Introduction
The temporomandibular joint (TMJ) is a synovial
joint that is composed of the mandibular fossa of the temporal bone
and the mandibular condyle (1).
TMJ-osteoarthritis (OA) symptoms include cartilage degeneration,
subchondral bone remodeling, and synovitis, which result in TMJ
dysfunction (2). Intriguingly,
histological studies have shown the presence of extensive fibrosis
in the TMJ-OA synovial tissue (3,4),
suggesting that fibrotic tissue formation may be responsible for
restricted joint movements (5).
We have previously established a fibroblast-like
synoviocyte (FLS) cell line, FLS1, from fibroblastic cells derived
from a mouse TMJ and found that these cells exhibit myofibroblast
(MF)-like fibrogenic characteristics (6). We have also demonstrated that
fibroblast growth factor (FGF)-1 alone significantly suppresses the
MF differentiation markers α-smooth muscle actin (α-SMA) and type I
collagen in FLS1 cells (6). The FGF
family consists of 24 members that share 13-71% amino acid identity
(7). Although FGF-11-15 are
generally considered to belong to the FGF family, they do not
activate any FGF receptor (FGFR) (8). However, four FGFRs belong to the
receptor tyrosine kinase (RTK) family (9). In general, FGF-1 binds to
FGFR1-4(10) to activate various
intracellular signaling factors, including phosphoinositide
3-kinase (PI3K)/Akt and mitogen-activated protein kinases (MAPKs),
such as extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun
N-terminal kinase (JNK), and p38 MAPK (11). Intriguingly, human synovial
fibroblasts derived from the knee synovial tissues express FGF-1
and FGF-R1 proteins (12). However,
it remains to be determined which FGF-1-induced intracellular
signaling pathway negatively controls the fibrogenic activity in
FLSs.
Epidermal growth factor (EGF) was first purified
from the mouse salivary gland as a soluble factor that accelerated
corneal wound healing (13);
however, EGF was soon after found to be a general growth factor
that affected various cellular functions involving cell
proliferation and differentiation (14). The EGF receptor (EGFR) family
consists of four members, namely EGFR/ErbB1/HER1, ErbB2/HER2,
ErbB3/HER3, and ErbB4/HER4, all of which belong to the RTK family
(15). EGFR dimerizes upon its
association with EGF. The cytoplasmic tyrosine kinase domains of
EGFR can be autophosphorylated to relay extracellular signals to
various intracellular signaling proteins. The carboxy terminal
tyrosine residues on EGFR, Tyr1068 and Tyr1173, are the major sites
of the autophosphorylation, which occurs as a result of EGF-binding
and converts the extracellular EGF signal to intracellular signals
(16,17). Autophosphorylated EGFR activates
various types of intracellular signaling molecules, including MAPKs
and PI3K/Akt (18). Intriguingly,
EGF has been detected in the human knee synovial fluid (19). In addition, the EGFR signaling is
critical for maintaining the superficial layer of the articular
cartilage and preventing OA initiation (20). However, whether EGF-induced
intracellular signaling affects the fibrogenic activity in FLSs
remains elusive. Furthermore, the mechanism whereby EGF affects the
FGF-1-mediated suppression of the fibrogenic activity in FLSs
derived from the TMJ synovial tissues warrants investigation.
It is worth noting that inflammatory cytokines, such
as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) are
important factors participating in the pathogenesis of OA. However,
the roles of IL-1β and TNF-α in the onset of OA have not been
comprehensively studied yet (21).
IL-1β binds to the cell membrane receptors IL-1R1 (IL-1RI and
CD121a) and IL-1R2 (IL-1RII and CD121b) (22), whereas TNF-α binds to the cell
membrane receptors TNF-R1 (p55, CD120a, and TNFRSF1a) and TNFR-2
(p75, CD120b, and TNFRSF1b) (23).
In general, IL-1β and TNF-α use similar signal transduction
mechanisms to activate nuclear factor-kappa B (NF-κB) and MAPKs,
including ERK1/2, JNK, and p38 MAPK. Interestingly, IL-1β and TNF-α
both induce MF differentiation in mesenchymal cells in an
NF-κB-dependent manner (24,25). In addition, transforming growth
factor-β1 (TGF-β1) promotes MF differentiation in an
NF-κB-dependent manner as well (26), suggesting that NF-κB-mediated signals
positively regulate MF differentiation in mesenchymal cells.
Interestingly, IL-1β promotes TGF-β1-induced MF differentiation in
nasal fibroblasts in MAPK/ERK kinase (MEK)/ERK-, JNK-, and p38
MAPK-dependent manners (24),
whereas TNF-α attenuates TGF-β1-induced MF differentiation in
pulmonary fibroblasts in a MEK/ERK-dependent manner (27). These results suggest that
MAPK-mediated signals positively or negatively regulate the MF
differentiation of mesenchymal cells in a cell-type-specific
manner.
Here, we examined the mechanisms whereby the RTK
ligands FGF-1 and EGF affect the fibrogenic activity in the
myofibroblastic FLS cell line FLS1. We also investigated the
effects of FGF-1 and EGF on the activity of PI3K/Akt and MAPKs,
such as ERK, JNK, and p38 MAPK, in FLS1 cells and examined whether
FGF-1-, or EGF-activated PI3K/Akt or MAPKs affected the status of
myofibroblastic differentiation in FLS1 cells. In addition, we
examined the cooperative and non-cooperative effects of RTK ligands
and inflammatory cytokines, such as IL-1β and TNF-α, on the
myofibroblastic differentiation in FLS1 cells. Our study clarified
the molecular mechanisms underlying the development of OA-related
fibrosis in TMJ and may aid in identifying new therapeutic targets
for this condition.
Materials and methods
Reagents
Recombinant mouse EGF was purchased from PeproTech,
Inc. Recombinant human IL-1β and TNF-α were obtained from Miltenyi
Biotec, GmbH (Bergisch Gladbach). The MEK inhibitors U0126 and
PD98059, and the EGFR inhibitor PD153035 were purchased from
Calbiochem (Merck KGaA). The NF-κB inhibitor BAY 11-7085 was
obtained from Cayman Chemical. Recombinant human FGF-1 and the
NF-κB kinase-2 (IKK-2) inhibitor TPCA-1 were purchased from R&D
Systems, Inc. The FGFR1 inhibitor SU-5402 was obtained from Wako
Pure Chemical Industries, Ltd. We confirmed that dimethyl sulfoxide
(DMSO), the vehicle used for the U0126, PD98059, PD153035, BAY
11-7085, TPCA-1, and SU-5402 treatments, did not affect the
expression of the MF markers α-SMA and type I collagen (data not
shown). Heparin sodium salt was obtained from Merck KGaA. Heparin
was included to achieve the optimal FGF-1 activity (28).
