1. Introduction: Systemic and
neuroinflammation are both characteristic in certain forms of
post-stroke depression
Stroke is a medical emergency that frequently
results in severe neurological sequelae and other complex
dysfunctions. Among these, post-stroke depression (PSD) is
prevalent, being experienced by about one-third of stroke patients
(1,2). PSD diminishes the quality and
expectancy of life through multiple factors: Cognitive decline,
high rate of suicide, increased risk of falls, functional
impairment, and poor response to rehabilitation (3). Depression not only decreases the
quality of life of those affected, but it is also associated with a
shorter interval to recurrent stroke and higher mortality (4,5).
There are many hypotheses about the underlying
molecular pathways of depressive disorders: the monoamine
neurotransmitter hypothesis, the hypothalamus-pituitary-adrenal
axis dysfunction hypothesis, the neurotrophic hypothesis, and the
neuroinflammation hypothesis. These hypotheses are complementary
rather than contradictory and probably address distinct features of
the same multi-faceted disease.
Substantial evidence supports the dominant presence
of inflammation in depressive disorders (6), especially the so-called atypical
depression, which can be characterized by fatigue, increased
appetite, weight gain, hypersomnia, hypoactive
hypothalamus-pituitary-adrenal axis, altered metabolism of the
frontoparietal cortex, and a high association with fibromyalgia and
chronic fatigue syndrome (7). In
atypical depression, signs of systemic inflammation have been
detected: higher circulating levels of C-reactive protein,
interleukin-1β (IL-1β), and tumor necrosis factor α (TNFα) along
with the dominance of IL-2 positive Th1 lymphocytes. Along with
increased circulating proinflammatory cytokines, the presence of
the typical systemic acute-phase reaction has been described
(8). The activation of
proinflammatory mediators perpetuates the oxidative and nitrosative
stress in the central nervous system, with the consumption of n-3
polyunsaturated fatty acids, glutathione, coenzyme Q10, and, in
general, antioxidant capacity; this activity triggers pronounced
changes in 5-hydroxytryptamine and N-methyl-D-aspartate (NMDA)
signaling (6).
According to considerable evidence, pro-resolving
macrophages, often labeled as M2 type cells, play a distinct role
in the clearance of the perished tissue, exert tissue repair, and
may rewire survivor neurons. However, the phenotype of these cells
is yet incompletely characterized (9). Despite indirect evidence, the putative
role of M2 type macrophages in post-stroke depression has not been
analyzed previously. In this narrative review, we present mostly
experimental, but, in some cases, human data to describe the
regulatory processes of the post-stroke period. We concatenated
information clusters concerning the role of neuroinflammation in
purinergic stress, altered glutamatergic signaling,
neuroprogression, suggesting that M2 type macrophages might feature
as prominent scavenger and reparatory actors in post-stroke
depression.
2. Post-stroke depression, genetic factors
and lesion localization
PSD is characterized by mood disorders with
depressive features or major depressive-like episodes (10). Genetic factors, location of the
lesions, and grade of post-stroke physical and intellectual
disability influence the severity and duration of PSD. Among
genetic factors, the serotonin-transporter-linked polymorphic
region 5-HTTLPR and solute carrier family 6 member 4 STin2VNTR
polymorphisms of the serotonin transporter gene and
hypermethylation of the 5-HTTLPR s/s genes have been associated
with the onset and duration of PSD (10). Association of PSD with stroke
localized to specific brain regions was postulated many years ago
(11). According to some
observations, lesions proximal to or in the frontal pole, or the
limbic area, were more frequently associated with PSD (12,13), but
other studies failed to confirm these topological correlations
(14,15). Although lesion volume reportedly has
predictive value for the outcome, contradictory results have been
published (16-20).
Other studies correlated PSD with lesions of the medial prefrontal
cortex, thalamus, amygdala, or pallidum by defining the disruption
of frontal cortico-limbic neuronal circuits (18,21,22).
Recent pioneering studies performed by voxel-based symptom lesion
mapping could not resolve the ambiguity; however, two groups
confirmed the correlation of PSD severity with dorsolateral
prefrontal and left cerebellar hemispheric localization (20,23,24).
Physical disability may be in part caused by PSD,
but at least in some patients, the disability and PSD are
independently associated with the primary event (25).
There is poorly known, whether PSD, as an outcome
has common or different underlying molecular pathways in different
topological involvements. Neurotrophic factor-related findings
translate, at least in some degree, divergence dependent on lesion
localization.
3. Brain-derived neurotrophic factor: A link
between purinergic signaling, neuroinflammation and depression
Brain-derived neurotrophic factor (BDNF) is a growth
factor member of the neurotrophin family that is essential for
neuronal development, survival, and plasticity. BDNF plays an
important, differential modulatory role in the development and
evolution of mood disorders; depression and stress are accompanied
by low levels of neurotrophic factor and dendritic atrophy in the
hippocampus and prefrontal cortex whereas high-level, transient
stress-induced synthesis is characteristic of individual nuclei of
the amygdala and nucleus accumbens (26). From experimental work and clinical
studies (which focused on neurological disorders such as traumatic
injury, Parkinson's disease, multiple sclerosis, and neuropathic
pain), it is known that BDNF is synthesized not only in neurons and
astrocytes but also in microglia (27-31).
BDNF mRNA levels were reportedly decreased in the
dentate gyrus and hippocampus of rats exposed to the chronic stress
of prolonged immobilization (32).
The Val66→Met66 point-mutation that causes deficient BDNF secretion
increases susceptibility to anxiety, depression, and bipolar
disorder (33,34). It has also been found that
antidepressant treatment increases the synthesis of BDNF, and a
neurotrophic hypothesis of depression has been reported (35). In rat models, many kinds of older or
newer antidepressant medications, e.g., monoamine oxidase
inhibitors, selective serotonin reuptake inhibitors, tricyclic
agents, specific serotoninergic drugs, electroconvulsive shock
therapy, and transcranial magnetic stimulation, trigger increased
production of BDNF. In patients with major depression, low values
of BDNF in serum, plasma, and platelets have been reported
(36,37). Circulating BDNF is at least partially
produced in the brain, and it can pass the blood-brain barrier
(38). Treatment with selective
serotonin reuptake inhibitors as well as serotonin and
norepinephrine reuptake inhibitors has been reported to raise
plasma BDNF levels, and the molecule has been designated as a
biomarker for antidepressant therapy (37,39).
BDNF has differential effects in various regions of the brain in
depression: whereas it is diminished in the prefrontal cortex and
the hippocampus, it is also implicated in malfunctioning of the
depression-related mesolimbic reward center, which is the ventral
tegmental area-nucleus accumbens dopaminergic circuit;
administration of exogenous BDNF in this area induces
depressive-like symptoms. Phasic activity of dopaminergic neurons
in the ventral tegmental area-nucleus accumben region triggers BDNF
in stressed mice (40,41). In an experimental stroke, a 2-fold
rise of BDNF expression was found in the ischemic core region,
which gradually decreased to the reference level at seven days
(42).
The synthesis and release of BDNF in microglia
appear to be tightly associated with the activation of
ATP-sensitive purinergic receptors, especially P2X4R. During a
stroke, dying neurons and other cells from the ischemic region
release ATP, which addresses the transmembrane P2X4 purinergic
receptors of glial cells. Upregulation of these receptors
accompanies microglial activation, P2X4R-triggered resident
microglia, and infiltrating macrophages, which are important
sources of pro-inflammatory cytokines and mediators of the
post-stroke immune response (43,44).
Activated P2X4R facilitate the release of BDNF from these cells
(30). Verma et al (45) created global and myeloid-specific
P2X4R null mice and subjected them to transient occlusion of the
middle cerebral artery. The authors found upregulation of PX4R on
neurons in wild-type animals, especially on microglia. In female
mice with global receptor deletion, the volumes of cortical and
hemispheric infarcts were significantly smaller and the recovery
better than in controls and their male littermates. However, the
myeloid-specific P2X4R knock-out had different effects: besides a
quick sensorimotor recovery, the animals of both sex had anhedonia
and depressive-like behavior along with high expression of IL-1β,
IL-6 and TNFα; low BDNF mRNA in the perilesional cortex; and low
plasma titers of the cytokines (45). These results indicate that the global
deletion of the P2X4 receptor is neuroprotective and suspends
neuroinflammation, but when deletion affects only the microglia
favors a depression-prone and pro-inflammatory phenotype. One
possible reason for this dichotomy is that microglia lacking P2X4
possesses low BDNF synthesizing capacity.
4. The post-stroke immune pathways and
tissue repair
A short overview of the post-stroke
immune response
The immune system is thought to play a critical role
in the course and the main outcomes in PSD (46). Recent data suggest that PSD is
related to complex immune deregulation: immunosuppression,
neuroinflammation, and a characteristic shift in
microglia/macrophage phenotype in the lesional area. After the
onset of stroke, injured neurons and glial cells quickly activate
the neighboring astrocytes through the expression of
damage-associated molecular patterns (high mobility group box-1,
HMGB1; peroxiredoxins, PRX; galectin-3) (47). These cells, along with resident
microglia, secrete a set of pro-inflammatory cytokines (IL-1β,
IL-6, IFNγ, TNFα), chemokines, and matrix metalloproteinases, like
MMP-9, contributing to the disruption of the blood-brain barrier
(48). Neuronal-derived fractalkine
(CX3CL1) further amplifies microglia activation. IL-1β, TNFα, and
complement C1q secreted from microglia trigger reactive
astrogliosis with the appearance of A1 type cells, which manifest
high expression of genes associated with neuronal damage and death,
such as Neutrophil gelatinase-associated lipocalin (Lcn2) and
Serine protease inhibitor A3N (Serpina3n) (49). Oligodendrocytes are also affected by
ischemia, losing their capacity to remyelinate neuronal axons.
Neutrophils are attracted by various chemokines, i.e.
CCL2,9,10,11,20; their accumulation in the lesional zone reportedly
worsens the clinical outcomes (47).
With a close shift, monocyte-derived macrophages pass through the
injured blood-brain barrier, penetrate the core lesion and also
deploy at the perilesional zone, the penumbra. Peripheral monocytes
are recruited through monocyte chemoattractant protein (MCP-1 or
CCL-2), and due to danger signals received from the environment,
switch to a pro-inflammatory phenotype, releasing various
metalloproteinases and reactive oxygen species (48). In the chronic recovery phase, in an
IL-4, IL-10, and transforming growth factor β (TGFβ) containing
milieu, infiltrating macrophages may switch their differentiation
path to an anti-inflammatory phenotype. IL-4 released from injured
neurons mediates this transition through the interferon regulatory
factor (IRF)-4 signaling (50).
Astrocytes also orchestrate the adaptive immune
response and T-cell invasion of the ischemic region, occurring 3
days to 1 month after stroke. IL-15 signaling increases the number
of CD8+ cytotoxic T cells and also the invasion of
natural killer lymphocytes, which is a detrimental effect (51). Th17 cells are also over-represented
and activated (52). Recent studies
underscore the central role of
CD4+/CD25+/Foxp3+ or
CD4+/CD25+/CD127- regulatory T
cells, which proliferate and are detectable in ischemic lesions up
to 30 days (48,53). A high number of Tregs at 48 h is
associated with the right functional outcome; reversely, a
decreased number indicates a higher probability for early
neurological deterioration (53,54).
Tregs also provide neurovascular protection through the
downregulation of MMP-9, but this effect depends on their IL-10
synthesizing capacity. IL-10 producer Tregs are susceptible to
antagonize IFNγ and TNFα (55). B
lymphocytes are also detectable in the invader cell populations; as
B-cell deficient mice show larger infarct volumes and more severe
neurological deficit, their role seems to be somewhat protective,
especially in the presence of IL-10(48). However, it was also documented that
B-cells and autoantibodies probably induce delayed cognitive
deficit and dementia (56).
