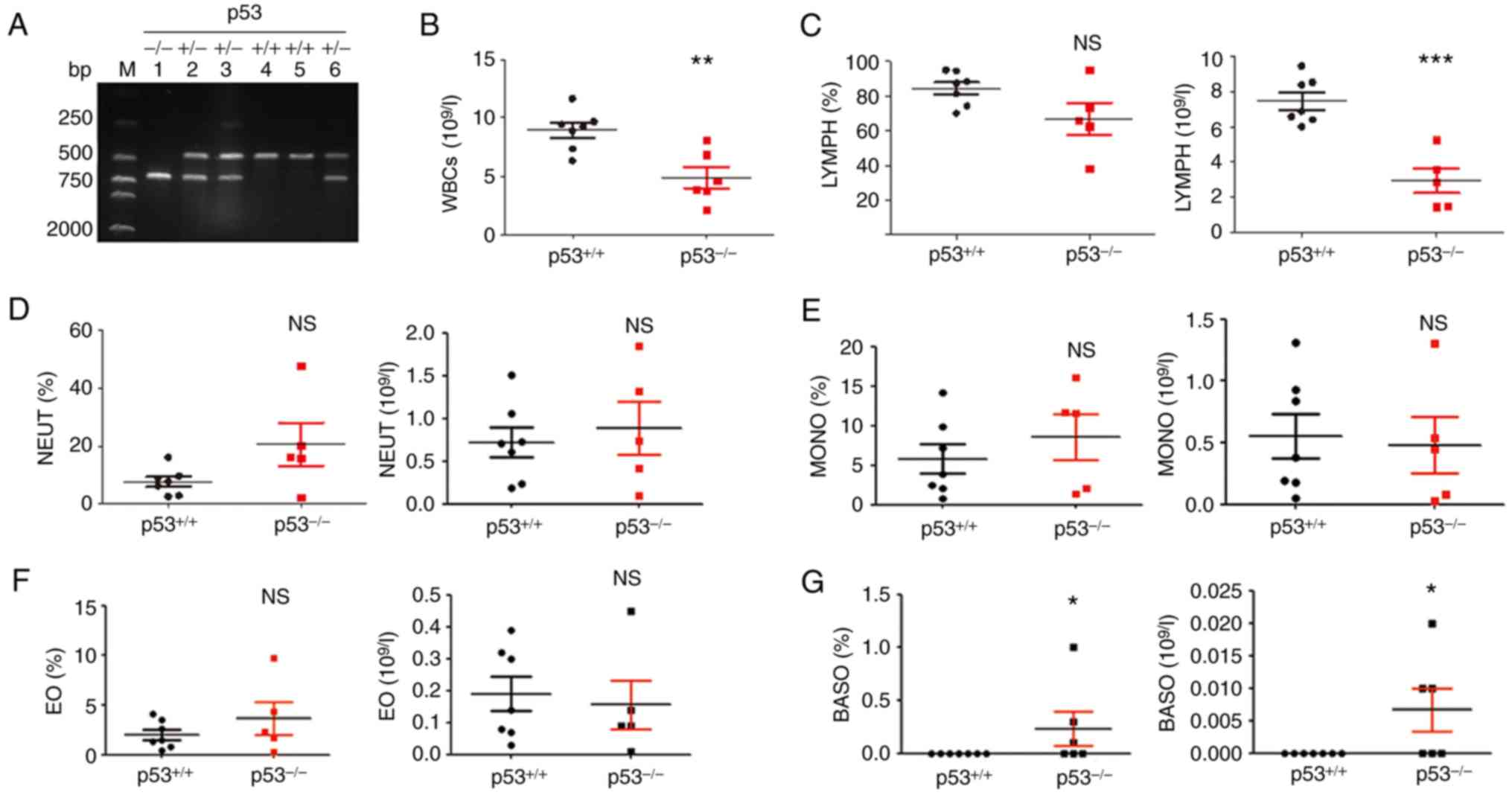 | Figure 1Genotype identification and routine
blood test results. (A) p53 knockout mice was identified by PCR.
Genotypes of p53-/-, p53+/- and
p53+/+ mice are shown. The PCR product size of the
wild-type mice was 450 bp, whereas that of the p53-/-
mice was 615 bp. (B) Absolute counts of white blood cells. (C)
Percentage of lymphocytes (left) and number of lymphocytes (right).
(D) Percentage of neutrophils (left) and number of neutrophils
(right). (E) Percentage of monocytes (left) and number of monocytes
(right). (F) Percentage of eosinophils (left) and number of
eosinophils (right). (G) Percentage of basophils (left) and number
of basophils (right). p53+/+ mice, n=6;
p53-/-mice, n=6. *P<0.05,
**P<0.01 and ***P<0.001. NS, no
significance; EO, eosinophils; MONO, monocytes; BASO, basophils;
NEUT, neutrophils; WBC, white blood cell; LYMPH, lymphocyte. |
To explore whether blood cell subsets
were altered in p53-/- mice, routine blood tests were
performed
Results showed that the absolute count of white
blood cells was decreased in p53-/- mice compared with
that in p53+/+ mice (**P<0.01; Fig. 1B). Lymphocytes percentage showed no
significant difference; however, lymphocyte absolute number was
decreased compared with that in p53+/+ mice
(***P<0.001; Fig. 1C).