Cell culture
The FLS cell line FLS1 was previously established
and reported (6): Briefly, to
prepare FLSs derived from the mouse TMJ, TMJ synovial tissue was
obtained from eight-week-old female mice (C57BL/6J). The tissue was
then immersed in digestion solution composed of 20 ml of Ham's F-12
containing 2 mg/ml collagenases consisting of class I and class II
collagenases (Collagenase NB4; Wako), at 37˚C for 30 min with
continuous vigorous rocking. The cells released from the tissue
were transfected with pBABE-puro-simian virus 40 large T antigen
(SV40LT) expression plasmid (cat. no. 13970) obtained from Addgene,
Inc., with Lipofectamine LTX Reagent (ThermoFisher Scientific,
Inc.) according to the manufacturer's protocol. The immortalized
FLSs, FLS1 cells were maintained in culture with Ham's F-12
supplemented with 2 mM glutamine, 10% FBS, and
penicillin-streptomycin (Invitrogen). These cells were then
sub-cultured at a ratio of 1:4 when they reached
sub-confluency.
RNA isolation and RT-qPCR
FLS1 cells were seeded into 12-well tissue culture
plates at a density of 1x105 cells/well in FLS1 growth
medium and maintained for 24 h. The growth medium was replaced with
Ham's F-12 containing 0.5% FBS for 24 h for cell starvation.
Subsequently, the cells were cultured with or without FGF-1,
heparin, EGF, IL-1β, or TNF-α for the indicated periods. Total RNA
was isolated from FLS-1 cells using ISOGEN reagent (Nippon Gene)
according to the manufacturer's protocol. First-strand cDNA was
synthesized from total RNA using the PrimeScript RT reagent Kit
(Takara-Bio). PCR was subsequently performed on a Thermal Cycler
Dice Real Time System (Takara-Bio) using SYBR Premix Ex Taq II
(Takara-Bio), with the following specific oligonucleotide primers:
Mouse α-SMA, 5'-CAGATGTGGATACAGCAAACAGGA-3' (forward) and
5'-GACTTAGAAGCATTTGCGGTGGA-3' (reverse); mouse α1 chain of collagen
type I (colIα1), 5'-GACATGTTCAGCTTTGTGGACCTC-3' (forward)
and 5'-GGGACCCTTAGGCCATTGTGTA-3' (reverse); and mouse GAPDH,
5'-TGTGTCCGTCGTGGATCTG-3' (forward) and 5'-TTGCTGTTGAAGTCGCAGGAG-3'
(reverse). The mRNA levels of α-SMA and colIα1 were
normalized to GAPDH mRNA levels, and the relative expression
levels were calculated as the fold increase or decrease relative to
the control.
Western blot analysis
Cells were seeded into 6-well tissue culture plates
at a density of 2x105 cells/well in FLS1 growth medium
and maintained for 24 h. Afterward, the cells were starved for 24 h
as indicated above and cultured with or without FGF-1 plus heparin,
EGF, IL-1β, or TNF-α for the indicated periods. Eventualy, the
cells were lysed in RIPA buffer [Sigma; 50 mM Tris-HCl (pH 7.2),
150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS] or
lysis buffer [20 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1%
Triton X-100] containing protease and phosphatase inhibitor
cocktails (Sigma). The protein contents of the cell extracts were
measured using BCA reagent (Pierce). Extracts containing equal
amounts of protein were separated on 10% SDS-polyacrylamide gels
and transferred onto polyvinylidenedifluoride membranes
(Millipore). After blocking the membranes with 1% BSA or 1% skim
milk in T-TBS (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, and 0.05% Tween
20), they were incubated with the appropriate primary antibody. The
primary antibodies used included rabbit anti-p44/42 (ERK1/2; cat.
no. 9102), rabbit anti-p38 MAPK (cat. no. 9212), rabbit
anti-SAPK/JNK (cat. no. 9252), rabbit anti-Akt (cat. no. 9272),
rabbit anti-phospho-p44/42 (ERK1/2, Thr202/Tyl204; cat. no. 9101),
rabbit anti-phospho-p38 MAPK (Thr180/Tyr182; cat. no. 9211), rabbit
anti-phospho-SAPK/JNK (Ther183/185; cat. no. 9251), rabbit
anti-phospho-Akt (Ser473; cat. no. 9271) polyclonal antibodies
(1:1,000; Cell Signaling Technology), and anti-β-actin antibody
(cat. no. sc-47778, 1:1,000; Santa Cruz Biotechnology). The blots
were then incubated with the appropriate alkaline
phosphatase-conjugated secondary antibody, and signals were
detected using an alkaline phosphatase substrate kit (BCIP/NBT
Substrate Kit; Vector Laboratories Inc.). Especially, β-actin blots
ware obtained from the same membrane as the total ERK1/2 blots
after stripping anti-total ERK1/2 antibody from the membranes
according to the manufacturer's protocol.
Statistical analysis
Data were presented as mean ± standard deviation
(SD; n=4) and statistically analyzed by Tukey's multiple comparison
test except for the data analysis in Fig. S1. In Fig. S1, the data were statistically
analyzed by Student's t-test. Values of *P<0.01 and
**P<0.05 were considered to indicate a statistically
significant difference. The results shown in all the experiments
are representatives of at least two separate experiments.