Molecular processes and tissue repair
in immediate and subacute phase of post-stroke recovery
The timeline of the post-stroke immunological
response has been sketched by Rayasam et al (57). They proposed four consecutive but
functionally interweaving phases: Innate immune response, adaptive
immune response, and resolution. Immediately after the ischemic
event, reactive oxygen species, heat shock proteins, and nucleoside
triphosphates (ATP, UTP) are released from the ischemic tissue. Two
important danger signals appear: HMGB1 promotes the breakdown of
the blood-brain barrier, and peroxiredoxins quickly activate the
Toll-like receptors of myeloid cells (58). As a result of these actions, the
innate immune response is initiated, and post-synaptic NMDA and
α-amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA) receptors
are stimulated. NMDA receptors are cation influx regulators, which
support neuronal health, especially synaptic plasticity, that is
necessary for learning and memory. In ischemia, in contrast to
these useful activities, a dichotomous behavior was observed:
overactivation of NMDA receptors mediates glutamate excitotoxicity;
however, AMPA receptors foster synaptic plasticity and long-term
potentiation (40,59). Some heat shock proteins, such as
Hsp70 and Hsp27, seem to promote neuronal survival, whereas Hsp32,
known as heme oxygenase (HO-1), might have a dual role reacting
with ferrous ions and production of hydroxyl radicals
(pro-oxidant), but inhibiting lipid-peroxidation and lowering
neuronal apoptosis, if overexpressed (anti-oxidant) (60,61).
These proteins, together with other free radicals, induce
mitochondrial failure and apoptosis. Immediately after the injury,
resident microglia and astrocytes are activated, followed by the
infiltrating neutrophils (which likely appear in the first hour)
(62) and an increasing proportion
of mononuclear/macrophage cells of myeloid origin. Microglia and
infiltrating macrophages are receptive to distinct chemotactic
signals: the former react to CX3CL1 (fractalkine) and the latter to
CCL2 proteins. In the past, the two species could not be
phenotypically differentiated, but nowadays, they can be
distinguished based on their Ly6C, CD45, or transmembrane protein
119 expression (63,64). In the subacute phase of recovery, M2
type microglia secrete neurotrophic factors, such as TGFβ or IGF,
remove disabled synapses and prevent the degradation of the
extracellular matrix through arginase-1 (Arg1). Pro-resolving
macrophage subspecies expressing the macrophages scavenger
receptor-1 (MSR-1 or CD206) are responsible for the clearance of
HMGB1 and PRX (65).
Cell survival and salvage signals in
the ischemic region
Surprising results were disclosed concerning
distinct ways of cell death and survival in stroke. Jiang et
al (42) applied a focal
ischemic and embolic stroke model to identify survivor cells in the
core lesion of strokes. Using TUNEL assay and caspase-3 staining,
they found a mixed form of cell death, with the emergence of
intense apoptosis and autophagy in addition to necrosis. They also
found that >80% of the cells in the core were
macrophage/microglia and that among ionized calcium-binding adapter
molecule-1 (Iba-1) positive cells, viable NeuN-positive elements
coexisted even days after the ischemic event (42). It has been proposed that early
regulatory signals, such as IL-4 and miRNA-124 (66,67),
which are emitted by damaged neurons, favor the rapid formation of
M2 type resolving microglia/macrophages. Phosphatidylserine
exteriorization of neurons is a strong phagocytotic signal, that is
divisive for M1 and M2 type cells. The first, emitting high
concentrations of reactive oxygen species, oxidize membrane
phosphatidylserine molecules often destroying not only dead, but
also viable neurons; the second react instead to chemically
unmodified, but exteriorized phosphatidylserine, recognizing it
like a classical, ‘eat me signal’ (66).
M2 type microglia produce higher levels of F-actin,
being more inclined to phagosome formation, clear the irreversibly
damaged structures by phagocytosis, while M2 type infiltrating
cells also show intense phagocytosis, releasing anti-inflammatory
cytokines, mainly IL-4, IL-10 and TGFβ (57,66).
These cytokines exert direct and indirect protective roles: IL-10
is a negative regulator of IL-1β, IL-6 and TNFα, whereas TGFβ
manifests neuroprotection through the synthesis of nerve growth
factor (NGF) and upregulation of anti-apoptotic proteins, such as
Bcl-2 and Bcl-x1(66).
In C57BL IL-4 knockout mice, the lack of IL-4 turned
on M1 type differentiation: When IL-4 was administered, the balance
was reversed and M2 type cells dominated; moreover, IL-4 was
beneficial in long-term functional recovery (68). Liesz et al (55) proposed a strong interaction between
invader leucocytes and resident microglia. They observed that in
perforin deleted mice, anti-CD49d therapy inhibited not only
leukocyte migration in the lesional area but also a significant
reduction of Iba-1+ microglia cell counts and IFNγ
secretion, along with a robust reduction of the infarct volume.
IL-10-expressing regulatory T cells also help M2 type
macrophage/microglia differentiation through activation of glycogen
synthase kinase 3β and a phosphatase and tensin homolog (69).
The essential signaling interactions and the
multicellular modulation of tissue repair are shown in Fig. 1. Titova et al (70), using functional neurological tests
and magnetic resonance imaging, investigated the late-phase effects
of middle cerebral artery occlusion (MCAO) on proton-irradiation
preconditioned rats. On post-stroke day 7 (which by extrapolation
corresponds to 7-8 months for humans), T2 tissue relaxation scores
and neurological severity scores were correlated and characterized
the late-stage brain recovery (intensive neovascularization, the
presence of Von Willebrand factor/glial fibrillary acidic
protein-positive glio-vascular complexes, enhanced neuronal
viability, and decreased numbers of phagocytes).
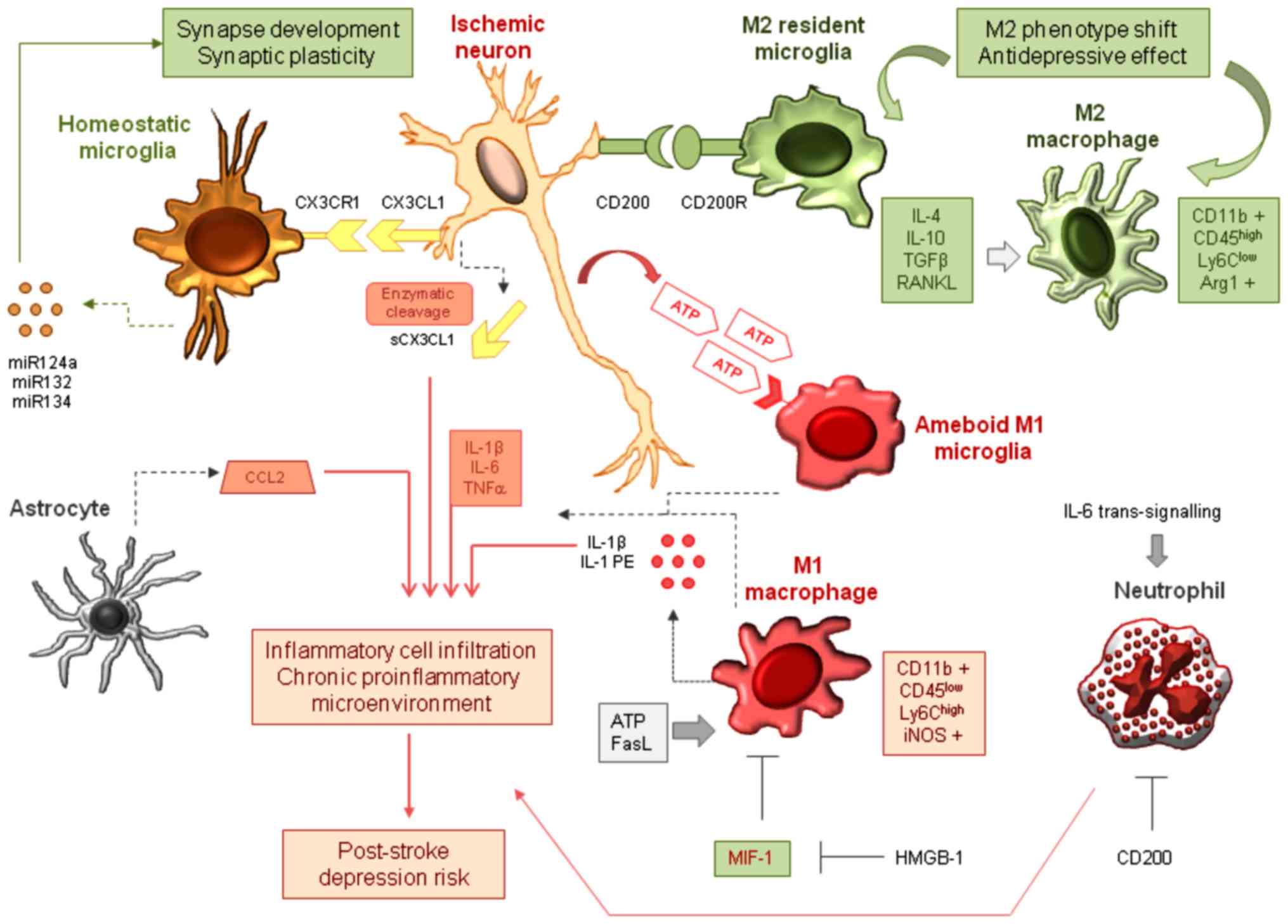 | Figure 1Generation and multi-cellular
modulation of the proinflammatory environment in post-stroke
depression. ATP released from injured neurons acts as a danger
signal and alerts the surrounding microglia via P2X4 purinergic
receptors, triggering M1-type microglial activation and ameboid
transformation. These cells, together with the infiltrating M1-type
macrophages release IL-1β, IL-6, TNFα, and other pro-inflammatory
mediators, partially in microvesicles and exosomes. Soluble CX3CL1
generated by metalloproteinase-mediated cleavage enhances the
invasion of inflammatory cells, along with CCL2 released by
astrocytes. Two receptor-ligand interactions between neurons and
microglia control neuronal survival and regeneration.
Membrane-bound CX3CL1 coupling to CX3CR1 maintains synaptic
plasticity, contributes to synapse development and keeps microglia
in a surveyor state. CD200-CD200R signaling determines a phenotype
shift towards M2-type microglia. miR-124a, miR-132, and miR-134 are
released from homeostatic microglia and facilitate neuronal
survival. M2 phenotype switch is protective, but insufficient M2
activity together with MIF-1 may cause the perpetuation of the
pro-inflammatory microenvironment and may contribute to the
development of post-stroke depression. IL, interleukin; TNFα, tumor
necrosis factor α. |
5. The microglia/macrophage network in
post-stroke depression
The macrophage theory of depression,
polarization and the classification dilemma
Applying histochemical analysis and
computer-assisted stereological cell counting, Ongür et al
(71) discovered that microglial
density in the subgenual prefrontal cortex, especially in the
Brodman's 24, is reduced in patients with major depressive and
bipolar disorder. Resident microglia constitute ~5-12% of all
central nervous system cells; they derive from myeloid precursors
of the embryonic yolk sac. Studies performed with in vivo
two-photon imaging technology confirmed that in the resting form,
microglia continuously monitor the microenvironment, emitting
numerous protrusions (72).
Microglia are especially susceptible to danger signals, which
transform them to a reactive phenotype, characterized by the
shortening of cellular processes and swelling and enlargement of
the soma; in the extreme form they have an ameboid appearance,
completely lacking cellular processes (59).
