Introduction
Deafness is the most common sensory deficit
disorder, accounting for a high incidence of disability (1). According to a survey from the World
Health Organization (WHO) in 2013, there are ~360 million people
with varying degrees of deafness worldwide (2). According to epidemiological data, ~1 in
1,000 newborns is diagnosed with congenital deafness (3). Deafness directly affects cognition,
thinking and memory, resulting in a decline in the quality of life,
thereby contributing to the burden of families and society
(4). The progression and development
of modern medicine have gradually reduced the proportion of
deafness cases caused by environmental factors (5). However, the proportion of patients with
hearing impairment caused by genetic factors has largely remained
to be determined (6). Based on
deafness combined with the presence or absence of malformations of
the external ear, hereditary deafness may be classified into
syndromic deafness and non-syndromic deafness (7). Nearly 70% of cases of hereditary
deafness may be attributed to non-syndromic hearing loss (NSHL) and
50% of cases of NSHL have Mendelian disease (8). At present, ~145 chromosomal loci are
known to be associated with non-syndromic deafness (9). The coagulation factor C homology (COCH)
gene was the first gene to be identified to cause non-syndromic
deafness (Online Mendelian Inheritance in Man ID, 603196) (10). Patients with a mutation in this gene
may present with a series of symptoms caused by cochlear and
vestibular dysfunction (11). At
present, detection of the COCH mutation is a subject of intense
research (12). Screening for gene
mutations using traditional Sanger sequencing is time-consuming and
expensive (13).
Recent developments in whole-exome sequencing (WES)
technology have shifted this paradigm and currently, rapid
sequencing of exomes, transcriptomes and genomes may be completed
at a relatively low cost (14). Of
note, the application of this technology to catalog the mutational
landscapes of genetic disorders has revealed a novel approach to
explore single-gene diseases (15,16). The
use of WES technology in patients with hereditary deafness and
targeted next-generation sequencing of genes associated with
deafness in hearing-impaired individuals has enabled the
identification of informative mutations (17). WES is not only able to provide an
efficient diagnosis for known deafness genes but also unravel novel
gene mutations that may help us understand the molecular mechanism
underlying deafness or hearing loss (18).
The present study aimed to identify genetic factors
associated with hearing loss in a Chinese pedigree with hearing
loss. WES was employed to identify a novel gene mutation in the
gene crystallin µ (CRYM) that was accountable for the disease.
Materials and methods
Subjects
The present study reported on familial non-syndromic
autosomal dominant sensorineural deafness in four generations of a
Chinese pedigree. The genealogical proband was a 35-year-old
female, recruited from Xinqiao Hospital (Chongqing, China) of the
Army Medical University in September 2015. This proband was
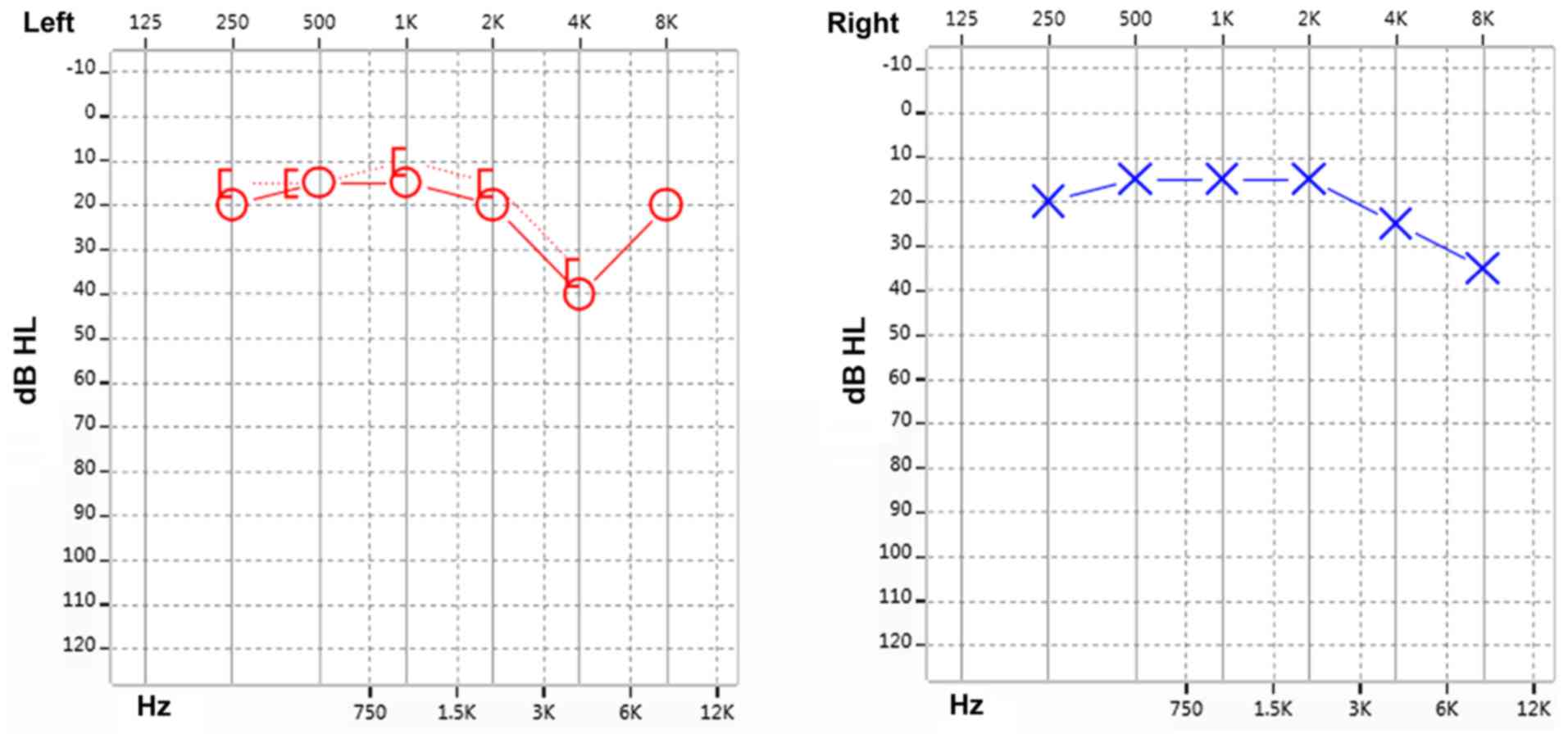
subjected to pure-tone audiometry, Distortion Product Otoacoustic
Emission (DPOAE) and Auditory Brainstem Response (ABR) evaluation.