Results
FGF-1 suppressed the expression of
myofibroblast markers in FLSs
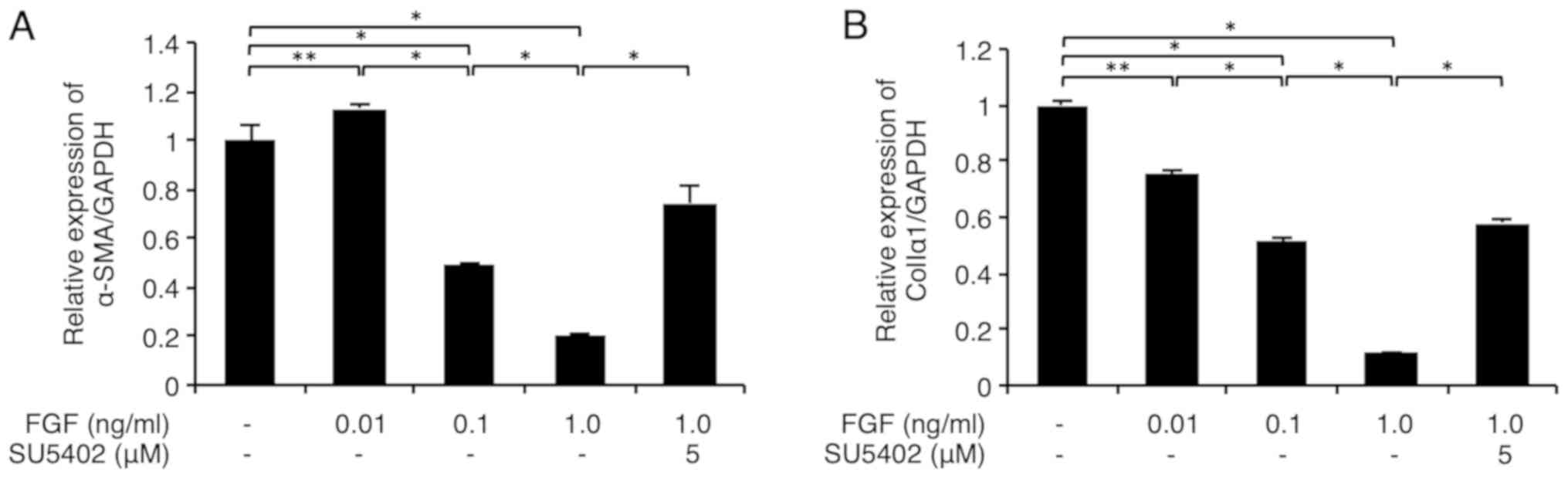
As shown in Fig. 1A,
FGF-1 (0.1-1 ng/ml) with heparin (15 µg/ml) significantly
downregulated the α-SMA mRNA level in FLS1 cells in a
dose-dependent manner. In addition, FGF-1 (0.01-1 ng/ml) with
heparin (15 µg/ml) significantly downregulated the colIa1
mRNA level in FLS1 cells in a dose-dependent manner (Fig. 1B). Importantly, we confirmed that the
FGFR1 inhibitor SU-5402 (5 µM) significantly abrogated this
FGF-1-mediated suppression of α-SMA and colIa1
expression (Fig. 1A and B, respectively). We also confirmed that 15
µM of heparin alone did not significantly affect the mRNA levels of
the MF markers α-SMA and type I collagen (Fig. S1) relative to the control cells.
EGF suppressed the expression of
myofibroblast markers in FLSs
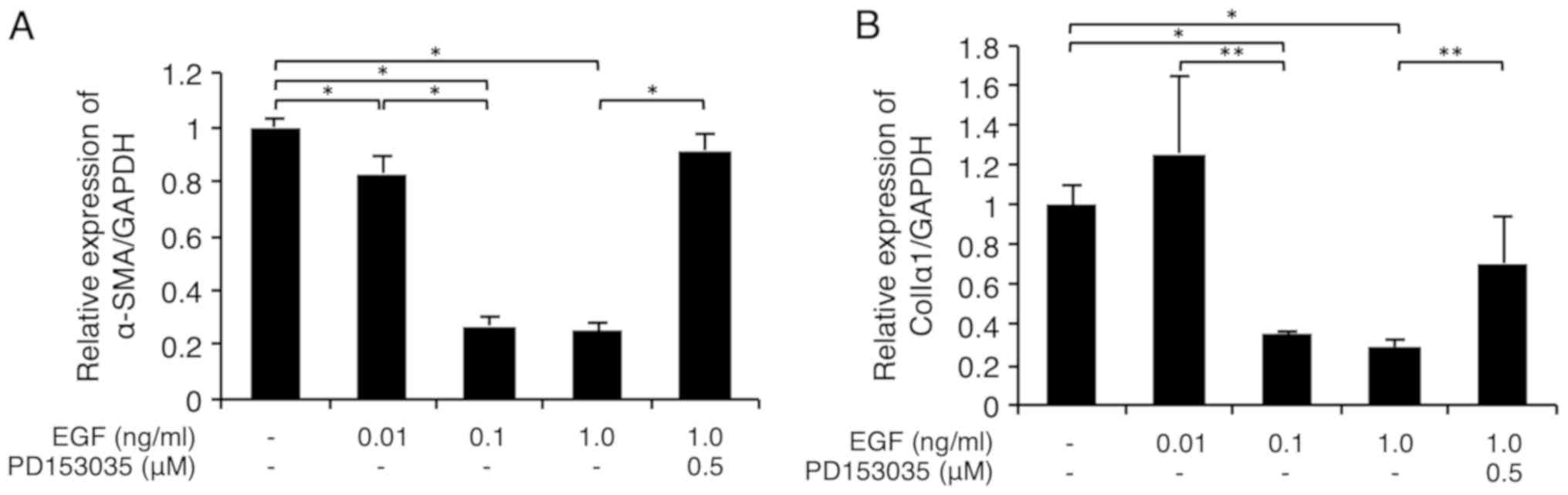
As shown in Fig. 2A,
EGF (0.01-0.1 ng/ml) significantly downregulated the α-SMA
mRNA level in FLS1 cells in a dose-dependent manner. In addition,
EGF (0.1-1 ng/ml) significantly downregulated the colIa1
mRNA level in FLS1 cells (Fig. 2B).
Importantly, we confirmed that the EGFR inhibitor PD153035 (0.5 µM)
significantly abrogated this EGF-mediated suppression of
α-SMA and colIa1 expression (Fig. 2A and B, respectively).