Smith (73), in 1991,
proposed the macrophage theory of depression, invoking the role of
pro-inflammatory signals, cytokines, hormones and various causes of
macrophage activation, such as infection, allergy or systemic
autoimmune disease. In his medical hypothesis, he mentioned the
high prevalence of depression in post-stroke patients with
atherosclerotic background, and he proposed a primary role for the
quickly activated IL-1 producers, monocytes and microglia. He also
argued for the strong inhibitory role of n-3 polyunsaturated fatty
acids on IL-1 and TNFα secretion by macrophages, cardiovascular
events and depression in Japan (73).
The M1-M2 concept of macrophage differentiation
originates from observations made in macrophages obtained from
C57BL/6 and Balb/c mice (9). The two
sub-species can be differentiated by their arginine metabolism: M1
cells convert L-arginine to nitric oxide by inducible nitric oxide
synthase, whereas the M2 type expresses Arg1 and produces mainly
citrulline. Other relevant end-products in the latter are
polyamines and proline, necessary for tissue repair and
extracellular matrix regeneration. The M2 type cells downregulate
their MHCII molecules, thus decreasing antigen presentation
(74). These observations suggest an
overt pro-inflammatory role for M1, and, in contrast, pro-resolving
abilities for M2 cells.
Macrophage polarization nowadays seems to be a more
complicated process than it was initially modeled. According to a
newer classification, M2 type cells include three functionally
different subtypes: M2a produces anti-inflammatory and trophic
factors, M2b has transitional phenotype between M1 and M2a, whereas
M2c are primarily phagocytes and suppress the innate immune system
(74). Xue et al (75) differentiated macrophages in the
presence of GM-CSF or M-CSF, applying in the second phase of
differentiation 28 different stimuli to analyze their transcription
patterns. They found that when IFNγ + TNFα or IL-10 were added, the
cells maintained their differentiation paths along the M1/M2 axis.
However, when adding stimuli not directly linked to the M1/M2
polarization, they registered a whole spectrum of transcriptional
signatures and suggested that many different macrophage phenotypes
may exist (75). Also, it turned out
that relevant markers of M1 or M2 type activation are somewhat
overlapping: Arg1 expression is strongly upregulated after IL-4
stimulation, which also elicits weak iNOS2 expression, whereas
(lipopolysaccharide) LPS or LPS + IFNγ administration also provoke
Arg1 slightly (9). Moreover, the
gene expression signatures of the in vivo LPS-non-responsive
M2 and in vitro alternatively (IL-4) stimulated macrophages
are only partially shared. Common regulatory pathways comprise
proliferation, apoptosis, differentiation, and arginine metabolism
(both express Arg1) (76). Two
transcription factors competing for the same activators are
responsible for the M1/M2 phenotype switch: cAMP response
element-binding protein (CREB) and nuclear factor-κB (NF-κB) both
link to C/EBP and CBP/p300; CREB mediates the M2 type, while NF-κB
transmits M1 type differentiation. Gsk3β activates NF-κB, but
inhibits CREB; PI3Akt suspends the activation of Gsk3β, thus
pushing the differentiation balance to the M2 phenotype (66).
Some authors even argue that the M1 vs. M2
nomenclature is useless since there is a significant overlap in
their transcriptional profiles, which show no hint of
differentiation-organizing value (77). The nomenclature guidelines now
recommend the exact definition of activators and the use of
multiple markers of activation to characterize the phenotype
(9).
Homeostatic regulatory loops and
imbalance in ischemia
Growing evidence highlights the existence of
bidirectional, versatile communication between neurons and resident
microglia in the animal and human brain. Neurons regulate microglia
through specific ligands, such as CD200 and CX3CL1 (fractalkine),
which attach to their receptors, CD200R, and CX3CL1R. Neurons seem
to influence both the basic and activated state of microglia also
by classical neurotransmitters, e.g., glutamate, and γ-aminobutyric
acid (59,78). Modulation of CD200 signaling was
proposed to promote a phenotype shift of macrophages towards the
arginase-1 producer, reparatory M2 subtype, through the
cAMP-responsive element-binding protein-C/enhancer-binding
protein-β (CREB-C/EBP-β) signaling pathway. Ligand-binding to the
CD200 receptor tyrosine-based inhibitory ITIM-motifs suppresses
downstream signaling through Src homology 2 domain-containing
phosphatase 1 and inhibits the danger-signaling through the pattern
recognition receptors, thus suppressing the development of the
inflammatory medium. In parallel, microglia release neurotrophic
factors and downregulate the expression of MHC II, CD45, and Fc
receptors (74). In this scene,
microglia show the M2 phenotype, halt antigen presentation and stay
in quiescent surveillance of their microcosmos. CD200-Fc treatment
of lipopolysaccharide-triggered rat macrophages upregulates M2
cells while downregulating the M1 subtype and IL-1β, IL-6 and G-CSF
(79).
Cerebral ischemia changes this equilibrium
dramatically. In ischemic conditions, CD200 expression of neurons
is reversely related to their viability, while CD200R is
upregulated in microglia (59). This
time, the monocyte-macrophage network work as a reliable sensor for
many damage-associated, homeostasis-spoiling messengers. Xu et
al (80) recently reported that
elevated levels of plasma macrophage migration inhibitory factor-1
(MIF-1) are a risk for post-stroke depression. MIF-1 is produced in
the pituitary gland constitutively, but especially in response to
the stress of inflammatory stimuli (81,82). The
transcription factor of a danger signal, HMGB1, is a repressor of
MIF-1 and can be targeted by microRNA19a, which has similar effects
of MIF-1, i.e., promotion of vascular inflammation and foam-cell
formation in the atherosclerotic vessel wall (83). Ischemic stroke is associated with the
emergence of neoantigens in the brain, and the myelin-specific,
T-cell-mediated immune response is detrimental in the long-term
post-stroke period (57). According
to Meng et al (85), ATP and
FasL in parallel induce the M1 macrophage phenotype, with the
secretion of IL-1β and MMP-3/MMP-9, and activated microglia
contribute to the inflammatory microenvironment with the production
of TNFα and reactive oxygen species (84). Pro-inflammatory signals also act via
positive feed-back loops: extracellular ATP stress induces the
pattern recognition receptors and triggers NF-κB signaling, which
results in IL-1β, IL-18, and Nod-like receptor pyrin 3 (NLRP3)
transcription. Further, inflammasome NLRP3 facilitates the
caspase-1-mediated enzymatic cleavage of pro-IL-1, amplifying the
pro-inflammatory milieu (74).
Microglia, M1- and M2 type macrophages
can be differentiated in brain tissue
Zarruk et al (64) applied permanent MCAO on LysM-EGFP
(bearing EGFP inserted into the lysosome M locus) knock-in
transgenic mice. According to their setting, microglia can be
identified as
CD11+/CD45+/Ly6G-/LysM-EGFP-
cells, while macrophages are
CD11+/CD45+/Ly6G-/LysM-EGFP+
elements. The authors showed differential expression of Arg1 (1000X
higher in macrophage) and IL-1β (90X increased in macrophages
compared with microglia). Wattananit et al (86) performed interesting long-term
follow-up experiments to differentiate between resident microglia
and infiltrating macrophage effects in cerebral ischemia: They
generated chimeric CX3CR1-GFP mice and used them as bone-marrow and
monocyte donors for transplantation to whole-body irradiated
(except the head) CD57BL animals subjected to MCAO. They found that
GFP-positive bone marrow-derived macrophages invaded the ischemic
lesion and expressed a predominant pro-inflammatory
Ly6Chigh phenotype at three days, then switched to an
anti-inflammatory Ly6Clow preponderance at seven and 14
days, when the cells also highly expressed BDNF. At seven days,
both pro-inflammatory (IL-1β, IL-6, TNFα and NOS) and
anti-inflammatory (TGFβ, CXCL13 and CD163) gene expressions were
high in hemicerebral tissue homogenates, but at 14 days only the
anti-inflammatory set remained upregulated. Selective monocyte
depletion with the anti-MCP-1 antibody at a late stage (seven
weeks) impaired behavior in functional-staircase and corridor
tests, while repressing TGFβ and CD163 at the late stage. These
results indicated that early infiltration of monocytes in the
injured brain is critical for efficient long-term recovery
(86). Horváth et al
(87) recently reported that
microglia/macrophages participate in the cellular infiltrate of the
ischemic rat brain 24 h after transient MCAO, both in the ischemic
core and the surrounding penumbra zone; in the core lesion,
inducible nitric oxide synthase 2-positive microglia/macrophages
dominated, whereas Arg1-positive cells in the penumbra were
upregulated.
Microglial microvesicles and exosomes
in neuroinflammation
Exosomes are membrane-derived, secreted globular
complexes of small diameter, typically 30-100 nm. Microvesicles are
larger, 100-1,000-nm particles that are shed from a variety of
cells (88). Microvesicles have been
proposed as biomarker vehicles in stroke; however, due to their
heterogeneity and methodological limitations, they have been, until
recently, the subject of only small-scale clinical studies.
Endothelial microvesicles reportedly generate in vitro
angiogenesis in an oxidative stress-dependent manner (89). Both exosomes and microvesicle deliver
a complex molecular burden: membrane and cytosolic proteins,
messenger RNA, and miRNA. Leroyer et al (90) demonstrated that microvesicles
obtained from the apoptotic or IL-1β-triggered endothelial cells of
the ischemic limb promote endothelial differentiation of
bone-marrow-derived mononuclear cells, whereas particles obtained
from atherosclerotic plaques are ineffective. In a stroke, reactive
microglia secrete exosomes and microvesicles containing IL-1β,
IL-1β processing enzyme, and the ATP-sensitive P2X7 receptor.
Besides containing soluble factors, these vesicle-packed mediators
also propagate neuroinflammation. Microvesicles possess a complex
signature of miRNAs, consisting of hundreds of regulatory
molecules, among which miR-132 and miR-134 supervise synaptic
plasticity, and miR-124a responds for axonal outgrowth in
regenerating neurons. In the prefrontal cortex of depressive
patients, the vesicle-encapsulated miR-1202 powerfully interferes
with glutamatergic/dopaminergic signaling (88). Moreover, in the peripheral monocytes
of major depressive patients' specific signatures were described
with increased miRNA-26b, miRNA-1972, miRNA-4485, miRNA-4498 and
miRNA-4743; the predicted target genes were linked to biological
processes probably involved in depression, including axon guidance
and extension, synaptic transmission, learning and memory (88).
ATP triggered increased secretion of microvesicles
containing specific mRNA transcript sets for the parental pool in
IL-4 vs. IFNγ and lipopolysaccharide-treated M1- and M2 type
peritoneal macrophages (91). Upon
ATP stimulation, macrophages and microglia released IL-1β and
elements of the inflammasome (91,92).
6. Mediators of neuroinflammation are key
elements in the post-ischemic response
Pro-inflammatory cytokines and
neuroprogression in cerebral ischemia
Substantial evidence indicates that classic
pro-inflammatory cytokines induce neuroprogression
(neurodegeneration, enhanced apoptosis, and low regeneration
capacity of neurons). IL-1β exacerbates neural cell death and
upregulates NMDA receptors (93),
thus impairing hippocampal neurogenesis. Also, IL-1β decreases the
hippocampal expression of a survival factor-neurotrophin tyrosine
kinase and induces the production of free radicals and degradative
MMP in astrocytes and endothelial cells. Other inflammatory
cytokines, e.g., IL-6, seem to have a dual role in neuronal
homeostasis: overexpression of IL-6 in mice causes
neurodegeneration (94). Grønhøj
et al (95) investigated the
effects of intravenously administered IL-6 in parallel with the
soluble IL-6 receptor in mice with permanent MCAO. They found that
IL-6 alone improved motor and sensory functions and did not affect
the size of infarction in wild-type C57BL/6 animals, but it reduced
infarct size in IL-6-/- counterparts.