In addition, there were no significant differences in the
percentage and absolute number of neutrophils, monocytes and
eosinophils (Fig. 1D-F). The
percentage and absolute number of basophils were significantly
higher in p53-/- mice than those in the control
(*P<0.05; Fig. 1G).
There was no difference in the number of red blood cells,
hemoglobin concentration, hematocrit, mean corpuscular hemoglobin
concentration and other red blood cell parameters between the two
groups (data not shown).
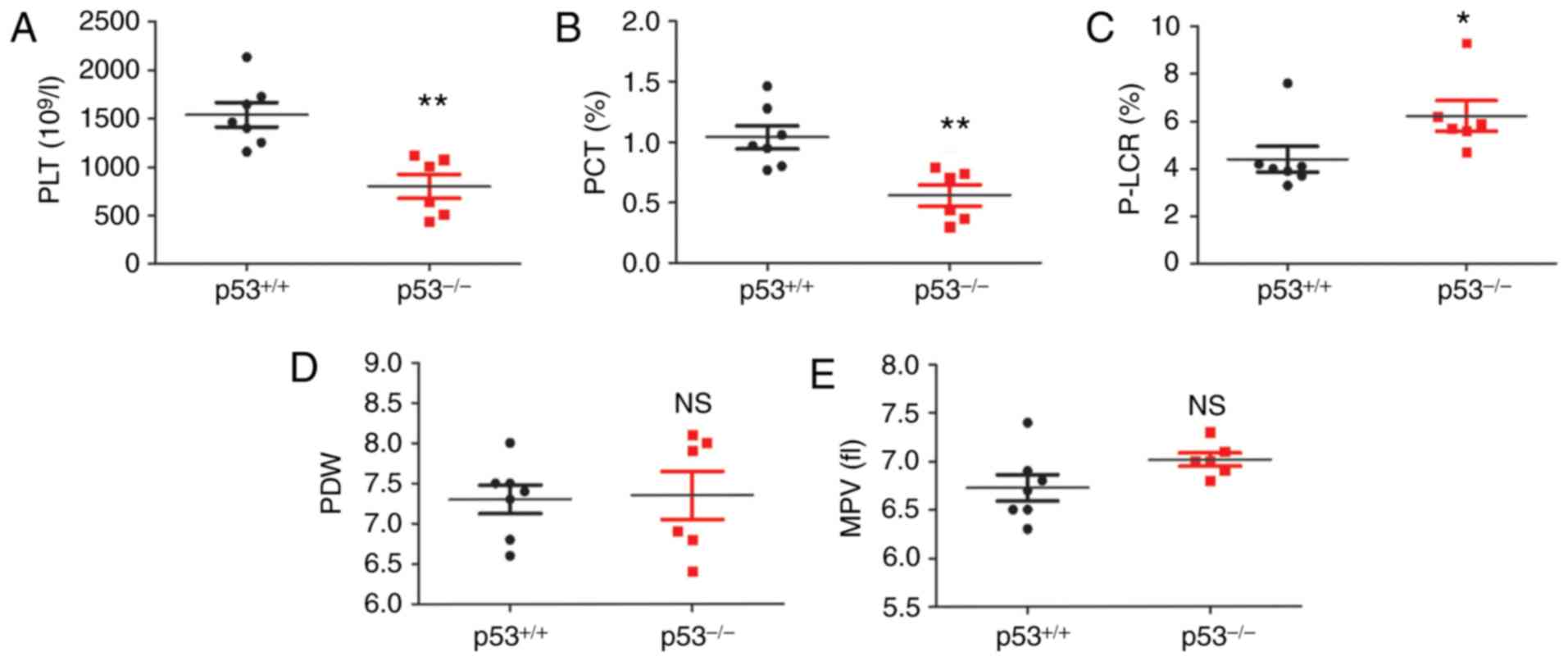
PLT number and PLT parameters are
altered in p53-/- mice
PLT counts an important diagnostic index for the
clinical diagnosis and treatment of thrombocytopenia of various
causes (24). The analysis of the
blood cell test results by a blood cell counter demonstrated a
significant alteration in the number of PLTs in the peripheral
blood of p53-/- mice, as shown in Fig. 2. The PLT count and plateletcrit (PCT)
of p53-/- mice were significantly lower than those of
wild-type mice (**P<0.01; Fig. 2A and B). The PLT-large cell ratio (P-LCR) was
significantly increased (Fig. 2C;
*P<0.05). Nevertheless, there was no significant
change in PLT distribution width (Fig.
2D). Mean PLT volume showed an increased tendency in
p53-/- mice; however, there was no significant
difference compared with the control group (wild-type mice, n=6;
p53-/- mice, n=6; Fig.
2E).