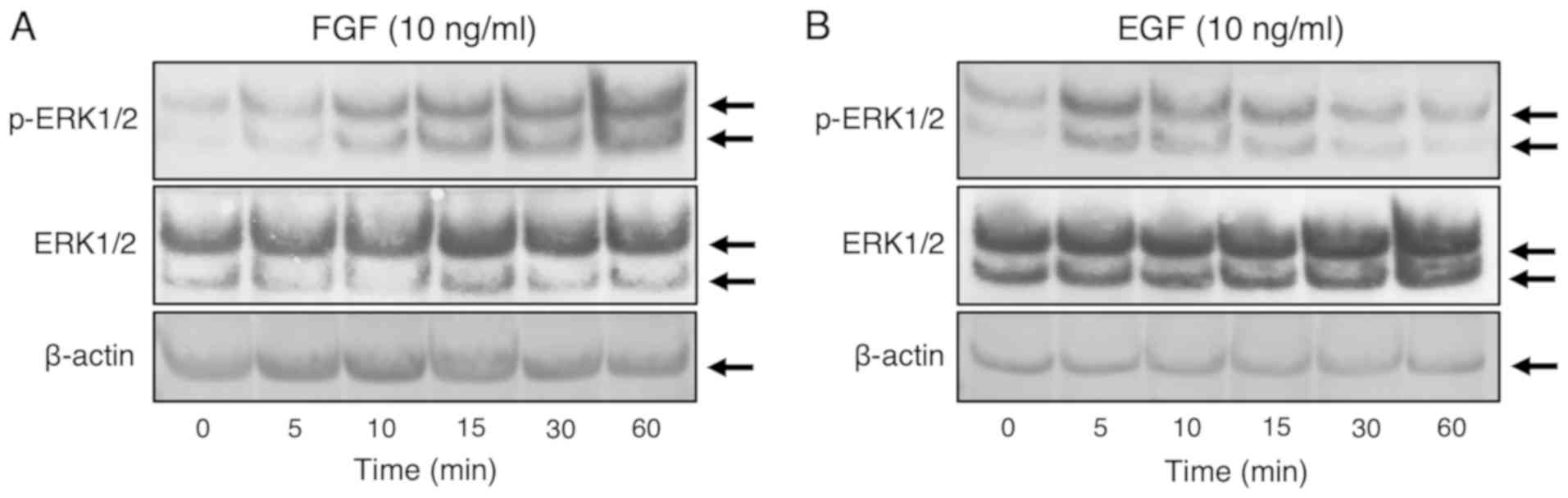
FGF-1 and EGF promoted phosphorylation
of ERK1/ERK2 in FLSs
We used western blotting to evaluate the
phosphorylation statuses of ERK1/2, p38 MAPK, JNK, and AKT after
the stimulation of FLS1 cells with EGF or FGF-1. As shown in
Fig. 3A, strong phosphorylation of
ERK1/2 was observed between 5 and 60 min after stimulation with
FGF-1 (10 ng/ml) with heparin (15 µg/ml). On the other hand, strong
phosphorylation of ERK1/2 was observed at 5-15 min after EGF (10
ng/ml) treatment (Fig. 3B). However,
phosphorylated p38 MAPK, JNK, or AKT were not at detectable levels
even after treatment with FGF-1 (10 ng/ml) and heparin (15 µg/ml)
or EGF (10 ng/ml) alone (data not shown). We also confirmed that
β-actin expression was unaffected by the administrations of FGF-1
(10 ng/ml) with heparin (15 µg/ml) or EGF (10 ng/ml) alone at any
time points of the treatments (Fig.
3A and B).
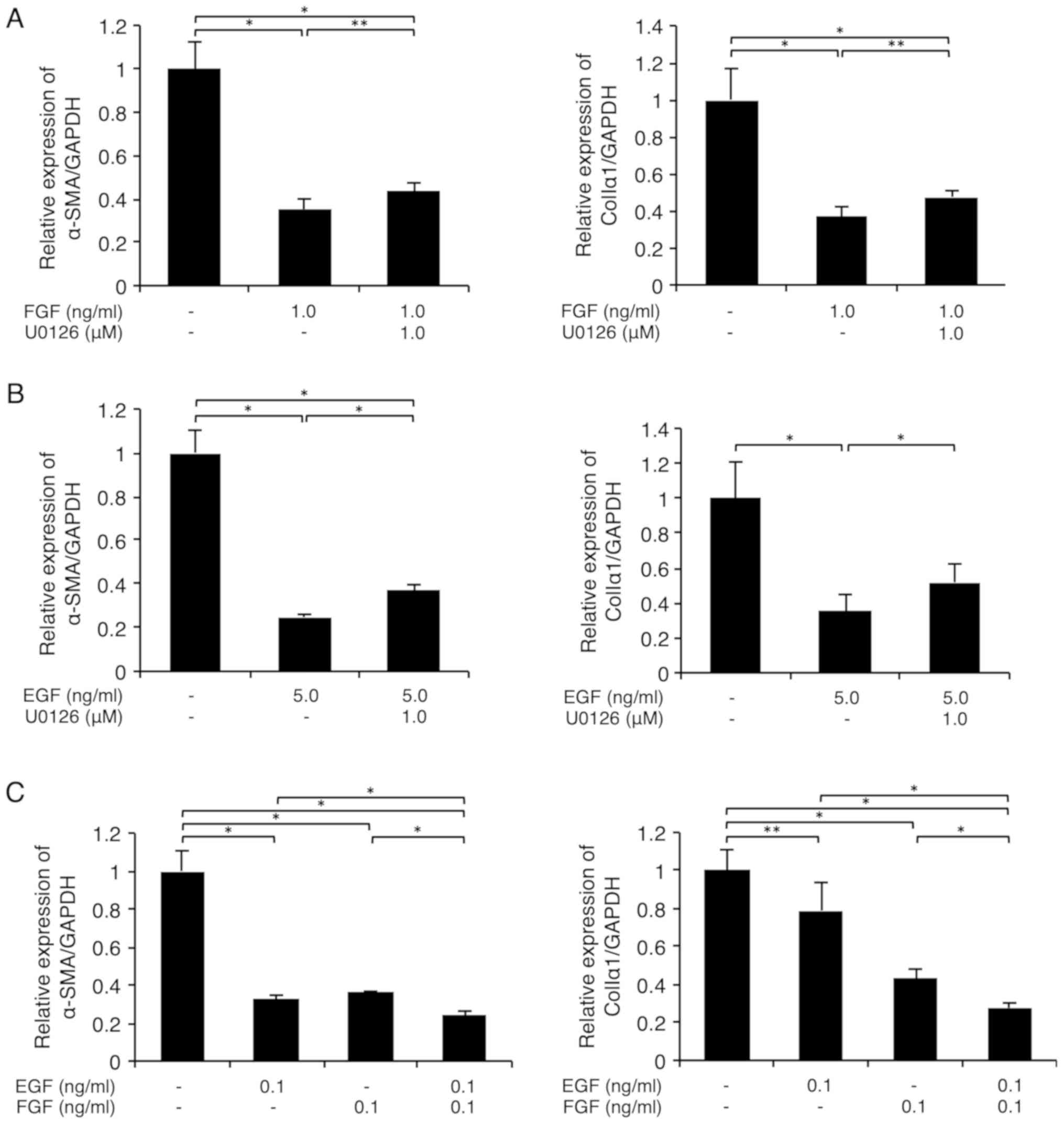
FGF-1 and EGF downregulated the mRNA
levels of the myofibroblast markers α-SMA and colIa1 in FLSs in a
MEK-dependent manner
As shown in Fig. 4A,
the MEK inhibitor U0126 (1 µM) partially and significantly reversed
the FGF-1 (1 ng/ml) plus heparin (15 µg/ml)-mediated suppression of
α-SMA (left graph) and colIa1 (right graph)
expression in FLS1 cells, respectively. In addition, U0126 (1 µM)
partially and significantly reversed the EGF (5 ng/ml)-mediated
suppression of α-SMA (left graph) and colIa1 (right
graph) expression, respectively, in FLS1 cells (Fig. 4B). We also found that the MEK
inhibitor PD98059 similarly abrogated the FGF-1 (0.25 ng/ml) and
heparin (15 µg/ml)-, or EGF (5 ng/ml)-mediated suppression of MF
marker expression at concentrations of 5 and 1 µM, respectively
(data not shown). Interestingly, FGF-1 (0.1 ng/ml) plus heparin (15
µg/ml) and EGF (0.1 ng/ml) additively downregulated the mRNA levels
of α-SMA (left graph) and colIa1 (right graph) in
FLS1 cells (Fig. 4C).