Moreover, co-administration of IL-6 and IL-6R
increased infarct size 24 h after permanent MCAO, worsened the
motor functions, and triggered the invasion of polymorphonuclear
leucocytes. Increased expression of IL-6 and IL-6R has been
detected in the surviving cortical neurons 72 h after permanent
MCAO (95). A putative pathway of
IL-6 trans-signaling to promote depression is the downregulation of
methyl CpG-binding protein 2 and local melatonin production in
macrophage/microglia (96). TNFα is
involved in the perpetuation of ischemic brain damage by inducing
apoptosis via caspase-mediated pathways and glutaminase
upregulation together with inhibition of glutamine transporter
activity (97). IFNγ is a potent
inducer of indoleamine 2,3-dioxygenase (IDO), with the generation
of multiple neurodegenerative effects through tryptophan catabolic
products (6). In acute human
ischemic stroke, plasma concentrations of IL-6, IL-8 and TNFα were
significantly higher at 72 h, but reduced quantities of IL-1β,
IL-6, IL-8 and TNFα mRNA were detected in peripheral leukocytes,
and the only variation of IL-6 was correlated with the severity and
outcome of stroke (98). IL-1 and
TNFα act synergistically on the stimulation of NF-κB (99). Pro-inflammatory cytokines act via
mitogen-activated protein kinase targets, among which the ERK1/2
kinase pathway is implicated in neuronal functions, such as
plasticity, maintenance, survival, and immune responses of neurons
(100,101).
It has been demonstrated in an endothelin-induced
focal cerebral ischemia model, that peripheral administration of
TNFα can induce IL-4 and IL-10 and suppress IFNγ in the brain if
applied on the innate immune system training background (repeated
administration of LPS) (102). This
striking result indicates that cytokine production in resident
microglia is contextual and should always be interpreted in
conjunction with external inductors and inhibitors. Apart from
this, prolonged elevation of pro-inflammatory mediators determines
neurodegeneration and neuronal loss, and probably preconditions
PSD.
The pathogenic role of proinflammatory cytokines in
post-stroke neuroprogression is synthesized in Fig. 1.
TNFα and IFNγ interfere with
glutamatergic signaling in post-stroke depression
The association between elevated concentrations of
pro-inflammatory cytokines and major depression has often been
proposed (103,104). A meta-analysis found that blood
concentrations of IL-6, TNFα and IL-2R are significantly higher in
major depressive disorder patients than in healthy controls, but
the analysis did not evaluate the relationship between these
cytokines and the severity of depression (105). An early study that linked high
IL-6, TNFα and IFNγ levels with post-stroke depression (106) found a 12-fold increase of IL-6 and
a 40-fold increase of IFNγ in stroke patients who developed
depression in a 12-month follow-up but had no symptoms at the time
of first hospital admission. The association of IL-6 and IFNγ has
an essential functional consequence, since these cytokines both
upregulate the tryptophan-degrading enzyme IDO, thus increasing the
metabolization of tryptophan and reducing the synthesis of
serotonin (107). Neurons,
microglia and infiltrating monocytes all express IDO, but their
catabolic end products are different; neurons metabolize tryptophan
to 5-HT, and astrocytes produce kynurenic acid, whereas the main
end-product in microglia is quinolinic acid, which provokes
glutamate release (108) and
suspends glutamate uptake, resulting in increased quantities of
extrasynaptic glutamate (109).
Quinolinic acid is an NMDA receptor agonist that enhances the
extrasynaptic NMDA receptor response and decreases BDNF production
(7).
Moreover, quinolinic acid increases
nicotinamide-adenine-dinucleotide (NAD+), induces
sirtuin-1 and -3, and finally, inflicts mitochondrial dysfunction
(96). Interestingly, the dispersion
of NMDA receptor and quinolinic acid-producing microglia cells
overlap in the subgenual and dorsal anterior cingulate cortex
(7). In summary, pro-inflammatory
cytokines inhibit the 5-HT synthesis and its neurotrophic effects
in the brain while upregulating tryptophan metabolites, among which
quinolinic acid mainly generates harmful effects: Hippocampal cell
death and neuronal degeneration; excitotoxicity via the NMDA
receptors; inhibition of glutamate uptake; increased production of
reactive oxygen species; and mitochondrial dysfunction (7,110).
Individuals who have acute depressive episodes have higher
quinolinic acid concentrations in their blood and cerebrospinal
fluid (111). On the other hand,
BDNF and 5-HT are ambassadors of synaptic plasticity and neuronal
survival, and they positively influence each other: BDNF stimulates
the growth and survival of 5-HT neurons, whereas 5-HT facilitates
the expression of BDNF in the ischemic brain. The selective 5-HT1A
agonist 8-hydroxy-2-(di-n-propylamino) tetralin suppresses
the phosphorylation of NMDA receptor subunit NR1 at Ser897 and thus
prevents excessive NMDA receptor activation (112).
The alteration of tryptophan metabolism and the
regulatory pathways of glutamate excitotoxicity are shown in
Fig. 2.
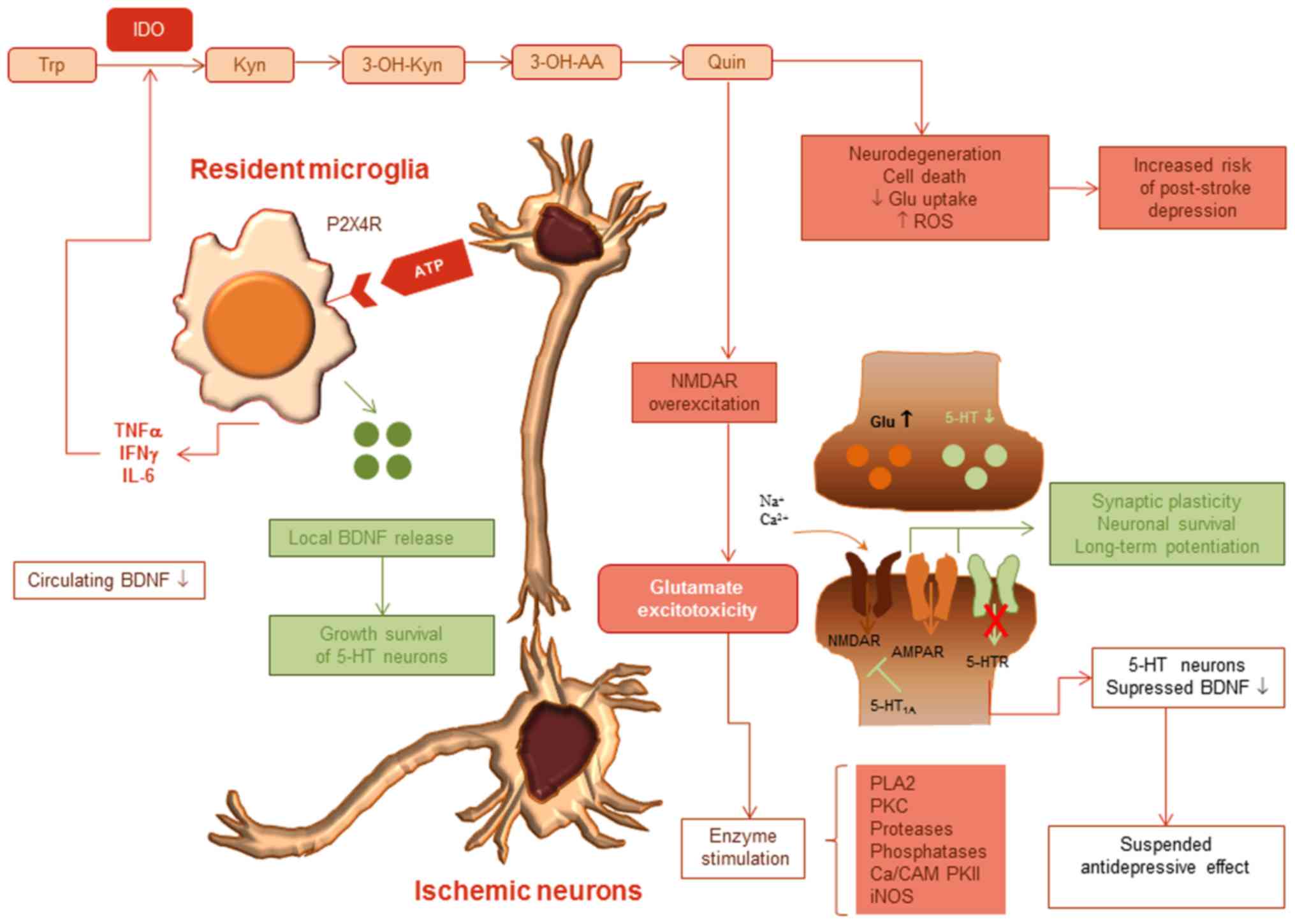 | Figure 2Ischemic neuron-glia cell
interactions alter tryptophane metabolism and induce glutamate
excitotoxicity. ATP-triggered resident microglia releases
pro-inflammatory cytokines IL-6, IFNγ and TNFα. These activate
indoleamine 2,3-dioxygenase, degrading tryptophan to kynurenine,
3-OH kynurenine, 3-OH anthranilic acid and finally, quinolinic
acid. Increased quinolinic acid in the region of injury provokes
neurodegeneration, apoptosis, whereas reactive oxygen species
production excites NMDAR, and mediates glutamate excitotoxicity,
increasing the risk of post-stroke depression. Microglia also
secrete BDNF, which shows high concentration in the ischemic core,
probably in a reparative endeavor, but is downregulated in the
systemic circulation. AMPAR and 5-HT receptors are suppressed,
decreasing synaptic plasticity and neuronal survival. IL,
interleukin; TNFα, tumor necrosis factor α; IFNγ, interferon γ;
BDNF, brain-derived neurotrophic factor. |
The role of other well-known inflammatory cytokines
in PSD is not clear. The link between IL-17 and depression is
contradictory: Davami et al (113) found no relationship of depression
with serum IL-17, whereas Liu et al (105) found elevated levels, but only in
depressed patients with rheumatoid arthritis as a co-morbidity.
Th17 cells and retinoic acid receptor-related orphan receptors, a
transcription factor crucial for the development of depression, can
increase susceptibility to the disorder (114). IL-17-expressing lymphocytes are
elevated as early as one hour after the ischemic trigger in
transient MCAO (115). A study that
profiled the IL-17 mRNA elevation curve in cerebral ischemia found
an elevation after one day, a peak at day 3, and high protein
concentrations persisting at day 6(116).
Osteoprotegerin and RANKL modulate the
post-ischemic inflammatory response and the pro-resolving
microglia/macrophage activation
In a hypoxia-ischemia cellular model, oxygen and
glucose deprivation stimulated TNFα- and IFNγ-mediated cytotoxicity
on neurons and oligodendrocytes. TNF also exerted toxicity on
neurons, but this effect was modulated through its decoy receptors,
osteoprotegerin (OPG), and TNF-related apoptosis-inducing ligand
R2(117). OPG is also a decoy
receptor for the receptor activator of nuclear κB ligand (RANKL),
making part of the regulatory OPG/RANKL/RANK triad and is
significantly increased in acute stroke and other atherosclerotic
manifestations of atherosclerotic polyvascular disease (118,119).
In wild-type mice, OPG, RANKL and RANK, all are overexpressed at
the ischemic border region. In OPG-/- mice, the
RANKL/RANK signaling reduces cerebral edema and infarction volume,
but the effect is the opposite when these signals are repressed.