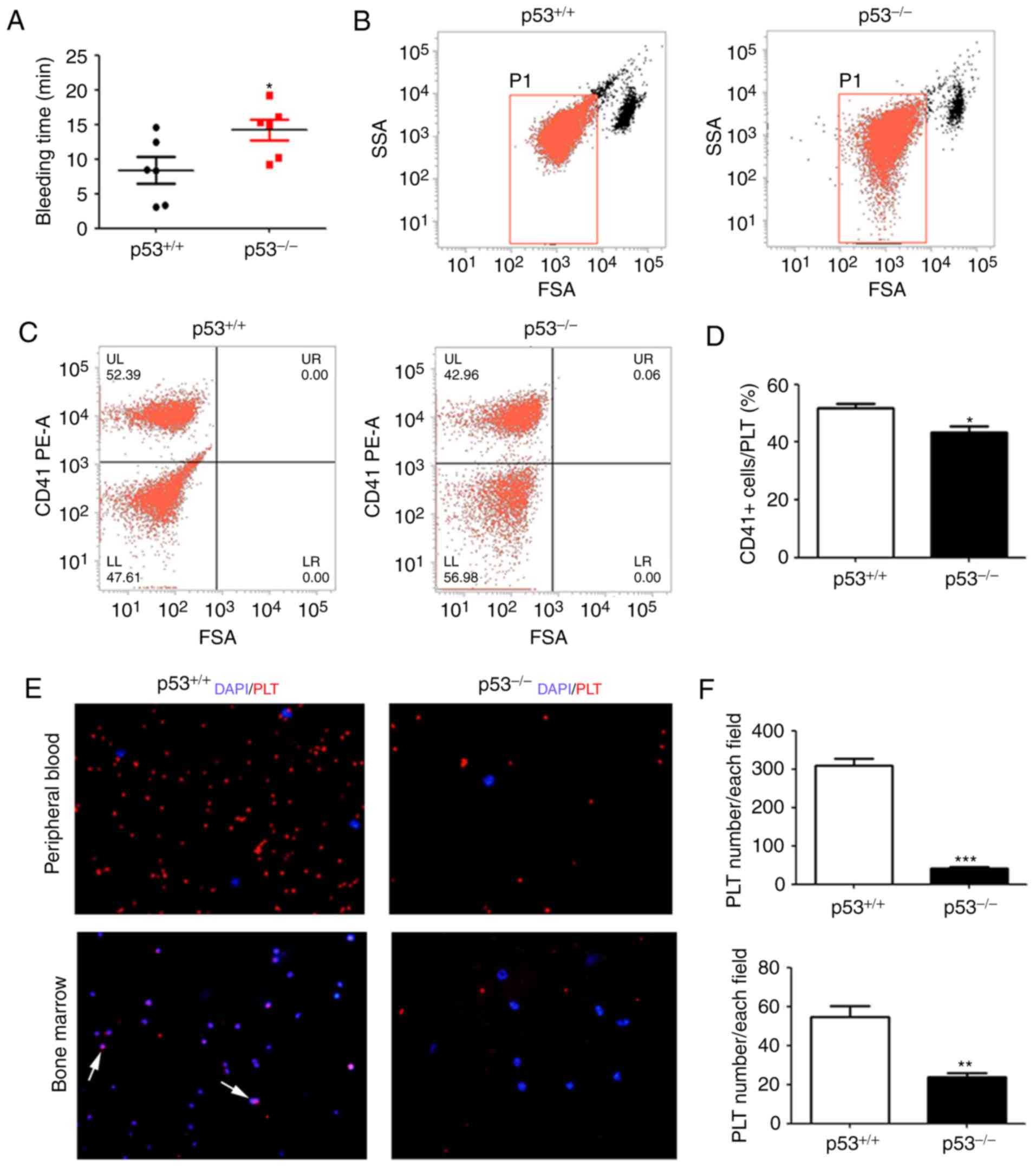
CD41-positive PLTs are decreased in
the peripheral blood of p53-/- mice
As aforementioned, the number of PLTs in
p53-/- mice was significantly lower compared with that
in wild-type mice. To investigate the function of PLTs, the
bleeding time test was performed. The results indicated that the
bleeding time of p53-/-mice was prolonged compared with
that of the control (*P<0.05; Fig. 3A). To further validate the alteration
in PLTs, flow cytometry was conducted for the detection of PLTs,
using the CD41 antibody (as a PLT-specific marker) which is used to
specifically identify PLTs and detect changes in their quantity
(25). The peripheral blood of
p53-/- and wild-type mice were labeled and analyzed by
flow cytometry. Next, a group of cells was gated between
lymphocytes and cell debris (26),
as shown in Fig. 3B. The results
showed that the percentage of CD41-positive cells in the
p53-/- group was significantly lower than that in the
wild-type mice (*P<0.05; Fig. 3C and D). The results of flow cytometry were
consistent with the routine blood test results obtained by an
automatic blood cell counter. The results demonstrated that the
number of PLTs in p53-/- mice was significantly lower
than that in the control group. Furthermore, CD41 was detected in
the blood and bone marrow by immunofluorescence, and the results
also showed that the PLT number in p53-/- mice was
significantly decreased in the bone marrow and blood
(***P<0.001 and **P<0.01; Fig. 3E and F).