The test results were indicative of sensorineural deafness in the
right ear and a high-frequency hearing loss in the left ear
(Fig. 1). The results presented a
progressive aggravation of hearing loss with increasing frequency
illustrated by a downsloping of the hearing curve. The patient was
diagnosed with sensorineural deafness according to the guidelines
for clinical evaluation and etiologic diagnosis of hearing loss by
the American College of Medical Genetics and Genomics (19). The patient and her family members
were enrolled in the present study. The study protocol was approved
by the Ethics Committee of Xinqiao Hospital (Chongqing, China) and
the study was performed in accordance with the Declaration of
Helsinki. Written informed consent was obtained from the proband
and the remaining family members. Furthermore, all of the
participants agreed to the publication of their results on clinical
characteristics and genetic data with protection of their privacy.
Subsequently, the detailed medical history was collected, including
family history of deafness and consanguineous marriages, year of
onset of deafness, age, progression, and history of ototoxic drugs
and noise exposure.
DNA extraction
Whole blood samples of all subjects were collected
and subjected to genomic DNA extraction using the RelaxGene Blood
DNA System (Tiangen Biotech) as per the manufacturer's
instructions. The extracted DNA was stored at -20˚C until further
analysis.
Audiology function examination
An audiology function examination was performed
using pure tone audiometry, DPOAE and ABR on all family members
using standard procedures as described previously (20).
Gene chip detection analysis
A total of 15 mutations of four deafness-associated
genes were detected, including those in gap junction protein beta 2
(GJB2; 35delG, 176del16, 235delC and 299delAT), GJB3 (538C>T),
solute carrier family 26 member 4 (2168A>G, IVS7-2A>G,
1174A>T, 122G>A, 1229C>T, 1975G>C, 2027T>A and
IVS15+5 G>A) and mitochondrial 12S ribosomal RNA (1494C>T and
1555A>G) using the Heredity Hearing Loss Array Detection Kit
purchased from Capital Bio Corp. The chip was imaged using a
LuxScan™ 10 KB Microarray Scanner (Capital Bio Corp.) (21). Data evaluation was performed using
SPSS 19.0 (IBM Corp.).
WES analysis
Specific primer sequences were designed by Primer
Premier 5.0 (Premier Biosoft International). The Ion PI™ Hi-Q™
Sequencing 200 Kit (Thermo Fisher Scientific, Inc.) was employed
for target gene mutation detection by computational mapping
analysis. Genomic DNA samples were sheared by sonication. The
sheared genomic DNA was then hybridized with the NimbleGen 2.0
probe sequence capture array from Roche (http://www.nimblegen.com/products/seqcap/ez/v2/index.html)
to enrich the exonic DNA (Joy Orient). The libraries were first
tested for exon-enrichment by quantitative PCR and for size
distribution and concentration using the Agilent Bioanalyzer 2100
(Agilent Technologies, Inc.). The samples were then sequenced on an
Illumina Hiseq 2500 platform (Illumina, Inc.). Each sample was
tested in two parallel reactions.