Baseline mRNA levels of IL-6, TNFα, IL-1β, monocyte chemoattractant
protein-1 MCP-1, iNOS and Arg1, obtained from neuronal-glial mixed
cultures, are low in OPG-/- mice (120). When these animals suffered brain
infarction, IL-6, TNFα, IL-1β, MCP-1, and iNOS concentrations
remained lower, and Arg1 values were higher than in wild-type
counterparts. Thus, via Arg1, RANKL probably is a positive
regulator of M2 type resolution-promoting macrophages. In wild-type
controls, RANKL also reduced IL-1β and MCP-1 but did not affect the
expression of iNOS and Arg1. A possible explanation of this finding
is a more robust alternative activation of macrophages in the
absence of OPG (120). Table I summarizes evidence suggesting the
specific microenvironments and M2 type differentiations with roles
in post-stroke depression.
 | Table ISuggesting evidence for the role of
pro-resolving, M2 type microglia/macrophage in post-stroke
depression. |
Table I
Suggesting evidence for the role of
pro-resolving, M2 type microglia/macrophage in post-stroke
depression.
| Experimental
model | Cytokine/mediator
environment | Host | Effect | Author/Refs. |
|---|
| Middle cerebral
artery occlusion | Anti-CD49d
antibodies |
Perforin-/- mice | Anti-CD49d therapy
depleted infiltrating lymphocytes, Iba-1+ microglia, and
IFNγ secretion | Liesz et al
(55) |
| Transient middle
cerebral artery occlusion | OPG deletion
RANKL? | OPG-/-
mice and wild-type mice | Baseline levels of
IL-6, TNFα, IL-1β, MCP-1, iNOS and Arg1 low After brain infarction,
IL-6, TNFα, IL-1β, MCP-1 remained low, ↑ Arg1 expression in
OPG-/- animals RANKL positive regulator of M2 type
differentiation | Shimamura et
al (120) |
| Peripheral
monocytes of major depression patients | miRNA-26b,
miRNA-1972, miRNA-4485, miRNA-4498, and miRNA-4743
upregulation | Human | Predicted effects:
Impaired axon guidance and extension, synaptic transmission,
learning and memory | Brites and
Fernandes (88) |
| Synthesis on
different animal models and human findings | CD200 | Mice/human | CD200R is
upregulated on microglia, but CD200 signaling fades in ischemic
neuronal injury | Szepesi et
al (59) |
| Peritoneal
macrophage culture | CD200-Fc | Rat | CD200R signaling
via CREB-C/EBP-β promotes Arg1+ microglia
differentiation, silent state, anti-inflammatory phenotype
Upregulation TGFβ, downregulation of IL-1β, IL-6 and G-CSF | Hayakawa et
al (79) |
| Cerebral
ischemia | IL-4 | Mice | Amplification of M2
type differentiation, but no attenuation of the functional outcome
and lesion size | Xia et al
(66) |
| Cerebral
ischemia | IL-4, IL-10,
TGFβ | Mice | M2 type myeloid
cells present intense phagocytosis and increased secretion of IL-4,
IL-10, TGFβ | Xia et al
(66) |
| Transient middle
cerebral artery | IL-4 | C57BL
IL-4-/- | Reversal of M1
type, differentiation | Liu et al
(68) |
| occlusion/permanent
distal middle cerebral artery occlusion | | mice | by IL-4
administration IL-4 beneficial in the long-term recovery | |
| Focal cerebral
ischemia | miRNA-124 | C57BL/6 | Increased number of
Arg1+ microglia/macrophage, increased neuronal
survival | Taj et al
(67) |
| Middle cerebral
artery occlusion | Bone marrow-derived
monocyte transplantation | Chimeric CX3CR1-GFP
and whole body-irradiated C57BL mice | Ly6Clow,
BDNF+ bone-marrow-derived macrophages in the ischemic
lesion at 14 days High TGFβ, CXCL13 and CD163 expression in
hemicerebral tissue homogenate | Wattananit et
al (86) |
| Middle cerebral
artery occlusion | Anti-MCP-1 | Chimeric CX3CR1-GFP
and whole body-irradiated C57BL mice | Selective monocyte
depletion with anti-MCP1 antibody resulted in impaired functional
tests in the late-stage recovery (at 7 days post-stroke) | Wattananit et
al (86) |
| Intracerebral
hemorrhage | IL-10 CD28SA | C57BL/6 male
mice | IL-10-treated Tregs
support M2 microglia/macrophage differentiation through glycogen
synthase kinase 3β and phosphatase and tensin homolog | |
| | | | Boosting Tregs with
CD28 agonist enhances | Zhou et al
(69) |
| | | | M2 type
differentiation, TGFβ, and IL-10 producer cells | |
| Murine ischemic
stroke | MSR-1 deletion in
infiltrating myeloid cells | Mice | HMGB1 and PRX are
internalized in vitro through MSR-1 MSR-1 and Marco
deficiency impairs clearance of DAMPs, exacerbates inflammation and
neuronal injury | Shichita et
al (65) |
7. Experimental evidence for pharmacological
checkpoints of inflammation
MMPs are critical regulators of neuronal
death/survival and tissue regeneration. The overexpression of MMP-9
was documented in the ischemic brain, and a causal relationship
with the disruption of the blood-brain barrier and hemorrhagic
transformation has been proposed (121). In a global cerebral ischemia model,
the cyclooxygenase 2 inhibitor robenacoxib, when administered
alone, enhanced neuronal death rather than protecting the cells
(122); however, when administered
together with salubrinal, an endoplasmatic reticulum stress
inhibitor, robenacoxib diminished neuronal loss and glial
activation. 2-Hydroxyarachidonic acid, a cyclooxygenase inhibitor,
acts suspending also on phospholipase A2, a mediator of membrane
phospholipid cleavage, and decreases infarct volume in rats
(123). In a carotid embolism
stroke model, atorvastatin and meloxicam reduced the inflammatory
process, neurodegeneration, and morphological changes specific for
astrocytes and microglia (124).
Llorente et al (125)
examined whether meloxicam protects against glutamatergic
excitotoxicity in cultures of organotypic hippocampal slices. They
found decreased mRNA synthesis of glutamatergic transporters
VGLUT1, VGLUT2, GLAST-1A, GLT-1 and EAAC-1 and some receptor
subunits, but not of membrane transporters. They concluded that the
drug could selectively modulate the expression of the glutamatergic
signaling system components along the NMDA: AMPA receptor
stoichiometry and repress glutamate excitotoxicity, leading to
neuronal death. Meloxicam represses low-grade inflammation, even in
non-vascularized tissues (126),
and has a dual beneficial effect on extracellular matrix repair.
Through its anti-fibrotic effect, downregulates the hydroxyproline
production and collagen deposition, and it is angiostatic and
anti-oxidant through stimulation of glutathione peroxidase,
catalase, superoxide dismutase, and decrease of lipid peroxidation
and myeloperoxidase activity (127,128).
In a model of kidney interstitial fibrosis, meloxicam inhibited
type IV collagen mRNA together with the heat shock protein hsp47,
which are indispensable for collagen synthesis, ERK and JNK kinases
(129). The anti-fibrotic and
anti-collagenolytic effects were also evident in rat liver and
cartilage, where meloxicam downregulated collagen type I and II
degradation, smooth muscle actin α, hydroxyproline, and,
interestingly, TGFβ along with tissue inhibitor of MMP-1 (TIMP-1)
(128,130). Linalool, a monoterpene, was
effective in treating glutamate excitotoxicity by improving the
altered profiles of mono-/polyunsaturated fatty acids in membrane
phospholipids, reducing microgliosis and cyclooxygenase-2 (Cox-2)
expression, and by improving motor and cognitive performances of
ischemic Wistar rats (131). Our
recent research (132,133) revealed that fish oil with high
eicosapentaenoic acid content triggered an M2/M1 macrophage
phenotype shift in the spleen and bone marrow of rats exposed to
transient MCAO. C-phycocyanin and phycocyanobilin, the
chromoproteins of cyanobacteria Spirulina platensis, exert
beneficial effects in ischemic animal models. Besides inhibition of
Cox-2 and upregulation of BDNF expression, they limit the
proinflammatory mediators IL-17A, IL-1β and TNF-α, promote the
differentiation of oligodendrocytes, and support the repair of
ischemic demyelination (134).
CR2-Crry, an overall inhibitor of complement
pathways, and CR2-fH, an inhibitor of the alternative pathway,
reduce microglia/macrophage activation, infarct size, and improve
neurological scores in mice in the acute post-stroke phase. In the
subacute phase, only the alternative pathway inhibitor improved
neurological deficit, neurogenesis, and neuronal migration and also
downregulated neutrophil infiltration, IL-1α, and a series of
matrix metalloproteinases; protection against cell death was
especially evident in the hippocampus (135). Cox-2 inhibition with rofecoxib
increases 5-HT levels in the prefrontal and parietal cortex.
Another Cox-2 inhibitor, celecoxib, efficiently decreases the
hypothalamus proinflammatory cytokine levels and the behavioral
impairment in experimental models (136). Celecoxib, combined with
antidepressants, such as fluoxetine or sertraline, exerts a more
substantial anti-depressive effect than the antidepressant alone
(137,138). Anti-TNF therapy is effective in
reducing neuroinflammation (139).
Suppression of inflammatory signs was also observed in HIV-1
transgenic adolescent rats, with signs of depression and high
expression of MCP-1 in the hippocampus; oral meloxicam treatment
reduces MCP-1 but does not reverse the depressive behavior
(140). In an exciting new
approach, Liu et al (141)
applied 50% argon/50% oxygen on Wistar rats supposed to tMCAO, 3 h
after starting the procedure, and 1 h after reperfusion. They
observed the gas mixture improved significantly a 6-point
neuroscore, neuronal survival (measured as the intensity of NeuN
expression), and a robust polarization of macrophages to the
Iba-1+/Arg1+ M2 form.
8. Discussion
Post-stroke depression occurs in about one-third of
stroke patients; depression decreases the quality of life, predicts
the recurrence of stroke, and increases the mortality rate. In PSD,
many studies have reported disruption of frontal cortico-limbic
circuits, although this functional dysequilibrium is probably a
consequence of the post-stroke immune response and
neuroinflammation.
Brain-derived neurotrophic factor, synthesized in
neurons, astrocytes, and microglia, is essential for neuronal
survival, plasticity and may play an essential role in the recovery
phase of stroke. Antidepressant treatments restore BDNF levels,
which, interestingly, also are increased in the ischemic core of
rats with experimental stroke. The release of BDNF is tightly
linked to the activation of ATP-sensitive purinergic P2X4R
receptors, which are dense on resident microglia and infiltrating
macrophages. The importance of purinergic signaling in stroke and
PSD is reflected by the fact that global knock-out of P2X4R reduces
infarct size, whereas myeloid-specific deletion causes anhedonia
and depressive behavior in the late-stage.
Danger signals released in the acute phase of
stroke transmit glutamate excitotoxicity via NMDA receptors. In the
acute phase, ischemia turns on inflammatory mechanisms: neutrophils
and, later, monocytes invade the lesion due to chemotactic signals,
such as CX3CL1 and CCL2. Pro-inflammatory cytokines, especially
IL-1β, TNFα, IL-6 and IFNγ, elicit neuronal degeneration,
apoptosis, and low regeneration capacity, and interfere with
tryptophan catabolism. The main degradation product of tryptophan
in microglia is quinolinic acid, an NMDA receptor agonist and 5-HT
antagonist that exerts glutamate excitotoxicity, suppressing
glutamate uptake, reducing synaptic plasticity and neuronal
survival. Experimental studies have revealed that the
anti-inflammatory cytokines IL-4, IL-10 and TGFβ are protective in
stroke by limiting the lesions and determining macrophages to M2
type differentiation. In the chronic recovery phase, resident
microglia and macrophages probably have a central role in
modulating tissue repair, BDNF signaling, 5-HT neurotransmission
and thus the development of post-stroke depression. However,
according to recent observations, these cells differ in their
behavior. In essence, resident microglia in some circumstances seem
to be silent, other times pro-inflammatory, whereas
bone-marrow-derived infiltrating monocytes at the late recovery
stage are anti-inflammatory and may play a critical role in the
late-phase tissue repair and neurological rehabilitation. Some
ambiguity persists in the nomenclature of pro-resolving
macrophages, and there is a clear need for the accurate description
of their stimuli and extended phenotype. Moreover, since it has
been documented that repeated peripheral pro-inflammatory stimuli
can train microglia and downregulate their cytokine expression,
future studies dedicated to the mapping of brain-repair factors
need to address in parallel both the local and peripheric immune
response.