PI3K signaling pathway is changed in
bone marrow of p53-/- mice
p53 knockout in mice with changes in PLTs indicated
that certain proteins involved in PLT production were altered.
Therefore, ‘p53’ and ‘platelet’ were chosen as keywords for the
protein and protein action analysis via the biology information
website (http://version10.string-db.org/). The association of
p53 and molecules related to PLTs, including integrin subunit alpha
2b, von willebrand factor, platelet derived growth factor receptor
alpha and platelet derived growth factor receptor beta, were
analyzed and confirmed (Fig. 4A).
Next, the key genes involved in PLT production and p53-related
signaling pathways were further analyzed. The results suggested
that the PI3K signaling pathway may be a bridge linking p53 and
PLT-related factors (***P<0.001; Fig. 4B). This conclusion is consistent with
findings from a previous study (18). To further confirm whether p53
regulates PLT production through the PI3K signaling pathway, total
RNA was extracted from the bone marrow of p53-/- and
wild-type mice. In this pathway, Akt1 (AKT Serine/Threonine Kinase
1), Pdk2 (pyruvate dehydrogenase kinase 2) and Pdpk1
(3-phosphoinositide dependent protein kinase 1) mRNA levels were
increased in the bone marrow of p53-/- mice, where Prkca
(protein kinase C-α) mRNA expression level was decreased,
suggesting that the PI3K signaling pathway is involved in p53
knockout (*P<0.05, **P<0.01 and
***P<0.001; Fig. 4C).
In conclusion, the expression levels of the PI3K pathway-related
genes and the heat map of these genes suggest that the PI3K pathway
may change in the bone marrow of p53-/- mice (Fig. 4D).