Results
Family investigation and clinical
phenotype
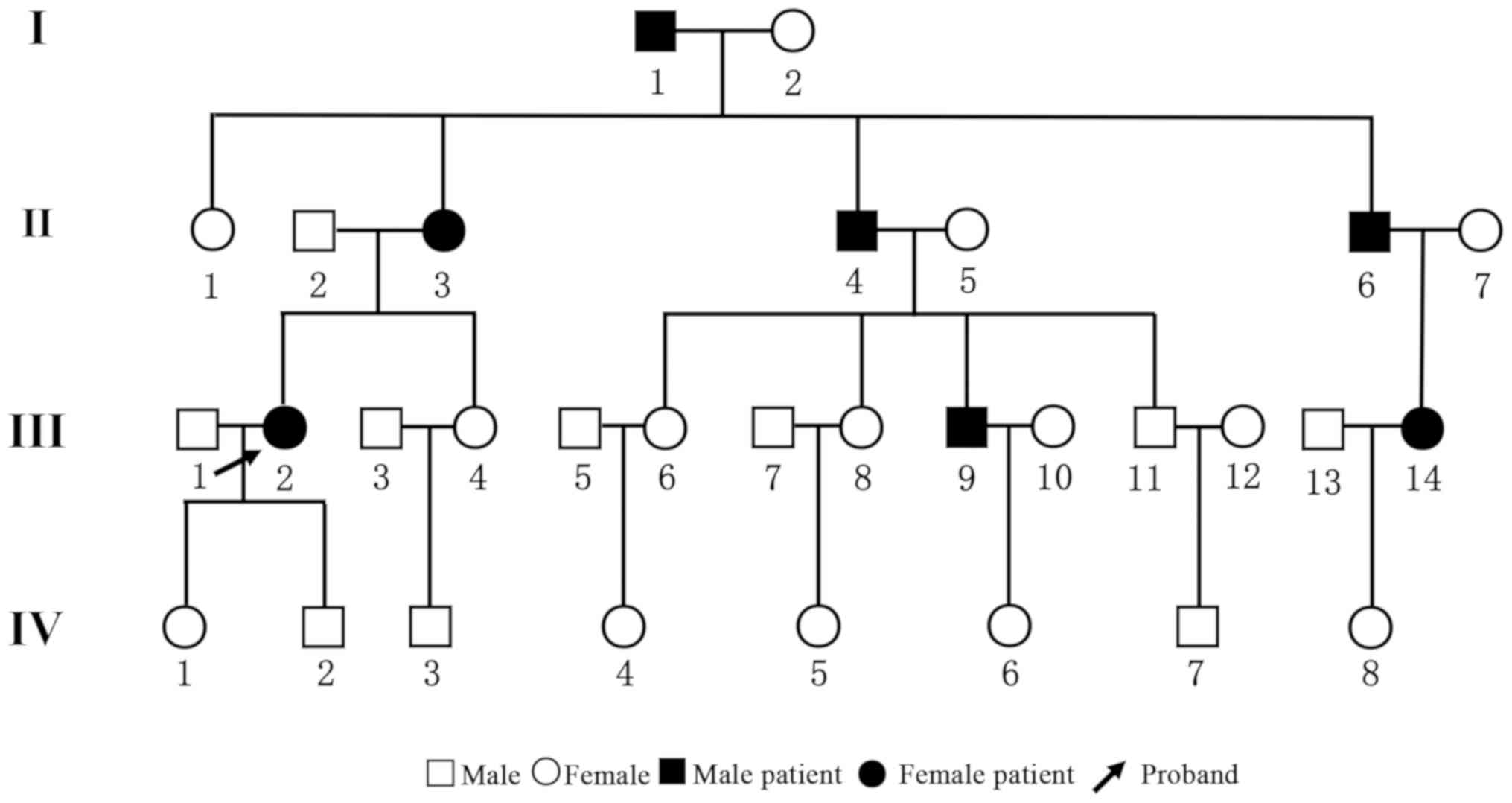
The pedigree chart is presented in Fig. 2. The family consisted of 31 members
belonging to four generations. There were 14 males and 17 females.
All family members underwent pure-tone audiometry examinations and
the results are listed in Table I. A
total of 7 patients, aged 31-79 years, were diagnosed with
sensorineural deafness of varying degrees. None of the patients had
a history of ototoxic drug exposure. Among them, cases
I1, II3 and II4 were diagnosed
with severe deafness on both ears. Cases III2,
III9 and III14 demonstrated mild
sensorineural deafness. Case II6 had mixed deafness
(conductive deafness and sensory deafness) on the right ear and
sensorineural deafness on the left ear. The results of the hearing
test for the remaining family members are listed in Supplementary
Table SI. There was no predilection
for sex and a significant phenomenon of continuous transmission was
noted. At least one parent of affected subjects was diagnosed with
deafness, suggesting that the disease had an autosomal dominant
pattern of inheritance.
 | Table IClinical characteristics of the
family members. |
Table I
Clinical characteristics of the
family members.
| Subject | Age (years) | Sex | Age at onset
(years) | Pure tone test | Degree of hearing
loss | DPOAE | Binaural ABR
threshold (dBnHL) |
|---|
| I1 | 78 | Male | 47 | Binaural full
frequency sensorineural deafness | High severity on
both sides | Binaural full
frequency extraction in abnormal range | 110 |
| II2 | 56 | Male | 54 | Binaural
sensorineural deafness | Moderate on both
sides | Binaural full
frequency extraction in abnormal range | 50 |
| II3 | 61 | Female | 52 | Binaural mixed
deafness | Severe on both
sides | Binaural full
frequency extraction in abnormal range | 70 |
| II4 | 56 | Male | 43 | Binaural
sensorineural deafness | Severe on both
sides | Binaural full
frequency extraction in abnormal range | 100 |
| II6 | 56 | Male | 50 | Binaural mixed
deafness | Severe on both
sides | Binaural full
frequency extraction in abnormal range | 70 |
| III2 | 35 | Female | 32 | Sensorineural
deafness in right ear, high-frequency hearing loss in left ear | Normal hearing
threshold | Binaural partial
frequency extraction in abnormal range | 30 |
| III9 | 31 | Male | 31 | Binaural
sensorineural deafness with high frequency | Normal hearing
threshold | Binaural partial
frequency extraction in abnormal range | 30 |
| III14 | 32 | Female | 31 | Binaural
sensorineural deafness with high frequency | Normal hearing
threshold | Binaural partial
frequency extraction in abnormal range | 40 |
Gene chip identification of
deafness-associated genes
The routine diagnosis of deafness genes was
performed by microarray in order to identify hot mutations in the
study subjects. The gene chip contained 4 genes and 15 loci. The
details of the gene chip and the mutations it is able to detect are
listed in Table II. However, none
of these mutations was identified by the gene chip in any of the
participants.
| Table IIGeneral deafness gene chip for target
capture. |
Table II
General deafness gene chip for target
capture.