RTK ligands and inflammatory cytokines
cooperatively inhibited the fibrogenic activity in FLSs in a
MEK/ERK-dependent manner
As shown in Fig. 5A,
the inflammatory cytokines IL-1β (10 ng/ml) and TNF-α (10 ng/ml)
significantly suppressed α-SMA (left graph) and
colIa1 (right graph) expression in FLS1 cells.
Interestingly, the suppression of α-SMA expression by the
combination of the inflammatory cytokines IL-1β (10 ng/ml) and
TNF-α (10 ng/ml) was evidently abrogated by U0126 (1 µM) or PD98059
(5 µM; Fig. 5B, left graph). In
addition, the suppression of colIa1 expression by the
combinatorial stimulation of the inflammatory cytokines was
partially but significantly abrogated by U0126 (1 µM), but not by
PD98059 (5 µM; Fig. 5B, right
graph). However, the NF-κB inhibitor BAY 11-7085 (5 µM) or the
IKK-2 inhibitor TPCA-1 (5 µM) did not abrogate the suppression of
MF marker expression caused by the combination of the inflammatory
cytokines and instead, further downregulated the levels of these MF
markers (Fig. S2). As shown in
Fig. 5C (left panels), strong
phosphorylation of ERK1/2 was observed at 5-15 min after IL-1β (10
ng/ml) stimulation. In addition, strong phosphorylation of ERK1/2
was observed at 30 min after TNF-α (10 ng/ml) stimulation (Fig. 5C, right panels). We also confirmed
that β-actin expression was unaffected by the administration of
IL-1β (10 ng/ml) or TNF-α (10 ng/ml) at any time point after the
stimulation (Fig. 5C).
Interestingly, FGF-1 (10 ng/ml) plus heparin (15 µg/ml)- or EGF (10
ng/ml)-mediated suppression of colIa1 expression was further
enhanced by the administration of IL-1β (10 ng/ml) or TNF-α (10
ng/ml) (Fig. 5D; left and right
graphs, respectively).
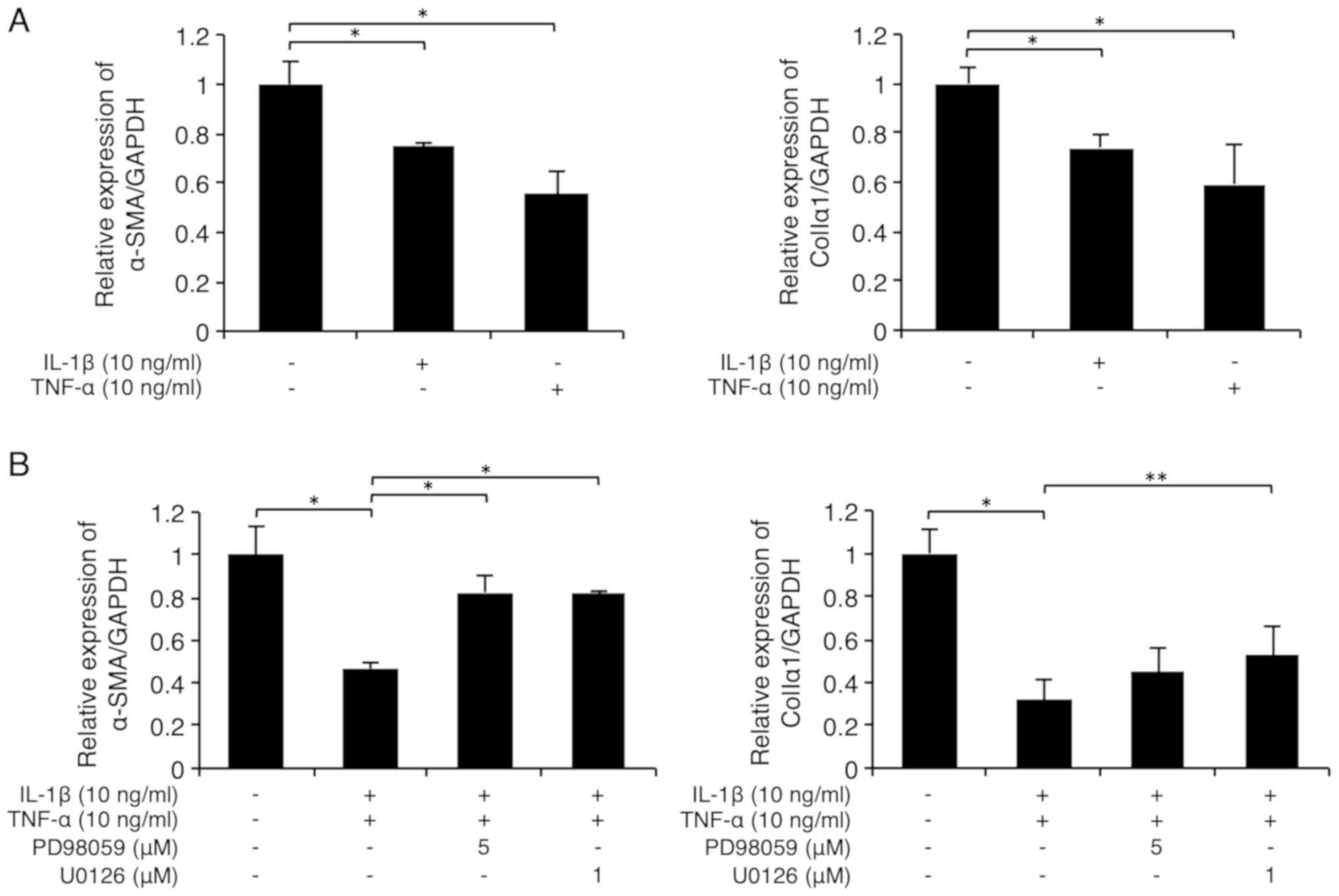 | Figure 5Receptor tyrosine kinase ligands and
inflammatory cytokines cooperatively inhibit the fibrogenetic
activity in fibroblast-like synoviocytes in a MEK/ERK-dependent
manner. (A and B) Cells were starved and then cultured with IL-1β
(10 ng/ml) and/or TNF-α (10 ng/ml) for 24 h. (B) Some cells were
pretreated with the specific MEK inhibitors PD98059 (5 µM) or U0126
(1 µM) for 30 min prior to stimulation. The relative expression
levels of the myofibroblast markers α-SMA and colIα1 were then
evaluated using RT-qPCR. (C) Cells were starved and then treated
with IL-1β (10 ng/ml; left) or TNF-α (10 ng/ml; right) for the
indicated times. ERK1/2 phosphorylation was evaluated using western
blot analysis. (D) Cells were starved and then cultured with IL-1β
(10 ng/ml) or TNF-α (10 ng/ml), with or without FGF-1 (10 ng/ml)
and heparin (15 µg/ml) or EGF (10 ng/ml) for 24 h. The relative
expression levels of the myofibroblast markers α-SMA and colIα1
were then evaluated using RT-qPCR. Data are presented as the mean ±
SD (n=4). *P<0.01, **P<0.05. α-SMA,
α-smooth muscle actin; colIα1, α1 chain of collagen type I; EGF,
epidermal growth factor; ERK, extracellular signal-regulated
kinase; FGF, fibroblast growth factor; IL, interleukin; MEK,
mitogen activated protein kinase; p-, phosphorylated; RT-qPCR,
reverse transcription-quantitative PCR; TNF-α, tumor necrosis
factor α. |
Discussion
Bernasconi et al (29), have investigated the morphological