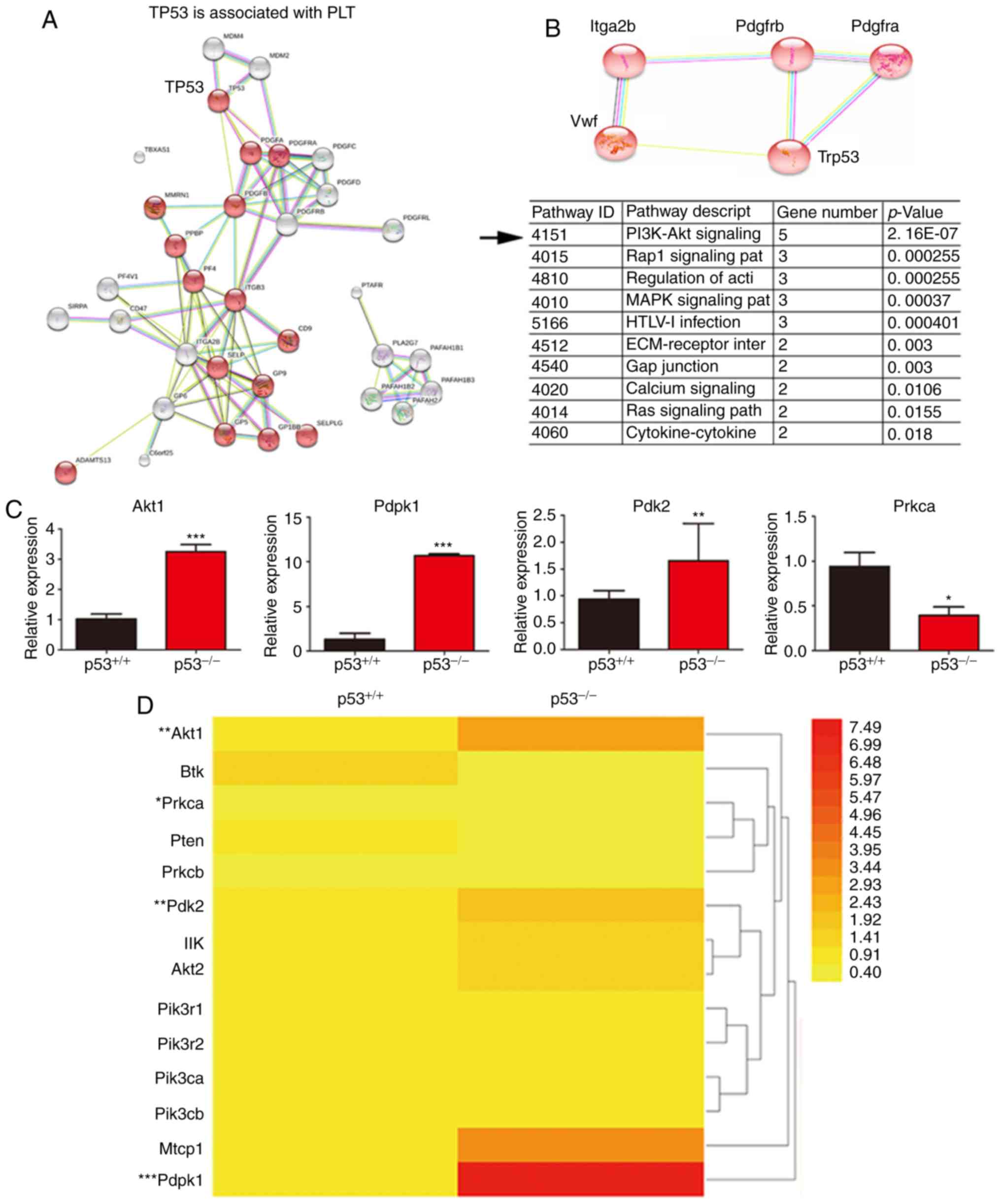 | Figure 4Bioinformatics analysis and detection
of PI3K pathway. (A) Detection of p53 and other genes related to
PLTs through online bioinformatics analysis (https://string-db.org/cgi/input.pl). The p53 gene is
shown to be closely related to PLT associated factors. (B) Data
analysis showed that the PI3K signaling pathways involved in the
regulation of p53 and PLTs. (C) Expression of genes related to the
PI3K signaling pathwayin the bone marrow of p53-/- and
p53+/+ mice as assessed by RT-qPCR. The PI3K pathway is
associated with p53 deficiency, which is consistent with the
results of the bioinformatics analysis. (D) Heat map showing that
the expression levels of few genes were altered in the PI3K
signaling pathway in p53-deficient mice. Including Akt1, Prkca,
Pdk2, and Pdpk1. *P<0.05, **P<0.01 and
***P<0.001. RT-qPCR, reverse
transcription-quantitative PCR; PLT, platelet; Akt1, AKT
serine/threonine kinase 1; Prkca, protein kinase C alpha; Pdk2,
pyruvate dehydrogenase kinase 2; Pdpk1, 3-phosphoinositide
dependent protein kinase 1. |
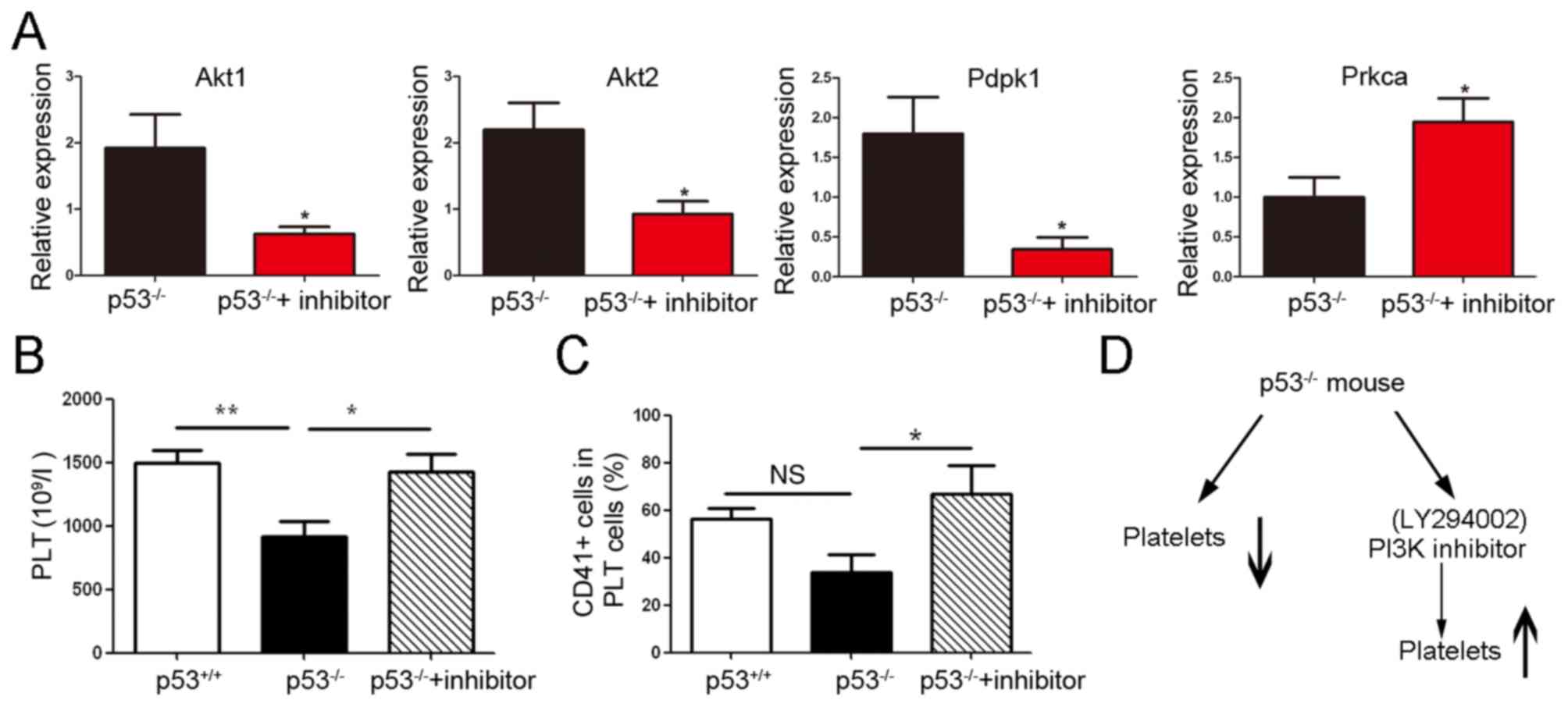
PI3K inhibitor reverts the PLT
downregulation in p53-/- mice
To investigate whether the inhibitor of PI3K
(LY294002, cat. no. S1105; Selleck Chemicals) could revert the
changes in PLTs in p53 knockout mice, a short-term assay was
conducted with intraperitoneal administration of a dose of PI3K
inhibitor once per day for 7 consecutive days. Akt1, Akt2 (AKT
Serine/Threonine Kinase 2), Pdpk1 and Prkca mRNA expression levels
were determined by RT-qPCR, and the results revealed that the Akt1,
Akt2 and Pdpk1 in PI3K signaling pathway were suppressed; however,
the Prkca mRNA level was increased (*P<0.05; Fig. 5A).