| Gene | Variant |
|---|
| GJB2 |
NM_004004.6:c.35delG |
| GJB2 | 176del16 |
| GJB2 | 235delC |
| GJB2 | 299delAT |
| GJB3 |
NM_024009.3:c.538C>T |
| SLC26A4 | 2168A>G |
| SLC26A4 |
NM_000441.2:c.919-2A>G |
| SLC26A4 | 1174A>T |
| SLC26A4 | 1226G>A |
| SLC26A4 | 1229C>T |
| SLC26A4 | 1975G>C |
| SLC26A4 | 2027T>A |
| SLC26A4 |
NM_000441.2:c.1707+5G>A |
| MT-RNR1 | 1494C>T |
| MT-RNR1 | 1555A>G |
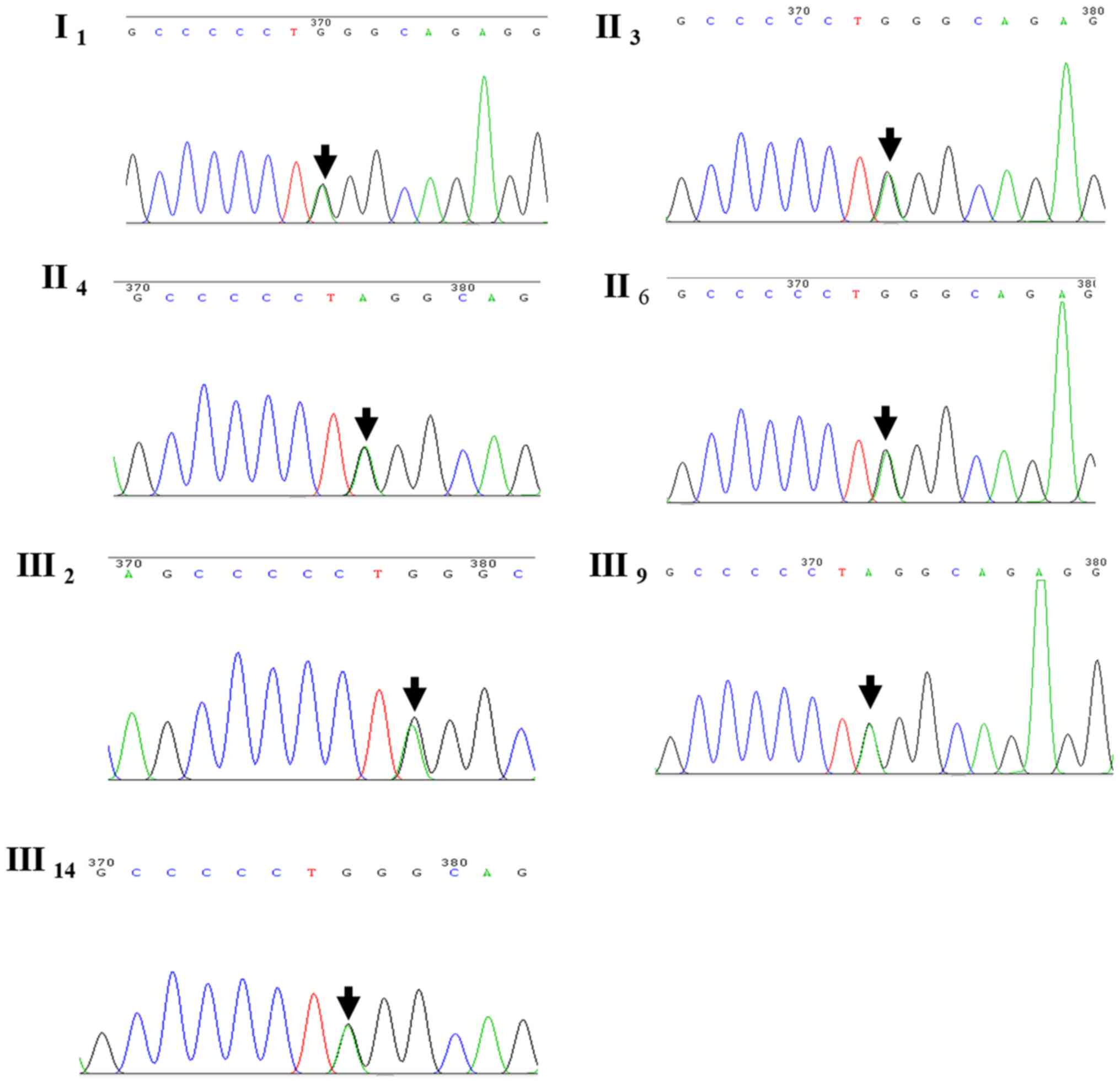
Mutation identified by WES
WES was performed in four family members, namely
cases III2, III14, IV2 and
IV8. A heterozygous mutation of CRYM was detected in
cases III2 and III14; however, it was absent
in subjects IV2 and IV8. The heterozygous
mutation detected was c.152C>T(Pro51Leu). However, no mutations
in other known genes associated with deafness were observed.
Whether the mutation was pathogenic and accounted for a familial
trait remain to be verified. Sanger sequencing was used to verify
the results obtained by WES in the remaining family members
participating in the study. Of note, the CRYM mutation was
identified in all other seven family members with deafness, as in
the proband III2 (Fig.
3).