modifications occurring in synovial tissue after severe derangement
of the articular structure, with dislocation or perforation of the
TMJ disks. After histological examination, they have reported
remarkable hyperplasia of the synovial tissue, with an increase in
the number of myofibroblastic fibroblast-like cells (29). Interestingly, post-traumatic joint
stiffness is characterized by an increase in the number of MFs in
the joint capsules (30,31), suggesting that MFs retain crucial
roles in the pathogenesis of joint stiffness.
MFs are the cells primarily responsible for inducing
fibrosis in scleroderma, renal fibrosis, pulmonary fibrosis, and
liver fibrosis (32). MFs retain
contractile properties and produce a large quantity of
extracellular molecules, such as type I collagen (33). The most widely recognized molecular
marker of differentiated and activated MFs is the de novo
expression of α-SMA (34). We have
previously established an FLS cell line, FLS1, which is derived
from a mouse TMJ. We have reported that FLS1 cells express higher
levels of MF marker molecules, such as α-SMA and type I collagen
than mouse NIH3T3 embryonic fibroblasts, which are frequently used
as a standard fibroblast control (6). Thus, the FLS1 cell line is a suitable
experimental model for the investigation of the molecular
mechanisms underlying the extensive fibrosis observed in the TMJ-OA
synovial tissue. We have previously demonstrated that
Rho-associated coiled-coil-forming kinase (ROCK)-mediated
actin-polymerization, which induces the translocation of
myocardin-related transcription factor (MRTF) from the cytoplasm to
the nucleus, promotes MF differentiation in FLS1 cells (6). However, it remains to be clarified what
intra-cellular signals other than the ROCK-mediated signal affect
the fibrogenic activity in FLS1 cells.
Here, we demonstrated that FGF-1 and EGF
dose-dependently downregulated the mRNA levels of the MF
differentiation markers α-SMA and type I collagen in FLS1
cells (Figs. 1 and 2). In addition, we found that
ERK1/2-mediated signaling played an important role in these
anti-fibrogenic effects of FGF-1 and EGF (Figs. 3 and 4). These results strongly suggested that
FGF-1 and EGF suppressed the fibrogenic activity in FLSs in a
MEK/ERK-dependent manner. However, U0126 only partially abrogated
the FGF-1- or EGF-mediated downregulation of MF differentiation
markers in FLS1 cells, suggesting that signaling pathways other
than MEK/ERK play important roles (Fig.
4). Interestingly, we have previously demonstrated that EGF
suppresses the expression of MF differentiation markers in
periodontal ligament-derived endothelial progenitor cells (EPCs,
SCDC2) through MEK- and JNK-dependent signaling pathways (35), whereas the activation of JNK was not
detected after the EGF stimulation of FLS1 cells (data not shown).
These results suggested that EGF differentially induced
intracellular signals in EPCs and FLSs and had a negative effect on
myofibroblastic differentiation. On the other hand, the combination
of the inflammatory cytokines IL-1β and TNF-α suppressed the
expression of the MF markers α-SMA (Fig.