Acknowledgements
Not applicable.
Funding
This study was supported by an internal research
grant of the ‘Emil Palade’ University of Medicine, Pharmacy,
Science and Technology of Targu Mures, Romania (no.
17803/1/22.12.2015) and partially funded by the Studium-Prospero
Foundation, Romania (contract no. 1547/18.12.2015).
Availability of data and materials
Not applicable.
Authors' contributions
EEN conceived the study, acquired the data and
wrote the manuscript. AF and JAS acquired the data and revised the
manuscript critically for important intellectual content. EH was
also involved in the conception of the study, was responsible for
the acquisition of funding and the critical revision of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Staub F and Bogousslavsky J: Post-stroke
depression or fatigue. Eur Neurol. 45:3–5. 2001.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gaete JM and Bogousslavsky J: Post-stroke
depression. Expert Rev Neurother. 8:75–92. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Paolucci S, Iosa M, Coiro P, Venturiero V,
Savo A, De Angelis D and Morone G: Post-stroke depression increases
disability more than 15% in ischemic stroke survivors: A
case-control study. Front Neurol. 10(926)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ayerbe L, Ayis S, Wolfe CD and Rudd AG:
Natural history, predictors and outcomes of depression after
stroke: Systematic review and meta-analysis. Br J Psychiatry.
202:14–21. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sibolt G, Curtze S, Melkas S, Pohjasvaara
T, Kaste M, Karhunen PJ, Oksala NK, Vataja R and Erkinjuntti T:
Post-stroke depression and depression-executive dysfunction
syndrome are associated with recurrence of ischaemic stroke.
Cerebrovasc Dis. 36:336–343. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Leonard B and Maes M: Mechanistic
explanations how cell-mediated immune activation, inflammation and
oxidative and nitrosative stress pathways and their sequels and
concomitants play a role in the pathophysiology of unipolar
depression. Neurosci Biobehav Rev. 36:764–785. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Woelfer M, Kasties V, Kahlfuss S and
Walter M: The role of depressive subtypes within the
neuroinflammation hypothesis of major depressive disorder.
Neuroscience. 403:93–110. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Maes M: A review on the acute phase
response in major depression. Rev Neurosci. 4:407–416.
1993.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Murray PJ, Allen JE, Biswas SK, Fisher EA,
Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence
T, et al: Macrophage activation and polarization: Nomenclature and
experimental guidelines. Immunity. 41:14–20. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Robinson RG and Jorge RE: Post-stroke
depression: A review. Am J Psychiatry. 173:221–231. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Folstein MF, Maiberger R and McHugh PR:
Mood disorder as a specific complication of stroke. J Neurol
Neurosurg Psychiatry. 40:1018–1020. 1977.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Narushima K, Kosier JT and Robinson RG: A
reappraisal of poststroke depression, intra- and inter-hemispheric
lesion location using meta-analysis. J Neuropsychiatry Clin
Neurosci. 15:422–430. 2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Spalletta G, Bossù P, Ciaramella A, Bria
P, Caltagirone C and Robinson RG: The etiology of poststroke
depression: A review of the literature and a new hypothesis
involving inflammatory cytokines. Mol Psychiatry. 11:984–991.
2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Carson AJ, MacHale S, Allen K, Lawrie SM,
Dennis M, House A and Sharpe M: Depression after stroke and lesion
location: A systematic review. Lancet. 356:122–126. 2000.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kutlubaev MA and Hackett ML: Part II:
predictors of depression after stroke and impact of depression on
stroke outcome: an updated systematic review of observational
studies. Int J Stroke. 9:1026–1036. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
MacHale SM, O'Rourke SJ, Wardlaw JM and
Dennis MS: Depression and its relation to lesion location after
stroke. J Neurol Neurosurg Psychiatry. 64:371–374. 1998.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Nys GM, van Zandvoort MJ, van der Worp HB,
de Haan EH, de Kort PL and Kappelle LJ: Early depressive symptoms
after stroke: Neuropsychological correlates and lesion
characteristics. J Neurol Sci. 228:27–33. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Terroni L, Amaro E Jr, Iosifescu DV,
Tinone G, Sato JR, Leite CC, Sobreiro MF, Lucia MC, Scaff M and
Fráguas R: Stroke lesion in cortical neural circuits and
post-stroke incidence of major depressive episode: A 4-month
prospective study. World J Biol Psychiatry. 12:539–548.
2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Morris PL, Robinson RG, de Carvalho ML,
Albert P, Wells JC, Samuels JF, Eden-Fetzer D and Price TR: Lesion
characteristics and depressed mood in the stroke data bank study. J
Neuropsychiatry Clin Neurosci. 8:153–159. 1996.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kim NY, Lee SC, Shin JC, Park JE and Kim
YW: Voxel-based lesion symptom mapping analysis of depressive mood
in patients with isolated cerebellar stroke: A pilot study.
Neuroimage Clin. 13:39–45. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nishiyama Y, Komaba Y, Ueda M, Nagayama H,
Amemiya S and Katayama Y: Early depressive symptoms after ischemic
stroke are associated with a left lenticulocapsular area lesion. J
Stroke Cerebrovasc Dis. 19:184–189. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang T, Jing X, Zhao X, Wang C, Liu Z,
Zhou Y and Wang Y and Wang Y: A prospective cohort study of lesion
location and its relation to post-stroke depression among Chinese
patients. J Affect Disord. 136:e83–e87. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gozzi SA, Wood AG, Chen J, Vaddadi K and
Phan TG: Imaging predictors of poststroke depression:
Methodological factors in voxel-based analysis. BMJ Open.
4(e004948)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Grajny K, Pyata H, Spiegel K, Lacey EH,
Xing S, Brophy C and Turkeltaub PE: depression symptoms in chronic
left hemisphere stroke are related to dorsolateral prefrontal
cortex damage. J Neuropsychiatry Clin Neurosci. 28:292–298.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shi YZ, Xiang YT, Yang Y, Zhang N, Wang S,
Ungvari GS, Chiu HF, Tang WK, Wang YL, Zhao XQ, et al: Depression
after minor stroke: The association with disability and quality of
life - a 1-year follow-up study. Int J Geriatr Psychiatry.
31:421–427. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yu H and Chen ZY: The role of BDNF in
depression on the basis of its location in the neural circuitry.
Acta Pharmacol Sin. 32:3–11. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Elkabes S, DiCicco-Bloom EM and Black IB:
Brain microglia/macrophages express neurotrophins that selectively
regulate microglial proliferation and function. J Neurosci.
16:2508–2521. 1996.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Knott C, Stern G, Kingsbury A, Welcher AA
and Wilkin GP: Elevated glial brain-derived neurotrophic factor in
Parkinson's diseased nigra. Parkinsonism Relat Disord. 8:329–341.
2002.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Stadelmann C, Kerschensteiner M, Misgeld
T, Brück W, Hohlfeld R and Lassmann H: BDNF and gp145trkB in
multiple sclerosis brain lesions: Neuroprotective interactions
between immune and neuronal cells? Brain. 125:75–85.
2002.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Trang T, Beggs S, Wan X and Salter MW:
P2X4-receptor-mediated synthesis and release of brain-derived
neurotrophic factor in microglia is dependent on calcium and
p38-mitogen-activated protein kinase activation. J Neurosci.
29:3518–3528. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Song X, Zhou B, Zhang P, Lei D, Wang Y,
Yao G, Hayashi T, Xia M, Tashiro S, Onodera S, et al: Protective
effect of silibinin on learning and memory impairment in
LPS-treated rats via ROS-BDNF-TrkB pathway. Neurochem Res.
41:1662–1672. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Smith MA, Makino S, Kvetnansky R and Post
RM: Stress and glucocorticoids affect the expression of
brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the
hippocampus. J Neurosci. 15:1768–1777. 1995.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lang UE, Hellweg R, Kalus P, Bajbouj M,
Lenzen KP, Sander T, Kunz D and Gallinat J: Association of a
functional BDNF polymorphism and anxiety-related personality
traits. Psychopharmacology (Berl). 180:95–99. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Monteggia LM, Luikart B, Barrot M,
Theobold D, Malkovska I, Nef S, Parada LF and Nestler EJ:
Brain-derived neurotrophic factor conditional knockouts show gender
differences in depression-related behaviors. Biol Psychiatry.
61:187–197. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Duman RS: Pathophysiology of depression:
The concept of synaptic plasticity. Eur Psychiatry. 17 (Suppl
3):306–310. 2002.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Fujimura H, Altar CA, Chen R, Nakamura T,
Nakahashi T, Kambayashi J, Sun B and Tandon NN: Brain-derived
neurotrophic factor is stored in human platelets and released by
agonist stimulation. Thromb Haemost. 87:728–734. 2002.PubMed/NCBI
|
|
37
|
Lee BH and Kim YK: The roles of BDNF in
the pathophysiology of major depression and in antidepressant
treatment. Psychiatry Investig. 7:231–235. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Pan W, Banks WA, Fasold MB, Bluth J and
Kastin AJ: Transport of brain-derived neurotrophic factor across
the blood-brain barrier. Neuropharmacology. 37:1553–1561.
1998.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Gonul AS, Akdeniz F, Taneli F, Donat O,
Eker C and Vahip S: Effect of treatment on serum brain-derived
neurotrophic factor levels in depressed patients. Eur Arch
Psychiatry Clin Neurosci. 255:381–386. 2005.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chaudhury D, Liu H and Han MH: Neuronal
correlates of depression. Cell Mol Life Sci. 72:4825–4848.
2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Jin Y, Sun LH, Yang W, Cui RJ and Xu SB:
The role of BDNF in the neuroimmune axis regulation of mood
disorders. Front Neurol. 10(515)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Jiang MQ, Zhao YY, Cao W, Wei ZZ, Gu X,
Wei L and Yu SP: Long-term survival and regeneration of neuronal
and vasculature cells inside the core region after ischemic stroke
in adult mice. Brain Pathol. 27:480–498. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
North RA and Jarvis MF: P2X receptors as
drug targets. Mol Pharmacol. 83:759–769. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Vázquez-Villoldo N, Domercq M, Martín A,
Llop J, Gómez-Vallejo V and Matute C: P2X4 receptors control the
fate and survival of activated microglia. Glia. 62:171–184.
2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Verma R, Cronin CG, Hudobenko J, Venna VR,
McCullough LD and Liang BT: Deletion of the P2X4 receptor is
neuroprotective acutely, but induces a depressive phenotype during
recovery from ischemic stroke. Brain Behav Immun. 66:302–312.
2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bravo-Alegria J, McCullough LD and Liu F:
Sex differences in stroke across the lifespan: The role of T
lymphocytes. Neurochem Int. 107:127–137. 2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Xu S, Lu J, Shao A, Zhang JH and Zhang J:
Glial cells: Role of the immune response in ischemic stroke. Front
Immunol. 11(294)2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zera KA and Buckwalter MS: The Local and
peripheral immune responses to stroke: Implications for therapeutic
development. Neurotherapeutics: Mar 19, 2020 (Epub ahead of print).
doi: 10.1007/s13311-020-00844-3.
|
|
49
|
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou
L, Giffard RG and Barres BA: Genomic analysis of reactive
astrogliosis. J Neurosci. 32:6391–6410. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhao X, Wang H, Sun G, Zhang J, Edwards NJ
and Aronowski J: Neuronal interleukin-4 as a modulator of
microglial pathways and ischemic brain damage. J Neurosci.