Next, the changes in PLT numbers were assessed using
the blood cell counter XT-2000i, and the CD41-positive PLTs were
further detected by flow cytometry. The results revealed that PLT
count was increased in p53-/- mice treated with PI3K
inhibitor (ANOVA followed by Dunnett's test, *P<0.05
and**P<0.01; Fig. 5B
and C), suggesting that the
reduction in PLTs was reverted by the PI3K inhibitor (Fig. 5D).
Discussion
p53 was originally regarded as an oncogene; however,
later on, it was designated as a tumor suppressor gene maintaining
genome stability. A previous study has shown that p53 expression is
high in megakaryocytes (27).
Further attention has been paid to the role of p53 in hematopoietic
stem cell differentiation (28). p53
has been found to regulate the differentiation of megakaryocytes
and macrophages (29-31),
specifically mediated megakaryocytic polyploidization and apoptosis
(16). A study published in 2012 has
shown that PLT count in p53-/- mice is slightly lower
than that in wild-type mice, indicating that p53 could alter PLT
function in p53-deficient mice (18). In the present study, a significant
reduction in PLTs and PCT was observed in the blood of
p53-/- mice, whereas an increase in P-LCR was presented.
Blood cell counter XT-2000i was used for routine blood tests. The
current veterinary software consists of settings and algorithms for
the analysis of specimens from rats, mice and rabbits, which may
explain the fact that PLT was slightly lower in p53-/-
mice (18), whereas in our study it
was observed to be significantly decreased. The results of the
bleeding time test also supported the alteration of the PLT
functions, in accordance with what has been previously reported
(18).
The mRNA expression of genes is involved in the PI3K
pathway in the bone marrow of p53-/- mice, as shown by
bioinformatics analysis, and further confirmed with RT-qPCR. Of
course, the protein of p-Akt should be detected by western blotting
analysis; however, western blotting analysis was not performed in
the present study, as there are only few megakaryocyte in bone
marrow, and this experiment would require the use of additional
mice. The expression of the related genes PI3K, Akt1, Pdpk1 and
Pdk2 was increased in the bone marrow of the p53-/-
mice. The aim of the present study was to identify the association
between these factors and PLT formation, and overexpression or
silencing experiments were required for this purpose. However, the
generation of PLTs is very complex in the bone marrow of mice,
therefore, these experiments were not performed in the present
study.
PI3K inhibitor was administered to p53-/-
mice. Akt1, Akt2 and Pdpk1 mRNA expression levels were decreased,
whereas the number of PLTs was shown to be increased. PLT-derived
growth factor receptor and signaling pathways of PLT have been
shown to be regulated by the PI3K pathway (32,33),
which supports the present findings that p53 deficiency may
down-regulate PLTs via the PI3K signaling pathway. In the present
study, it was confirmed that these genes down-regulate PLTs and
that PLT count could be reverted after the administration of PI3K
inhibitor in p53 knockout mice. There are probably two reasons for
the increase in PLTs by PI3K inhibitor; one depends on p53, and the
other is that PI3K directly controls plateletgenesis. A previous
study has shown that the PI3K pathway is associated with PLTs
(34).
In general, PLTs are derived from megakaryocytes in
the bone marrow. However, a recent study has demonstrated that
megakaryocytes in lung tissue could also produce PLTs, arousing
more research interest in PLTs from the lung (35). It would be interest to compare the
changes in PLTs from the lung between p53 knockout mice and
wildtype mice.
p53 has been shown to regulate macrophage
differentiation (30), indicating
that p53 could affect myeloid cell differentiation. Herein, we
speculated that p53 deficiency could induce megakaryocyte
abnormalities, leading to changes in PLT count and other PLT
parameters. A significant decrease in PLT count and abnormalities
in other PLT indices were observed as a result of p53 mutation.