Bioinformatics analysis
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) was used to
predict the possible impact of an amino acid substitution on the
structure and function of the mutation, which indicated that the
CRYM c.152C>T(Pro51Leu) mutation is probably damaging, with a
score of 1.000 (sensitivity, 0.00; specificity, 1.00), and the
Protein Variation Effect Analyzer (http://provean.jcvi.org/index.php) indicated a
deleterious effect with a score of -5.395 (score below -2.5 means
deleterious effect). Collectively, these results strongly indicated
that the p.P51L mutation was likely to be deleterious to the
protein. The result of a multiple sequence alignment suggested that
the proline at position 51 of CRYM is highly conserved among
various species (Fig. 4). In
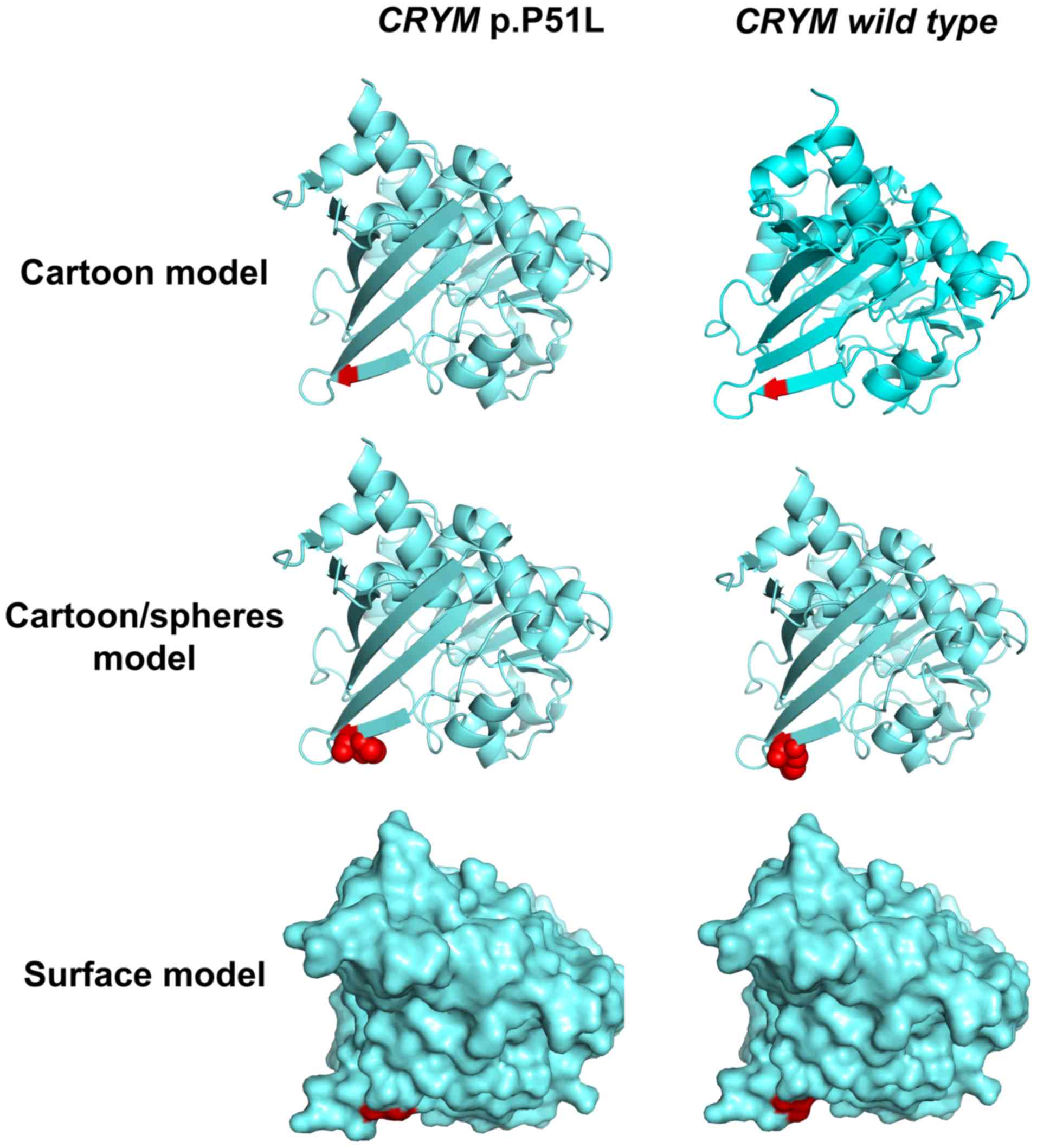
addition, the 3-dimensional protein structures were predicted and
displayed with a cartoon, a cartoon/spheres and a surface model,
which were generated using PyMOL software (version 2.3; Schrödinger
LLC; Fig. 5). The protein structure
parameters indicated that The Inertia Axis Aligned Bounding Box
(IABB) dimensions were (43.63, 47.33, 63.49) and (43.32, 47.22,
63.49) and the IABB volume was 131,101.14 and 129,861.34 for the
wild-type and muted type, respectively. This indicates that this
substitution is able to destabilize the protein conformation.
Follow-up
The only follow-up of the proband was by a phone
call after 4 years. The subjective sensation of her hearing was not
significantly changed according to the telephone survey and
answered a subjective questionnaire with the help of her family
member. No further hearing examinations were completed on the other
family members and no family members were prescribed hearing
aids.
Discussion
In the present study, a Chinese pedigree with
hereditary sensorineural hearing loss of unknown genetic etiology
was analyzed for a causative mutation using WES. A novel mutation,
NM_00188.5(CRYM): c.152C>T(Pro51Leu), was detected. This
mutation, not being a common variant, has a high likelihood of
being pathogenic in patients with sensorineural hearing loss.
Deafness is a hereditary disorder with a high degree
of genetic heterogeneity. DFNA is primarily the result of a single
gene mutation (22). To date, a
total of 105 genes have been reported to be associated with NSHL
(23). Among the genes linked to
this condition, COCH was the first gene to be reported to be
associated with vestibular function (24). Clinical symptoms of COCH mutation are
autosomal dominant, non-syndromic and characterized by progressive
sensorineural hearing loss (10). At
onset, the disease involves high-frequency hearing impairment. It
progresses with age to severe hearing loss affecting low and high
frequencies (25).