35:11281–11291. 2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Lee GA, Lin TN, Chen CY, Mau SY, Huang WZ,
Kao YC, Ma RY and Liao NS: Interleukin 15 blockade protects the
brain from cerebral ischemia-reperfusion injury. Brain Behav Immun.
73:562–570. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Dolati S, Ahmadi M, Khalili M, Taheraghdam
AA, Siahmansouri H, Babaloo Z, Aghebati-Maleki L, Jadidi-Niaragh F,
Younesi V and Yousefi M: Peripheral Th17/Treg imbalance in elderly
patients with ischemic stroke. Neurol Sci. 39:647–654.
2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Santamaría-Cadavid M, Rodríguez-Castro E,
Rodríguez-Yáñez M, Arias-Rivas S, López-Dequidt I, Pérez-Mato M,
Rodríguez-Pérez M, López-Loureiro I, Hervella P, Campos F, et al:
Regulatory T cells participate in the recovery of ischemic stroke
patients. BMC Neurol. 20(68)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Stubbe T, Ebner F, Richter D, Engel O,
Klehmet J, Royl G, Meisel A, Nitsch R, Meisel C and Brandt C:
Regulatory T cells accumulate and proliferate in the ischemic
hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab.
33:37–47. 2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Liesz A, Zhou W, Mracskó É, Karcher S,
Bauer H, Schwarting S, Sun L, Bruder D, Stegemann S, Cerwenka A, et
al: Inhibition of lymphocyte trafficking shields the brain against
deleterious neuroinflammation after stroke. Brain. 134:704–720.
2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Doyle KP, Quach LN, Solé M, Axtell RC,
Nguyen TV, Soler-Llavina GJ, Jurado S, Han J, Steinman L, Longo FM,
et al: B-lymphocyte-mediated delayed cognitive impairment following
stroke. J Neurosci. 35:2133–2145. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Rayasam A, Hsu M, Hernández G, Kijak J,
Lindstedt A, Gerhart C, Sandor M and Fabry Z: Contrasting roles of
immune cells in tissue injury and repair in stroke: The dark and
bright side of immunity in the brain. Neurochem Int. 107:104–116.
2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Nakamura K and Shichita T: Cellular and
molecular mechanisms of sterile inflammation in ischaemic stroke. J
Biochem. 165:459–464. 2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Szepesi Z, Manouchehrian O, Bachiller S
and Deierborg T: Bidirectional microglia-neuron communication in
health and disease. Front Cell Neurosci. 12(323)2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Sharp FR, Zhan X and Liu DZ: Heat shock
proteins in the bra in: Role of Hsp70, Hsp 27, and HO-1 (Hsp32) and
their therapeutic potential. Transl Stroke Res. 4:685–692.
2013.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Nitti M, Piras S, Brondolo L, Marinari UM,
Pronzato MA and Furfaro AL: Heme Oxygenase 1 in the nervous system:
Does it favor neuronal cell survival or induce neurodegeneration?
Int J Mol Sci. 19(E2260)2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Hermann DM, Kleinschnitz C and Gunzer M:
Role of polymorphonuclear neutrophils in the reperfused ischemic
brain: Insights from cell-type-specific immunodepletion and
fluorescence microscopy studies. Ther Adv Neurol Disord.
11(1756286418798607)2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Bennett ML, Bennett FC, Liddelow SA, Ajami
B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A,
Tucker A, et al: New tools for studying microglia in the mouse and
human CNS. Proc Natl Acad Sci USA. 113:E1738–E1746. 2016.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Zarruk JG, Greenhalgh AD and David S:
Microglia and macrophages differ in their inflammatory profile
after permanent brain ischemia. Exp Neurol. 301:120–132.
2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Shichita T, Ito M, Morita R, Komai K,
Noguchi Y, Ooboshi H, Koshida R, Takahashi S, Kodama T and
Yoshimura A: MAFB prevents excess inflammation after ischemic
stroke by accelerating clearance of damage signals through MSR1.
Nat Med. 23:723–732. 2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Xia CY, Zhang S, Gao Y, Wang ZZ and Chen
NH: Selective modulation of microglia polarization to M2 phenotype
for stroke treatment. Int Immunopharmacol. 25:377–382.
2015.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Taj HS, Kho W, Riou A, Wiedermann D and
Hoehn M: MiRNA-124 induces neuroprotection and functional
improvement after focal cerebral ischemia. Biomaterials.
91:151–165. 2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai
M, Ji X, Leak RK, Gao Y, Chen J, et al: Interleukin-4 is essential
for microglia/macrophage M2 polarization and long-term recovery
after cerebral ischemia. Stroke. 47:498–504. 2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Zhou K, Zhong Q, Wang YC, Xiong XY, Meng
ZY, Zhao T, Zhu WY, Liao MF, Wu LR, Yang YR, et al: Regulatory T
cells ameliorate intracerebral hemorrhage-induced inflammatory
injury by modulating microglia/macrophage polarization through the
IL-10/GSK3β/PTEN axis. J Cereb Blood Flow Metab. 37:967–979.
2017.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Titova EM, Ghosh N, Valadez ZG, Zhang JH,
Bellinger DL and Obenaus A: The late phase of post-stroke
neurorepair in aged rats is reflected by MRI-based measures.
Neuroscience. 283:231–244. 2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Ongür D, Drevets WC and Price JL: Glial
reduction in the subgenual prefrontal cortex in mood disorders.
Proc Natl Acad Sci USA. 95:13290–13295. 1998.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Nimmerjahn A, Kirchhoff F and Helmchen F:
Resting microglial cells are highly dynamic surveillants of brain
parenchyma in vivo. Science. 308:1314–1318. 2005.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Smith RS: The macrophage theory of
depression. Med Hypotheses. 35:298–306. 1991.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Dey A and Hankey Giblin PA: Insights into
macrophage heterogeneity and cytokine-induced neuroinflammation in
major Ddepressive disorder. Pharmaceuticals (Basel).
11(E64)2018.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Xue J, Schmidt SV, Sander J, Draffehn A,
Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L,
et al: Transcriptome-based network analysis reveals a spectrum
model of human macrophage activation. Immunity. 40:274–288.
2014.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Orecchioni M, Ghosheh Y, Pramod AB and Ley
K: Macrophage polarization: Different gene signatures in M1(LPS+)
vs. classically and M2(LPS-) vs. alternatively activated
macrophages. Front Immunol. 10(1084)2019.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Ransohoff RM: A polarizing question: Do M1
and M2 microglia exist? Nat Neurosci. 19:987–991. 2016.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Eyo UB and Wu LJ: Bidirectional
microglia-neuron communication in the healthy brain. Neural Plast.
2013(456857)2013.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Hayakawa K, Wang X and Lo EH: CD200
increases alternatively activated macrophages through cAMP-response
element binding protein-C/EBP-beta signaling. J Neurochem.
136:900–906. 2016.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Xu T, Pu S, Ni Y, Gao M, Li X and Zeng X:
Elevated plasma macrophage migration inhibitor factor as a risk
factor for the development of post-stroke depression in ischemic
stroke. J Neuroimmunol. 320:58–63. 2018.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Yang DB, Yu WH, Dong XQ, Zhang ZY, Du Q,
Zhu Q, Che ZH, Wang H, Shen YF and Jiang L: Serum macrophage
migration inhibitory factor concentrations correlate with prognosis
of traumatic brain injury. Clin Chim Acta. 469:99–104.
2017.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Leyton-Jaimes MF, Kahn J and Israelson A:
Macrophage migration inhibitory factor: A multifaceted cytokine
implicated in multiple neurological diseases. Exp Neurol.
301:83–91. 2018.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Chen H, Li X, Liu S, Gu L and Zhou X:
MircroRNA-19a promotes vascular inflammation and foam cell
formation by targeting HBP-1 in atherogenesis. Sci Rep.
7(12089)2017.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Ritzel RM, Patel AR, Grenier JM, Crapser
J, Verma R, Jellison ER and McCullough LD: Functional differences
between microglia and monocytes after ischemic stroke. J
Neuroinflammation. 12(106)2015.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Meng HL, Li XX, Chen YT, Yu LJ, Zhang H,
Lao JM, Zhang X and Xu Y: Neuronal soluble Fas ligand drives
M1-Microglia polarization after cerebral ischemia. CNS Neurosci
Ther. 22:771–781. 2016.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Wattananit S, Tornero D, Graubardt N,
Memanishvili T, Monni E, Tatarishvili J, Miskinyte G, Ge R,
Ahlenius H, Lindvall O, et al: Monocyte-derived macrophages
contribute to spontaneous long-term functional recovery after
stroke in mice. J Neurosci. 36:4182–4195. 2016.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Horváth E, Huțanu A, Chiriac L, Dobreanu
M, Orădan A and Nagy EE: Ischemic damage and early inflammatory
infiltration are different in the core and penumbra lesions of rat
brain after transient focal cerebral ischemia. J Neuroimmunol.
324:35–42. 2018.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Brites D and Fernandes A:
Neuroinflammation and depression: Microglia activation,
extracellular microvesicles and microRNA dysregulation. Front Cell
Neurosci. 9(476)2015.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Mezentsev A, Merks RM, O'Riordan E, Chen
J, Mendelev N, Goligorsky MS and Brodsky SV: Endothelial
microparticles affect angiogenesis in vitro: Role of oxidative
stress. Am J Physiol Heart Circ Physiol. 289:H1106–H1114.
2005.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Leroyer AS, Ebrahimian TG, Cochain C,
Récalde A, Blanc-Brude O, Mees B, Vilar J, Tedgui A, Levy BI,
Chimini G, et al: Microparticles from ischemic muscle promotes
postnatal vasculogenesis. Circulation. 119:2808–2817.
2009.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Garzetti L, Menon R, Finardi A, Bergami A,
Sica A, Martino G, Comi G, Verderio C, Farina C and Furlan R:
Activated macrophages release microvesicles containing polarized M1
or M2 mRNAs. J Leukoc Biol. 95:817–825. 2014.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Bianco F, Pravettoni E, Colombo A, Schenk
U, Möller T, Matteoli M and Verderio C: Astrocyte-derived ATP
induces vesicle shedding and IL-1 beta release from microglia. J
Immunol. 174:7268–7277. 2005.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Viviani B, Gardoni F, Bartesaghi S,
Corsini E, Facchi A, Galli CL, Di Luca M and Marinovich M:
Interleukin-1 beta released by gp120 drives neural death through
tyrosine phosphorylation and trafficking of NMDA receptors. J Biol
Chem. 281:30212–30222. 2006.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Allan SM and Rothwell NJ: Inflammation in
central nervous system injury. Philos Trans R Soc Lond B Biol Sci.
358:1669–1677. 2003.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Grønhøj MH, Clausen BH, Fenger CD,
Lambertsen KL and Finsen B: Beneficial potential of intravenously
administered IL-6 in improving outcome after murine experimental
stroke. Brain Behav Immun. 65:296–311. 2017.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Anderson G, Kubera M, Duda W, Lasoń W,
Berk M and Maes M: Increased IL-6 trans-signaling in depression:
Focus on the tryptophan catabolite pathway, melatonin and
neuroprogression. Pharmacol Rep. 65:1647–1654. 2013.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Zou JY and Crews FT: TNF alpha potentiates
glutamate neurotoxicity by inhibiting glutamate uptake in
organotypic brain slice cultures: Neuroprotection by NF kappa B
inhibition. Brain Res. 1034:11–24. 2005.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Zeng L, Wang Y, Liu J, Wang L, Weng S,
Chen K, Domino EF and Yang GY: Pro-inflammatory cytokine network in
peripheral inflammation response to cerebral ischemia. Neurosci
Lett. 548:4–9. 2013.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Kaminska B: MAPK signalling pathways as
molecular targets for anti-inflammatory therapy - from molecular
mechanisms to therapeutic benefits. Biochim Biophys Acta.