Although activation and aggregation of PLTs were not detected, the
change of PLT parameters and bleeding time was useful to gain
insight into p53 function in PLTs. The molecular mechanisms by
which p53 induces alterations in PLTs require further
investigation. It would be great significance to investigate the
mechanism by which p53 regulates PLTs via the PI3K pathway in order
to prevent PLTs from promoting tumor metastasis (36).
In conclusion, the present study results suggest
that p53 could alter PLT number, bleeding time and that the PI3K
signaling pathway is involved in this process, thus providing
useful insights into the study of thrombocytopoiesis and
controlling the amount of PLTs in the development of cancer. p53 is
also useful for investigating the manner in which PLTs affect tumor
metastasis or PLT diseases.
Supplementary Material
List of quantitative PCR primers.
Acknowledgements
The authors would like to thank Professor Tiebang
Kang (SunYat-Sen University Cancer Center, Guangzhou, China) for
providing the p53 knockout mice as a gift. Flow cytometry was
partly performed at Guangdong Provincial Key Laboratory of
Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen
Memorial Hospital, Sun Yat-Sen University. The authors would also
like to thank Mr. Ting Luo (Guangdong Laboratory Animals Monitoring
Institute) for assisting with the routine blood tests.
Funding
The current study was supported by grants from the
National Natural Science Foundation of China (grant nos 81773118
and 31471290), the Pearl River S&T Nova Program of Guangzhou
(grant no. 201610010045) and the Guangdong Pharmaceutical
University Innovative Foundation (grant no. 2016KCXTD019).
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL and LW conceived and designed the study. MY, QL
and TN acquired, analyzed and interpreted the data. JK, XZ, SL and
XH were responsible for the sample collection and treatment. MY and
LJ performed the statistical analysis. MY wrote the manuscript and
revised it critically. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experiments on mice were approved by the Animal
Ethics Committee of the Guangdong Pharmaceutical University
(Guangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lane DP and Crawford LV: T antigen is
bound to a host protein in SV40-transformed cells. Nature.
278:261–263. 1979.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Linzer DI and Levine AJ: Characterization
of a 54K dalton cellular SV40 tumor antigen present in
SV40-transformed cells and uninfected embryonal carcinoma cells.
Cell. 17:43–52. 1979.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kress M, May E, Cassingena R and May P:
Simian virus 40-transformed cells express new species of proteins
precipitable by anti-simian virus 40 tumor serum. J Virol.
31:472–483. 1979.PubMed/NCBI
|
|
4
|
Greenblatt MS, Bennett WP, Hollstein M and
Harris CC: Mutations in the p53 tumor suppressor gene: Clues to
cancer etiology and molecular pathogenesis. Cancer Res.
54:4855–4878. 1994.PubMed/NCBI
|
|
5
|
Vousden KH and Lane DP: P53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Puisieux A, Lim S, Groopman J and Ozturk
M: Selective targeting of p53 gene mutational hotspots in human
cancers by etiologically defined carcinogens. Cancer Res.
51:6185–6189. 1991.PubMed/NCBI
|
|
7
|
Belyi VA, Ak P, Markert E, Wang H, Hu W,
Puzio-Kuter A and Levine AJ: The origins and evolution of the p53
family of genes. Cold Spring Harb Perspect Biol.
2(a001198)2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Brown CJ, Lain S, Verma CS, Fersht AR and
Lane DP: Awakening guardian angels: Drugging the p53 pathway. Nat
Rev Cancer. 9:862–873. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Wandall HH, Rumjantseva V, Sørensen AL,
Patel-Hett S, Josefsson EC, Bennett EP, Italiano JE Jr, Clausen H,
Hartwig JH and Hoffmeister KM: The origin and function of platelet
glycosyltransferases. Blood. 120:626–635. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sacchetti C and Bianchini E: Biological
activity of human bone marrow megakaryocytes; genesis, in vivo and
in vitro maturation, platelet formation, phagocytosis and
pathological character. Arch Maragliano Patol Clin. 10:431–454.
1955.PubMed/NCBI(In Italian).
|
|
11
|
Werner H: Megakaryocytes in the bone
marrow of the white rat. Z Mikrosk Anat Forsch. 60:269–288.
1954.PubMed/NCBI
|
|
12
|
De La Fuente V: Megakaryocytic reaction
localized in the bone marrow; report of a new hematologic syndrome
with observations on the origin and development of megakaryocytes
and on the derivation of platelets. Arch Intern Med (Chic).
78:387–404. 1946.PubMed/NCBI
|
|
13
|
Vladareanu AM, Vasilache V, Bumbea H and
Onisâi M: Platelet dysfunction in acute leukemias and
myelodysplastic syndromes. Rom J Intern Med. 49:93–96.