Recently, with the advent of comprehensive genetic
testing for hereditary hearing loss using massively parallel
sequencing (26), further gene
mutations accounting for NSHL have been identified, thereby
highlighting its genetic heterogeneity (27). The application of a deafness
diagnostic screening panel based on a deafness mutations/gene
database has gained popularity (28). A gene diagnostic chip for hereditary
deafness has been used previously to detect gene mutations
associated with hearing impairment among high-risk pregnant females
(29) and newborns (30), and for pre-natal diagnosis of
deafness to avoid the birth of children with congenital deafness
(31).
At present, gene chip is used for rapid genetic
diagnosis and in epidemiological surveys of hearing loss. However,
it was originally devised for hot-spot mutations that have already
been reported. Therefore, it cannot be used to detect other
mutations in these genes. With the rapid development of WES
technology, the scientific community has begun to widely adopt
second-generation sequencing technology to solve biological
problems. This novel generation of sequencers is able to rapidly
sequence whole genomes and zoom in to deeply sequence the target
regions of interest (32). Numerous
genetic variants of unknown significance may be detected in
patients by WES. In the present study, a regular gene chip was used
to screen mutations for hearing loss, although no positive results
were obtained. However, with the application of NGS, a novel
mutation of CRYM, c.152C>T(Pro51Leu), responsible for hearing
impairment was identified.
CRYM, which is also termed Thyroid Hormone Binding
Protein (THBP) or Deafness, autosomal dominant, 40 (DFNA40), is
located on chromosome 16. Its transcript length is 1,482 bp with
eight exons encoding 314 amino acids. CRYM is mainly expressed in
the heart and brain. It is particularly abundant in the inner ear
(33). A previous study has
demonstrated that CRYM is expressed in the human cochlea (34). CRYM has a critical role in the
physiological regulation of the activity of thyroid hormone
(35). An experimental study has
indicated that CRYM may have a role in the development of cortical
and hippocampal pyramidal cells in the early postnatal period
(36). Furthermore, CRYM expression
is possibly upregulated through the activator protein-1 (AP-1) site
in the promoter (37). Another
important CRYM function has also been identified: When combined
with ketimine reductase, it may act as a classical imine reductase
(38). In addition, CRYM has a
critical role and is regarded as a novel androgen-regulated gene
whose expression is elevated in prostate cancer (39). It has been demonstrated in an animal
model that CRYM mutations cause auditory dysfunction through
thyroid hormone-binding effects on the cochlea (40). It is well known that thyroid hormone
is crucial for normal development, as well as maintenance of
hearing function (41). CRYM is
divided into two classes: Taxon-specific and ubiquitous. This gene
encodes a taxon-specific crystallin protein that binds NADPH and
has a sequence similarity to bacterial ornithine cyclodeaminases,
which is particularly abundant in kangaroo lenses (42).
To date, CRYM-null patients have not been reported
(35). Abe et al (43) were the first to demonstrate that CRYM
mutation is associated with hearing loss. In their study, they
indicated that one missense and one stop-lost variant in CRYM was a
pathogenic mutation for hearing loss in two different families of
Japanese origin. A search for the CRYM mutation in Clinvar was
performed (https://www.ncbi.nlm.nih.gov/clinvar/?term=CRYM%5Bgene%5D).
Most of the identified mutations in other loci of CRYM were benign,
of uncertain significance or had conflicting interpretations of
pathogenicity. The details of the mutations identified are listed
in Table III. The previously
unreported novel CRYM mutation identified in the present study is
likely to be pathogenic.
| Table IIISummary of mutations in the
crystallin µ gene and the clinical significance of pedigrees with
associated dominant sensorineural deafness. |
Table III
Summary of mutations in the
crystallin µ gene and the clinical significance of pedigrees with
associated dominant sensorineural deafness.
| Nucleic acid
mutation | Amino acid
variation | Molecular
consequence | Clinical
significance |
|---|
| c.945A>T | p.Ter315Tyr | Single nucleotide
variant | Pathogenic |
| c.941A>C | p.Lys314Thr | Missense | Pathogenic |
| c.907G>A | p.Ala303Thr | Missense | Uncertain
significance |
| c.864C>G | p.Thr288= | Single nucleotide
variant | Benign |
| c.807T>C | p.Phe269= | Single nucleotide
variant | Benign/likely
benign |
| c.761C>T | p.Ala254Val | Missense | Uncertain
significance |
| c.741C>T | p.Tyr247= | Single nucleotide
variant | Benign |
| c.662C>T | p.Ala221Val | Missense | Likely benign |
| c.580G>A | p.Ala194Thr | Missense | Uncertain
significance |
|
c.523_524delinsTT | p.Glu175Leu | Missense | Conflicting
interpretations of pathogenicity |
| c.490-12C>T | | Single nucleotide
variant | Benign |
| c.489+9A>G | | Single nucleotide
variant | Likely benign |
| c.480C>T | p.Ser160= | Single nucleotide
variant | Likely benign |
| c.479C>T | p.Ser160Phe | Missense | Uncertain
significance |
| c.474G>A | p.Gln158= | Single nucleotide
variant | Uncertain
significance |
| c.343A>G | p.Ile115Val | Missense | Uncertain
significance |
| c.325-12T>C | | Single nucleotide
variant | Benign |
| c.279G>A | p.Gln93= | Single nucleotide
variant | Likely benign |
| c.135C>A | p.Pro45= | Single nucleotide
variant | Likely benign |
| c.108C>A | p.Ser36Arg | Missense | Conflicting
interpretations of pathogenicity |
| c.8G>T | p.Arg3Leu | Missense | Uncertain
significance |
In conclusion, the present study identified a novel
mutation, NM_00188.5(CRYM): c.152C>T(Pro51Leu), which further
strengthened the association between CRYM mutation and NSHL.