1754:253–262. 2005.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Hetman M and Gozdz A: Role of
extracellular signal regulated kinases 1 and 2 in neuronal
survival. Eur J Biochem. 271:2050–2055. 2004.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Subramaniam S and Unsicker K: ERK and cell
death: ERK1/2 in neuronal death. FEBS J. 277:22–29. 2010.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Wendeln AC, Degenhardt K, Kaurani L,
Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A,
et al: Innate immune memory in the brain shapes neurological
disease hallmarks. Nature. 556:332–338. 2018.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Schiepers OJG, Wichers MC and Maes M:
Cytokines and major depression. Prog Neuropsychopharmacol Biol
Psychiatry. 29:201–217. 2005.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Dowlati Y, Herrmann N, Swardfager W, Liu
H, Sham L, Reim EK and Lanctôt KL: A meta-analysis of cytokines in
major depression. Biol Psychiatry. 67:446–457. 2010.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Liu Y, Ho RCM and Mak A: The role of
interleukin (IL)-17 in anxiety and depression of patients with
rheumatoid arthritis. Int J Rheum Dis. 15:183–187. 2012.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Su JA, Chou SY, Tsai CS and Hung TH:
Cytokine changes in the pathophysiology of poststroke depression.
Gen Hosp Psychiatry. 34:35–39. 2012.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Yirmiya R, Rimmerman N and Reshef R:
Depression as a microglial disease. Trends Neurosci. 38:637–658.
2015.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Myint AM, Kim YK, Verkerk R, Scharpé S,
Steinbusch H and Leonard B: Kynurenine pathway in major depression:
Evidence of impaired neuroprotection. J Affect Disord. 98:143–151.
2007.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Tilleux S and Hermans E: Neuroinflammation
and regulation of glial glutamate uptake in neurological disorders.
J Neurosci Res. 85:2059–2070. 2007.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Tavares RG, Tasca CI, Santos CE, Alves LB,
Porciúncula LO, Emanuelli T and Souza DO: Quinolinic acid
stimulates synaptosomal glutamate release and inhibits glutamate
uptake into astrocytes. Neurochem Int. 40:621–627. 2002.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Miller AH, Maletic V and Raison CL:
Inflammation and its discontents: The role of cytokines in the
pathophysiology of major depression. Biol Psychiatry. 65:732–741.
2009.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Salazar-Colocho P, Del Río J and Frechilla
D: Neuroprotective effects of serotonin 5-HT 1A receptor activation
against ischemic cell damage in gerbil hippocampus: Involvement of
NMDA receptor NR1 subunit and BDNF. Brain Res. 1199:159–166.
2008.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Davami MH, Baharlou R, Ahmadi Vasmehjani
A, Ghanizadeh A, Keshtkar M, Dezhkam I and Atashzar MR: Elevated
IL-17 and TGF-β serum levels: A positive correlation between
T-helper 17 cell-related pro-inflammatory responses with major
depressive disorder. Basic Clin Neurosci. 7:137–142.
2016.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Beurel E, Harrington LE and Jope RS:
Inflammatory T helper 17 cells promote depression-like behavior in
mice. Biol Psychiatry. 73:622–630. 2013.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Li HL, Kostulas N, Huang YM, Xiao BG, van
der Meide P, Kostulas V, Giedraitas V and Link H: IL-17 and
IFN-gamma mRNA expression is increased in the brain and
systemically after permanent middle cerebral artery occlusion in
the rat. J Neuroimmunol. 116:5–14. 2001.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Zhang J, Mao X, Zhou T, Cheng X and Lin Y:
IL-17A contributes to brain ischemia reperfusion injury through
calpain-TRPC6 pathway in mice. Neuroscience. 274:419–428.
2014.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Kichev A, Rousset CI, Baburamani AA,
Levison SW, Wood TL, Gressens P, Thornton C and Hagberg H: Tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling
and cell death in the immature central nervous system after
hypoxia-ischemia and inflammation. J Biol Chem. 289:9430–9439.
2014.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Heymann MF, Herisson F, Davaine JM,
Charrier C, Battaglia S, Passuti N, Lambert G, Gouëffic Y and
Heymann D: Role of the OPG/RANK/RANKL triad in calcifications of
the atheromatous plaques: Comparison between carotid and femoral
beds. Cytokine. 58:300–306. 2012.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Nagy EE, Varga-Fekete T, Puskas A, Kelemen
P, Brassai Z, Szekeres-Csiki K, Gombos T, Csanyi MC and Harsfalvi
J: High circulating osteoprotegerin levels are associated with
non-zero blood groups. BMC Cardiovasc Disord.
16(106)2016.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Shimamura M, Nakagami H, Osako MK,
Kurinami H, Koriyama H, Zhengda P, Tomioka H, Tenma A, Wakayama K
and Morishita R: OPG/RANKL/RANK axis is a critical inflammatory
signaling system in ischemic brain in mice. Proc Natl Acad Sci USA.
111:8191–8196. 2014.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Kim HY and Han SH: Matrix
metalloproteinases in cerebral ischemia. J Clin Neurol. 2:163–170.
2006.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Anuncibay-Soto B, Pérez-Rodriguez D,
Santos-Galdiano M, Font-Belmonte E, Ugidos IF, Gonzalez-Rodriguez
P, Regueiro-Purriños M and Fernández-López A: Salubrinal and
robenacoxib treatment after global cerebral ischemia. Exploring the
interactions between ER stress and inflammation. Biochem Pharmacol.
151:26–37. 2018.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Ugidos IF, Santos-Galdiano M,
Pérez-Rodríguez D, Anuncibay-Soto B, Font-Belmonte E, López DJ,
Ibarguren M, Busquets X and Fernández-López A: Neuroprotective
effect of 2-hydroxy arachidonic acid in a rat model of transient
middle cerebral artery occlusion. Biochim Biophys Acta Biomembr.
1859:1648–1656. 2017.PubMed/NCBI View Article : Google Scholar
|
|
124
|
De los Reyes LM and Céspedes AE:
Atorvastatin-meloxicam association inhibits neuroinflammation and
attenuates the cellular damage in cerebral ischemia by arterial
embolism. Biomedica. 34:366–378. 2014.PubMed/NCBI View Article : Google Scholar : (In Spanish).
|
|
125
|
Llorente IL, Landucci E,
Pellegrini-Giampietro DE and Fernández-López A: Glutamate receptor
and transporter modifications in rat organotypic hippocampal slice
cultures exposed to oxygen-glucose deprivation: The contribution of
cyclooxygenase-2. Neuroscience. 292:118–128. 2015.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Nagy E, Vajda E, Vari C, Sipka S, Fárr AM
and Horváth E: Meloxicam ameliorates the cartilage and subchondral
bone deterioration in monoiodoacetate-induced rat osteoarthritis.
PeerJ. 5(e3185)2017.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Arafa HM, Abdel-Wahab MH, El-Shafeey MF,
Badary OA and Hamada FM: Anti-fibrotic effect of meloxicam in a
murine lung fibrosis model. Eur J Pharmacol. 564:181–189.
2007.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Hassan MH and Ghobara MM: Antifibrotic
effect of meloxicam in rat liver: Role of nuclear factor kappa B,
proinflammatory cytokines, and oxidative stress. Naunyn
Schmiedebergs Arch Pharmacol. 389:971–983. 2016.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Honma S, Shinohara M, Takahashi N,
Nakamura K, Hamano S, Mitazaki S, Abe S and Yoshida M: Effect of
cyclooxygenase (COX)-2 inhibition on mouse renal interstitial
fibrosis. Eur J Pharmacol. 740:578–583. 2014.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Csifo E, Nagy EE, Horvath E, Farr A and
Muntean DL: Mid-term effects of meloxicam on collagen type II
degradation in a rat osteoarthritis model induced by iodoacetate.
Farmacia. 63:556–560. 2015.
|
|
131
|
Sabogal-Guáqueta AM, Posada-Duque R,
Cortes NC, Arias-Londoño JD and Cardona-Gómez GP: Changes in the
hippocampal and peripheral phospholipid profiles are associated
with neurodegeneration hallmarks in a long-term global cerebral
ischemia model: Attenuation by Linalool. Neuropharmacology.
135:555–571. 2018.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Horváth E, Oradan A and Chiriac L:
Dobreanu M, Nagy EE, Voidăzan S, Berei R, Muntean DL and Hutanu A:
Fish-oil preconditioning upregulates expression of splenic Arg1
positive M2 type macrophags and the Arg1/iNos2 ratio after
experimental induced transient cerebral ischemia. Farmacia.
67:820–829. 2019.
|
|
133
|
Horvath E, Hutanu A, Oradan A, Chiriac L,
Muntean DL, Nagy EE and Dobreanu M: N-3 polyunsaturated fatty acids
induce granulopoiesis and early monocyte polarization in the bone
marrow of a tMCAO rat model. Rev Rom Med Lab. 27:51–61. 2019.
|
|
134
|
Pentón-Rol G, Marín-Prida J and
Falcón-Cama V: C-phycocyanin and phycocyanobilin as remyelination
therapies for enhancing recovery in multiple sclerosis and ischemic
stroke: A preclinical perspective. Behav Sci (Basel).
8(E158)2018.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Alawieh A, Elvington A, Zhu H, Yu J, Kindy
MS, Atkinson C and Tomlinson S: Modulation of post-stroke
degenerative and regenerative processes and subacute protection by
site-targeted inhibition of the alternative pathway of complement.
J Neuroinflammation. 12(247)2015.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Kopschina Feltes P, Doorduin J, Klein HC,
Juárez-Orozco LE, Dierckx RA, Moriguchi-Jeckel CM and de Vries EF:
Anti-inflammatory treatment for major depressive disorder:
Implications for patients with an elevated immune profile and
non-responders to standard antidepressant therapy. J
Psychopharmacol. 31:1149–1165. 2017.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Akhondzadeh S, Jafari S, Raisi F, Nasehi
AA, Ghoreishi A, Salehi B, Mohebbi-Rasa S, Raznahan M and
Kamalipour A: Clinical trial of adjunctive celecoxib treatment in
patients with major depression: A double blind and placebo
controlled trial. Depress Anxiety. 26:607–611. 2009.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Abbasi SH, Hosseini F, Modabbernia A,
Ashrafi M and Akhondzadeh S: Effect of celecoxib add-on treatment
on symptoms and serum IL-6 concentrations in patients with major
depressive disorder: Randomized double-blind placebo-controlled
study. J Affect Disord. 141:308–314. 2012.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Tyring S, Gottlieb A, Papp K, Gordon K,
Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, et al:
Etanercept and clinical outcomes, fatigue, and depression in
psoriasis: Double-blind placebo-controlled randomised phase III
trial. Lancet. 367:29–35. 2006.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Nemeth CL, Glasper ER, Harrell CS, Malviya
SA, Otis JS and Neigh GN: Meloxicam blocks neuroinflammation, but
not depressive-like behaviors, in HIV-1 transgenic female rats.
PLoS One. 9(e108399)2014.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Liu J, Nolte K, Brook G, Liebenstund L,
Weinandy A, Höllig A, Veldeman M, Willuweit A, Langen KJ, Rossaint
R, et al: Post-stroke treatment with argon attenuated brain injury,
reduced brain inflammation and enhanced M2 microglia/macrophage
polarization: A randomized controlled animal study. Crit Care.
23(198)2019.PubMed/NCBI View Article : Google Scholar
|