2011.PubMed/NCBI
|
|
14
|
Velculescu VE and El-Deiry WS: Biological
and clinical importance of the p53 tumor suppressor gene. Clin
Chem. 42:858–868. 1996.PubMed/NCBI
|
|
15
|
Horiike S, Kita-Sasai Y, Nakao M and
Taniwaki M: Configuration of the TP53 gene as an independent
prognostic parameter of myelodysplastics yndrome. Leuk Lymphoma.
44:915–922. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fuhrken PG, Apostolidis PA, Lindsey S,
Miller WM and Papoutsakis ET: Tumor suppressor protein p53
regulates megakaryocytic polyploidization and apoptosis. J Biol
Chem. 283:15589–15600. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Luff SA, Kao CY and Papoutsakis ET: Role
of p53 and transcription-independent p53-induced apoptosis in
shear-stimulated megakaryocytic maturation, particle generation,
and platelet biogenesis. PLoS One. 13(e0203991)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Apostolidis PA, Woulfe DS, Chavez M,
Miller WM and Papoutsakis ET: Role of tumor suppressor p53 in
megakaryopoiesis and platelet function. Exp Hematol. 40:131–142.
2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jirouskova M, Shet AS and Johnson GJ: A
guide to murine platelet structure, function, assays, and genetic
alterations. J Thromb Haemost. 5:661–669. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Michelson AD: Flow cytometry: A clinical
test of platelet function. Blood. 87:4925–4936. 1996.PubMed/NCBI
|
|
21
|
Subramaniam M, Frenette PS, Saffaripour S,
Johnson RC, Hynes RO and Wagner DD: Defects in hemostasis in
P-selectin-deficient mice. Blood. 87:1238–1242. 1996.PubMed/NCBI
|
|
22
|
Liu Y, Jennings NL, Dart AM and Du XJ:
Standardizing a simpler, more sensitive and accurate tail bleeding
assay in mice. World J Exp Med. 2:30–36. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kafert S, Krauter J, Ganser A and Eder M:
Differential quantitation of alternatively spliced messenger RNAs
using isoform-specific real-time RT-PCR. Anal Biochem. 269:210–213.
1999.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kunicka JE, Fischer G, Murphy J and
Zelmanovic D: Improved platelet counting using two-dimensional
laser light scatter. Am J Clin Pathol. 114:283–289. 2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kunz D, Höffkes H, Kunz WS and Gressner
AM: Standardized flow cytometric method for the accurate
determination of platelet counts in patients with severe
thrombocytopenia. Cytometry. 42:284–289. 2000.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Schmitz G, Rothe G, Ruf A, Barlage S,
Tschöpe D, Clemetson KJ, Goodall AH, Michelson AD, Nurden AT and
Shankey TV: European Working group on clinical cell analysis:
Consensus protocol for the flow cytometric characterisation of
platelet function. Thromb Haemost. 79:885–896. 1998.PubMed/NCBI
|
|
27
|
Baccini V, Roy L, Vitrat N, Chagraoui H,
Sabri S, Le Couedic JP, Debili N, Wendling F and Vainchenker W:
Role of p21(Cip1/Waf1) in cell-cycle exit of endomitotic
megakaryocytes. Blood. 98:3274–3282. 2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
TeKippe M, Harrison DE and Chen J:
Expansion of hematopoietic stem cell phenotype and activity in
Trp53-null mice. Exp Hematol. 31:521–527. 2003.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Su W, Hopkins S, Nesser NK, Sopher B,
Silvestroni A, Ammanuel S, Jayadev S, Möller T, Weinstein J and
Garden GA: The p53 transcription factor modulates microglia
behavior through microRNA-dependent regulation of c-Maf. J Immunol.
192:358–366. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Matas D, Milyavsky M, Shats I, Nissim L,
Goldfinger N and Rotter V: P53 is a regulator of macrophage
differentiation. Cell Death Differ. 11:458–467. 2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Suraneni PK and Crispino JD: The hippo-p53
pathway in megakaryopoiesis. Haematologica. 101:1446–1448.
2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Gibbins JM: Platelet adhesion signalling
and the regulation of thrombus formation. J Cell Sci.
117:3415–3425. 2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Fantauzzo KA and Soriano P: PI3K-mediated
PDGFRα signaling regulates survival and proliferation in skeletal
development through p53-dependent intracellular pathways. Genes
Dev. 28:1005–1017. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Guidetti GF, Canobbio I and Torti M:
PI3K/Akt in platelet integrin signaling and implications in
thrombosis. Adv Biol Regul. 59:36–52. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lefrancais E, Ortiz-Muñoz G, Caudrillier
A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T,
Coughlin SR, et al: The lung is a site of platelet biogenesis and a
reservoir for haematopoietic progenitors. Nature. 544:105–109.
2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Cheng Z, Gao W, Fan X, Chen X, Mei H, Liu
J, Luo X and Hu Y: Extracellular signal-regulated kinase 5
associates with casein kinase II to regulate GPIb-IX-mediated
platelet activation via the PTEN/PI3K/Akt pathway. J Thromb
Haemost. 15:1679–1688. 2017.PubMed/NCBI View Article : Google Scholar
|