Further functional studies regarding mutations in this clinical
condition may assist in clarifying the pathogenic mechanism
underlying familial sensorineural deafness.
Supplementary Material
Hearing testing results of the family
members without clinical symptoms.
Acknowledgements
Not applicable.
Funding
No funding received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AD designed the study; QL performed all of the
experiments; XZ and JY carried out the audiology function
examination; MW collected the clinical data, analyzed the data, and
prepared the manuscript. All authors have read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of Xinqiao Hospital (Chongqing, China) and the study was
performed in accordance with the Declaration of Helsinki. Written
informed consent was obtained from the proband and the remaining
family members.
Patient consent for publication
All of the participants agreed to the publication of
their results on clinical characteristics and genetic data with
protection of their privacy.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sheffield AM and Smith RJH: The
epidemiology of deafness. Cold Spring Harb Perspect Med.
9(a033258)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chadha S and Cieza A: World Health
Organization and its initiative for ear and hearing care.
Otolaryngol Clin North Am. 51:535–542. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yao GD, Li SX, Chen DL, Feng HQ, Zhao SB,
Liu YJ, Guo LL, Yang ZM, Zhang XF, Sun CX, et al: Combination of
hearing screening and genetic screening for deafness-susceptibility
genes in newborns. Exp Ther Med. 7:218–222. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lin FR, Yaffe K, Xia J, Xue QL, Harris TB,
Purchase-Helzner E, Satterfield S, Ayonayon HN, Ferrucci L,
Simonsick EM, et al: Hearing loss and cognitive decline in older
adults. JAMA Intern Med. 173:293–299. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lasak JM, Allen P, McVay T and Lewis D:
Hearing loss: Diagnosis and management. Primary Care. 41:19–31.
2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Momi SK, Wolber LE, Fabiane SM, MacGregor
AJ and Williams FM: Genetic and environmental factors in
age-related hearing impairment. Twin Res Hum Genet. 18:383–392.
2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kremer H: Hereditary hearing loss; about
the known and the unknown. Hear Res. 376:58–68. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Vona B, Müller M, Dofek S, Holderried M,
Löwenheim H and Tropitzsch A: A big data perspective on the
genomics of hearing loss. Laryngorhinootologie. 98 (Suppl
1):S32–S81. 2019.PubMed/NCBI View Article : Google Scholar : (In English,
German).
|
|
9
|
Petersen M and Willems P: Non-syndromic,
autosomal-recessive deafness. Clin Genet. 69:371–392.
2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Robertson NG, Lu L, Heller S, Merchant SN,
Eavey RD, McKenna M, Nadol JB Jr, Miyamoto RT, Linthicum FH Jr,
Lubianca Neto JF, et al: Mutations in a novel cochlear gene cause
DFNA9, a human nonsyndromic deafness with vestibular dysfunction.
Nat Genet. 20:299–303. 1998.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Khetarpal U: DFNA9 is a progressive
audiovestibular dysfunction with a microfibrillar deposit in the
inner ear. Laryngoscope. 110:1379–1384. 2000.PubMed/NCBI View Article : Google Scholar
|
|
12
|
JanssensdeVarebeke S, Topsakal V, Van Camp
G and Van Rompaey V: A systematic review of hearing and vestibular
function in carriers of the Pro51Ser mutation in the COCH gene. Eur
Arch Otorhinolaryngol. 276:1251–1262. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ku C, Cooper DN, Iacopetta B and Roukos
DH: Integrating next-generation sequencing into the diagnostic
testing of inherited cancer predisposition. Clin Genet. 83:2–6.
2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Levy SE and Myers RM: Advancements in
next-generation sequencing. Annu Rev Genomics Hum Genet. 17:95–115.
2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Boycott KM, Vanstone MR, Bulman DE and
MacKenzie AE: Rare-disease genetics in the era of next-generation
sequencing: Discovery to translation. Nat Rev Genet. 14:681–691.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Mardis ER: The impact of next-generation
sequencing technology on genetics. Trends Genet. 24:133–141.
2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Vona B, Müller T, Nanda I, Neuner C,
Hofrichter MA, Schröder J, Bartsch O, Läßig A, Keilmann A, Schraven
S, et al: Targeted next-generation sequencing of deafness genes in
hearing-impaired individuals uncovers informative mutations. Genet
Med. 16:945–953. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang T, Wei X, Chai Y, Li L and Wu H:
Genetic etiology study of the non-syndromic deafness in Chinese
Hans by targeted next-generation sequencing. Orphanet J Rare Dis.
8(85)2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Alford RL, Arnos KS, Fox M, Lin JW, Palmer
CG, Pandya A, Rehm HL, Robin NH, Scott DA, Yoshinaga-Itano C, et
al: American College of Medical Genetics and Genomics guideline for
the clinical evaluation and etiologic diagnosis of hearing loss.
Genet Med. 16:347–355. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mehraei G, Gallardo AP, Shinn-Cunningham
BG and Dau T: Auditory brainstem response latency in forward
masking, a marker of sensory deficits in listeners with normal
hearing thresholds. Hear Res. 346:34–44. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yan D, Xiang G, Chai X, Qing J, Shang H,
Zou B, Mittal R, Shen J, Smith RJ, Fan YS, et al: Screening of
deafness-causing DNA variants that are common in patients of
European ancestry using a microarray-based approach. PLoS One.