5B, left graph) and type I collagen (Fig. 5B, right graph), and this effect was
significantly abrogated by the MEK1/2 inhibitor U0126. In contrast,
PD98059, which is known as a MEK1 inhibitor, but not a MEK1/2
inhibitor, in a cell-type specific manner (36), significantly abrogated the
suppression of α-SMA expression by the combinatorial
treatment with the inflammatory cytokines (Fig. 5B, left graph), but did not abrogate
the suppression of type I collagen expression (Fig. 5B, right graph), suggesting that
MEK2-mediated intracellular signaling played an important role in
the suppression of type I collagen expression in FLSs after
stimulation with inflammatory cytokines. Importantly, neither an
NF-κB inhibitor nor an IKK-2 inhibitor abrogated the IL-1β- and
TNF-α-mediated suppression of MF marker expression (Fig. S2), suggesting that these effects of
the inflammatory cytokines in FLSs were not mediated by NF-κB.
Interestingly, the NF-κB and IKK-2 inhibitors further decreased the
expression levels of the MF markers in FLS1 cells treated with the
inflammatory cytokines (Fig. S2),
suggesting that the NF-κB-mediated signaling possibly positively
regulated the MF marker expression in FLSs. In addition, we
confirmed that IL-1β and TNF-α induced ERK1/2 phosphorylation in
FLS1 cells (Fig. 5C). Moreover,
these inflammatory cytokines further enhanced the FGF-1- or
EGF-mediated suppression of type I collagen expression (Fig. 5D). These results strongly suggested
that RTK ligands and inflammatory cytokines cooperatively inhibited
the fibrogenic activity in FLSs in a MEK/ERK-dependent manner.
Interestingly, Ma et al (37)
have previously reported that EGF and IL-1β synergistically promote
ERK1/2-mediated invasive breast ductal cancer cell migration and
invasion. In addition, Ziv et al (38) and Kakiashvili et al (39) have reported that TNF-α activates
ERK1/2 through EGFR activation in epithelial cells. However, it
remains to be determined whether IL-1β and TNF-α activate ERK1/2
through EGFR activation in FLSs.
Our findings partially clarify the molecular
mechanisms underlying the development of OA-related fibrosis in the
TMJ and may aid in identifying novel therapeutic targets for this
condition. In addition, FGF-1 and EGF may be used to prevent
OA-related fibrosis around inflammatory TMJs.
Supplementary Material
Heparin itself does not affect the
expression of the myofibroblast markers α-SMA and colIα1 in
fibroblast-like synoviocytes. Cells were starved and then cultured
with heparin (15 μg/ml) alone for 24 h. The relative expression
levels of the myofibroblast markers α-SMA and colIα1 were then
evaluated using reverse transcription-quantitative PCR. Data
represent the mean ± SD (n=4). α-SMA, α-smooth muscle actin;
colIα1, α1 chain of collagen type I.
Neither the NF‑κB inhibitor BAY
11-7085 nor the IKK-2 inhibitor TPCA-1 abrogated the IL-1β- and
TNF-α-mediated suppression of the myofibroblast marker expression
in fibroblast-like synoviocytes. Cells were starved and then
cultured with IL-1β (10 ng/ml) and TNF-α (10 ng/ml), with or
without the NF-κB inhibitor BAY 11-7085 or the IKK-2 inhibitor
TPCA-1 for 24 h. The relative expression levels of the
myofibroblast markers α-SMA and colIα1 were then evaluated using
reverse transcription-quantitative PCR. Data are presented as the
mean ± SD (n=4). *P<0.01. α-smooth muscle actin; colIα1, α1
chain of collagen type I; IL, interleukin; TNF-α, tumor necrosis
factor α.
Acknowledgements
Not applicable.
Funding
The present study was supported by JSPS KAKENHI
(grant nos. JP16H05534 to AI, JP16K11654 to NC, JP17K11851 to MK
and JP19K19277 to SY).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SM, SY, NC, SK, HK, MK and KS performed RT-qPCR and
western blotting. SM and AI designed the present study. AI was a
major contributor in writing the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ibi M: Inflammation and temporomandibular
joint derangement. Biol Pharm Bull. 42:538–542. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wang XD, Zhang JN, Gan YH and Zhou YH:
Current understanding of pathogenesis and treatment of TMJ
osteoarthritis. J Dent Res. 94:666–673. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dijkgraaf LC, Liem RS and de Bont LG:
Synovial membrane involvement in osteoarthritic temporomandibular
joints: A light microscopic study. Oral Surg Oral Med Oral Pathol
Oral Radiol Endod. 83:373–386. 1997.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Dijkgraaf LC, Liem RS and de Bont LG:
Ultrastructural characteristics of the synovial membrane in
osteoarthritic temporomandibular joints. J Oral Maxillifac Surg.
55:1269–1279. 1997.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dijkgraaf LC, Zardeneta G, Cordewener FW,
Liem RS, Schmitz JP, de Bont LG and Milam SB: Crosslinking of
fibrinogen and fibronectin by free radicals: A possible initial
step in adhesion formation in osteoarthritis of the
temporomandibular joint. J Oral Maxillofac Surg. 61:101–111.
2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yokota S, Chosa N, Kyakumoto S, Kimura H,
Ibi M, Kamo M, Satoh K and Ishisaki A: ROCK/actin/MRTF signaling
promotes the fibrogenic phenotype of fibroblast-like synoviocytes
derived from the temporomandibular joint. Int J Mol Med.
39:799–808. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chen TM, Chen YH, Sun HS and Tsai SJ:
Fibroblast growth factors: Potential targets for regenerative
therapy of osteoarthritis. Chin J Physiol. 62:2–10. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Beenken A and Mohammadi M: The fgf family:
Biology, pathophysiology and therapy. Nat Rev Drug Discov.
8:235–253. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Liu F and Zhuang S: Role of receptor
tyrosine kinase signaling in renal fibrosis. Int J Mol Sci.
17(E972)2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Omitz DM, Xu J, Colvin JS, McEwen DG,
MacArthur CA, Couller F, Gao G and Goldfarb M: Receptor specificity
of the fibroblast growth factor family. J Biol Chem.
271:15292–15297. 1996.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Raju R, Palapetta SM, Sandhya VK, Sahu A,
Alipoor A, Balakrishnan L, Advani J, George B, Kini KR, Geetha NP,
et al: A network map of FGF-1/FGFR signaling system. J Signal
Transduct. 2014(962962)2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhu L, Weng Z, Shen P, Zhou J, Zeng J,
Weng F, Zhang X and Yang H: S100B regulates inflammatory response
during osteoarthritis via fibroblast growth factor receptor 1
signaling. Mol Med Rep. 18:4855–4864. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cohen S and Elliott GA: The stimulation of
epidermal keratinization by a protein isolated from the
submaxillary gland of the mouse. J Invest Dermatol. 40:1–5.
1963.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tsai CJ and Nussinov R: Emerging mechanism
of EGFR activation in physiological and pathological contexts.
Biophys J. 117:5–13. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lemmmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinase. Cell. 141:1117–1134.
2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dawnward J, Waterfield MD and Parker PJ:
Autophosphorylation and protein kinase C phosphorylation of the
epidermal growth factor receptor. Effect on tyrosine kinase
activity and ligand binding affinity. J Biol Chem. 260:14538–14546.
1985.PubMed/NCBI
|
|
17
|
Helin K, Velu T, Martin P, Vass WC,
Allevato G, Lowy DR and Beguinot L: The biological activity of the
human epidermal growth factor receptor is positively regulated by
its C-terminal tyrosines. Oncogene. 6:825–832. 1991.PubMed/NCBI
|
|
18
|
Wee P and Wang Z: Epidermal growth factor
receptor cell proliferation signaling pathways. Cancers (Basel).
9(E52)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ren G, Lutz L, Raiton P, Wiley JP,
McAllister J, Powell J and Krawetz RJ: Serum and synovial fluid
cytokine profiling in hip osteoarthritis: Distinct from knee
osteoarthritis and correlated with pain. BMC Musculoskelet Disord.
19(39)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jia H, Ma X, Tong W, Doyran B, Sun Z, Wang
L, Zhang X, Zhou Y, Badar F, Chandra A, et al: EGFR signaling is
critical for maintaining the superficial layer of articular
cartilage and preventing osteoarthritis initiation. Proc Natl Acad
Sci USA. 113:14360–14365. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wojdasiewicz P, Poniatowski ŁA and
Szukiewicz D: The role of inflammatory and anti-inflammatory
cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm.
2014(561459)2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Boraschi D and Tagliabue A: The
interleukin-1 receptor family. Semin Immunol. 25:394–407.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
MacEwan DJ: TNF receptor subtype
signaling: Differences and cellular consequences. Cell Signal.
14:477–492. 2002.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Shin JM, Kang JH, Lee SA, Park IH and Lee
HM: Baicalin downregulates IL-1β-stimulated extracellular matrix
production in nasal fibroblasts. PLoS One.
11(e0168195)2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hou J, Ma T, Cao H, Chen Y, Wang C, Chen
X, Xiang Z and Han X: TNF-α induced NF-κB activation promotes
myofibroblast differentiation of LR-MSCs and exacerbates
bleomycin-induced pulmonary fibrosis. J Cell Physiol.
233:2409–2419. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mia MM and Bank RA: The IκB kinase
inhibitor strongly attenuates TGFβ1-induced myofibroblast formation
and collagen synthesis. J Cell Mol Med. 19:2780–2792.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu X, Kelm RJ Jr and Strauch AR:
Transforming growth factor beta1-mediated activation of the smooth
muscle alpha-actin gene in human pulmonary myofibroblasts is
inhibited by tumor necrosis factor-alpha via mitogen-activated
protein kinase 1-dependent induction of the Egr-1 transcriptional
repressor. Mol Biol Cell. 20:2174–2185. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Harmer NJ: Insights into the role of
heparin sulphate in fibroblast growth factor signaling. Biochem Soc
Trans. 34:442–445. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bernasconi G, Marchetti C, Reguzzoni M and
Baciliero U: Synovia hyperplasia and calcification in the human TMJ
disk: A clinical, surgical, and histologic study. Oral Surg Oral
Med Oral Pathol Oral Radiol Endod. 84:245–252. 1997.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hildebrand KA, Zhang M and Hart DA:
Myofibroblast upregulators are elevated in joint capsules in
posttraumatic contractures. Clin Orthop Relat Res. 456:85–91.
2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Hildebrand KA, Zhang M, van Snellenberg W,
King GJ and Hart DA: Myofibroblast numbers are elevated in human
elbow capsules after trauma. Clin Orthop Relat Res. 189–197.
2004.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hinz B, Phan SM, Thannickal VJ, Plunotto
M, Desmouliére A, Varga J, De Wever O, Mareel M and Gabbiani G:
Recent developments in myofibroblast biology: Paradigms for
connective tissue remodeling. Am J Pathol. 180:1340–1355.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
van Caam A, Vonk M, van den Hoogen F, van
Lent P and van der Kraan P: Unraveling SSc pathophysiology; The
myofibroblast. Front Immunol. 9(2452)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hinz B, Phan SH, Thannickal VJ, Galli A,
Bochaton-Piallat ML and Gabbiani G: The myofibroblast: one
function, multiple organs. Am J Pathol. 170:1807–1816.
2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kimura H, Okubo N, Chosa N, Kyakumoto S,
Kamo M, Miura H and Ishisaki A: EGF positively regulates the
proliferation and migration, and negatively regulates myofibroblast
differentiation of periodontal ligament-derived endothelial
progenitor cells through MEK/ERK- and JNK-dependent signals. Cell
Physiol Biochem. 32:899–914. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lee HR, Lee J and Kim HJ: Differential
effects of MEK inhibitor on rat neural stem cell differentiation:
Repressive roles of MEK2 in neurogenesis and induction of
astrocytogenesis by PD98059. Pharmacol Res.
149(104466)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ma L, Lan F, Zheng Z, Xie F, Wang L, Liu
W, Han J, Zheng F, Xie Y and Huang Q: Epidermal growth factor (EGF)
and interleukin (IL)-1β synergistically promote ERK1/2-mediated
invasive breast ductal cancer cell migration and invasion. Mol
Cancer. 11(79)2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ziv E, Rotem C, Miodovnik M, Ravid A and
Koren R: Two modes of ERK activation by TNF in keratinocytes:
Different cellular outcomes and bi-directional modulation by
vitamin D. J Cell Biochem. 104:606–619. 2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kakiashvili E, Dan Q, Vandermeer M, Zhang
Y, Waheed F, Pham M and Száski K: The epidermal growth factor
receptor mediates tumor necrosis factor-alpha-induced activation of
the ERK/GEF-H1/RhoA pathway in tubular epithelium. J Biol Chem.
286:9268–9279. 2011.PubMed/NCBI View Article : Google Scholar
|