12(e0169219)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nance WE: The genetics of deafness. Ment
Retard Dev Disabil Res Rev. 9:109–119. 2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
DiStefano MT, Hemphill SE, Oza AM, Siegert
RK, Grant AR, Hughes MY, Cushman BJ, Azaiez H, Booth KT, Chapin A,
et al: ClinGen expert clinical validity curation of 164 hearing
loss gene-disease pairs. Genet Med. 21:2239–2247. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
de Kok YJ, Bom SJ, Brunt TM, Kemperman MH,
van Beusekom E, van der Velde-Visser SD, Robertson NG, Morton CC,
Huygen PL, Verhagen WI, et al: A Pro51Ser mutation in the COCH gene
is associated with late onset autosomal dominant progressive
sensorineural hearing loss with vestibular defects. Hum Mol Genet.
8:361–366. 1999.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kemperman MH, Bom SJ, Lemaire FX, Verhagen
WI, Huygen PL and Cremers CW: DFNA9/COCH and its phenotype. Nat
Genet. 61:66–72. 2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Shearer AE, DeLuca AP, Hildebrand MS,
Taylor KR, Gurrola J II, Scherer S, Scheetz TE and Smith RJ:
Comprehensive genetic testing for hereditary hearing loss using
massively parallel sequencing. Proc Natl Acad Sci USA.
107:21104–21109. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hilgert N, Smith RJ and Camp GV: Function
and expression pattern of nonsyndromic deafness genes. Curr Mol
Med. 9:546–564. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Abe S, Yamaguchi T and Usami SI:
Application of deafness diagnostic screening panel based on
deafness mutation/gene database using invader assay. Genet Test.
11:333–340. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Fang Y, Gu MS, Suo F, Wang CX, Liu XH and
Liu FM: Application of gene detection technique in the antenatal
diagnosis of hereditary hearing loss. Eur Rev Med Pharmacol Sci.
21:1452–1455. 2017.PubMed/NCBI
|
|
30
|
He X, Li X, Guo Y, Zhao Y, Dong H, Dong J,
Zhong L, Shi Z, Zhang Y, Soliman M, et al: Newborn screening of
genetic mutations in common deafness genes with bloodspot-based
gene chip array. Am J Audiol. 27:57–66. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Atik T, Bademci G, Diaz-Horta O, Blanton
SH and Tekin M: Whole-exome sequencing and its impact in hereditary
hearing loss. Genet Res (Camb). 97(e4)2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Margulies M, Egholm M, Altman WE, Attiya
S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et
al: Genome sequencing in microfabricated high-density picolitre
reactors. Nature. 437:376–380. 2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Schrauwen I, Hasin-Brumshtein Y,
Corneveaux JJ, Ohmen J, White C, Allen AN, Lusis AJ, Van Camp G,
Huentelman MJ and Friedman RA: A comprehensive catalogue of the
coding and non-coding transcripts of the human inner ear. Hear Res.
333:266–274. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Usami S, Takumi Y, Suzuki N, Oguchi T,
Oshima A, Suzuki H, Kitoh R, Abe S, Sasaki A and Matsubara A: The
localization of proteins encoded by CRYM, KIAA1199, UBA52, COL9A3,
and COL9A1, genes highly expressed in the cochlea. Neuroscience.
154:22–28. 2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Suzuki S, Mori JI and Hashizume K:
Mu-crystallin, a NADPH-dependent T(3)-binding protein in cytosol.
Trends Endocrinol Metab. 18:286–289. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hommyo R, Suzuki SO, Abolhassani N,
Hamasaki H, Shijo M, Maeda N, Honda H, Nakabeppu Y and Iwaki T:
Expression of CRYM in different rat organs during development and
its decreased expression in degenerating pyramidal tracts in
amyotrophic lateral sclerosis. Neuropathology. 38:247–259.
2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Suzuki S, Nishio SI, Ishii H, Sekido T,
Takeshige K, Ohkubo Y, Hiwatashi D, Takeda T and Komatsu M:
Possible roles of the AP-1 site in the cytosolic T3 binding protein
promoter and insights into its physiological significance. Horm
Metab Res. 45:501–506. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hallen A, Cooper AJ, Smith JR, Jamie JF
and Karuso P: Ketimine reductase/CRYM catalyzes reductive
alkylamination of α-keto acids, confirming its function as an imine
reductase. Amino Acids. 47:2457–2461. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Malinowska K, Cavarretta IT, Susani M,
Wrulich OA, Uberall F, Kenner L and Culig Z: Identification of
mu-crystallin as an androgen-regulated gene in human prostate
cancer. Prostate. 69:1109–1118. 2009.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Oshima A, Suzuki S, Takumi Y, Hashizume K,
Abe S and Usami S: CRYM mutations cause deafness through thyroid
hormone binding properties in the fibrocytes of the cochlea. J Med
Genet. 43(e25)2006.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Griffith AJ, Szymko YM, Kaneshige M,
Quiñónez RE, Kaneshige K, Heintz KA, Mastroianni MA, Kelley MW and
Cheng SY: Knock-in mouse model for resistance to thyroid hormone
(RTH): An RTH mutation in the thyroid hormone receptor beta gene
disrupts cochlear morphogenesis. J Assoc Res Otolaryngol.
3:279–288. 2002.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wistow G: Lens crystallins: Gene
recruitment and evolutionary dynamism. Trends Biochem Sci.
18:301–306. 1993.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Abe S, Katagiri T, Saito-Hisaminato A,
Usami S, Inoue Y, Tsunoda T and Nakamura Y: Identification of CRYM
as a candidate responsible for nonsyndromic deafness, through cDNA
microarray analysis of human cochlear and vestibular tissues. Am J
Hum Genet. 72:73–82. 2003.PubMed/NCBI View
Article : Google Scholar
|