Introduction
Atherosclerosis is a multifactorial and chronic
disease, which is generally hallmarked by chronic inflammation,
lipid storage, endothelial dysfunction and vascular calcification
in the vasculature (1-3).
This disease will lead to series of severe consequence, such as
heart attack, stroke and gangrene of the extremities (4). Many therapies have been used for this
disease, including nicotinic acids, fibrate derivatives, statins,
bile acid sequestrants and cholesterol absorption inhibitors
(5). Although contemporary
intervention and pharmacological agents have been applied to this
disease, because of the irreversible damage on tissues and organs,
the treatment on the atherosclerosis is not ideal. Therefore, it is
imperative to find effective biomarkers for early discovery and to
carry out preventive treatment.
Many scholars have focused on the relationships
between DNA methylation and atherosclerosis. DNA methylation is the
most studied epigenetic modification in human beings (6,7).
Simultaneously, a great number of human diseases, such as cancer
and Parkinson disease (8,9) have been confirmed to be related to
aberrant DNA methylation. As the important regulator of gene
transcription, DNA methylation is a heritable and stable epigenetic
marker (10). Therefore, confronting
the chronic disease with no specific therapeutics, research on DNA
methylation could be recognized as a novel direction for early
treatment and diagnosis of this disease.
In the present study, after analyzing the 15
atherosclerosis samples and 15 normal controls downloaded from the
ArrayExpress database, the differential DNA methylated genes were
extracted from the total samples. In order to verify the capacity
of these genes in differentiating atherosclerosis and normal
controls, clustering analysis was performed on these genes.
Subsequently, analysis of these differential methylated genes were
applied to the GO and pathway analysis. Finally, the related genes
were mapped to the relevant GO terms and biological pathways.
Through these significant GO terms and pathways, it was anticipated
that the related functions with atherosclerosis could be found to
elucidate the pathogenesis and identify molecular makers for early
diagnosing and treatment of the disease.
Materials and methods
DNA methylation data recruiting and
preprocessing
The genome-wide DNA methylation aberrations
(E-GEOD-46394) in human atheroscleosis were downloaded from the
ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/) and were deposited
on the A-AFFY-44 - Affymetrix GeneChip Human Genome U133 Plus 2.0
[HG-U133_Plus_2]. The E-GEOD-46394 was comprised of 30 samples,
including 15 atherosclerosis samples and 15 normal controls. The
age range of the samples is from 45 to 88 years on the basis of the
common age of this disease. Simultaneously, the sex of the
atherosclerosis samples and normal controls were randomly selected,
all containing men and women. The clinical characteristics of these
samples are shown in supplement Table SI. From the database, a
total of 485,577 CpGs in the DNA methylation data were obtained by
us. However, three types of probe should be removed, including the
single-nucleotide polymorphism (SNP) with the distance from CpG to
SNP ≤2, the probe with minor allele frequency (MAF) ≤0.05 and the
probe on cross-hybridising and sex chromosome. A total of 427,909
methylation data with CpGs were obtained for the following analysis
after the preprocessing.
Screening of differential methylated
genes
The value of the methylation expression profiles was
defined as the β-values with range between 0 (no methylation) to 1
(complete methylation) (11). In
order to identify the differential methylated CpGs, two stringent
criteria were strictly observed. The absolute β-values of the
difference between atherosclerosis and the healthy groups had to be
>0.5, and that the P-value should be <0.05 after t-test.
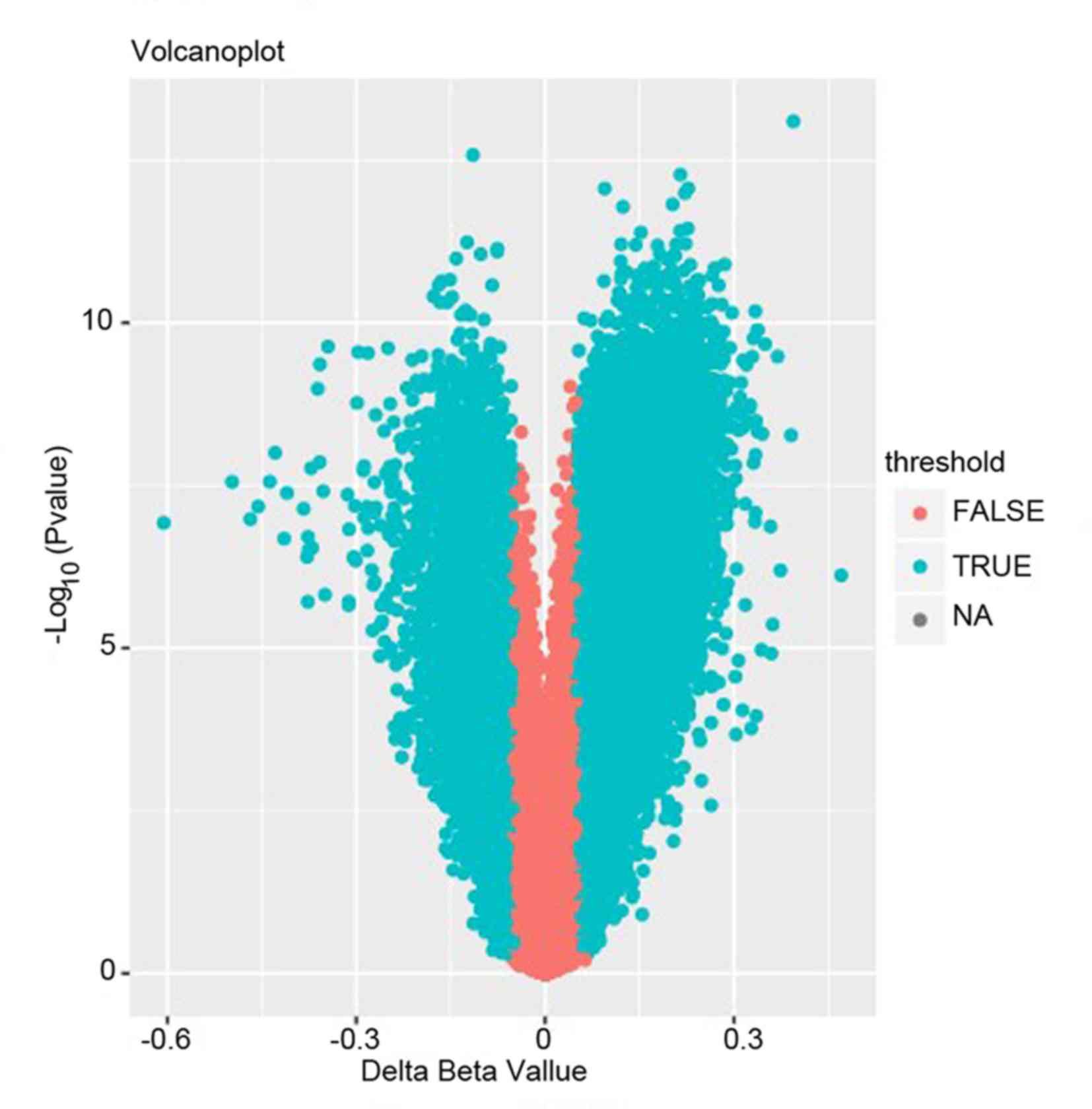
Through the preliminary screening, a total of 58,903 differential
methylated CpGs were extracted including 42,932 up-methylated CpGs
and 15,971 down-methylated CpGs (representing 13,245 genes).
In order to ensure that the data could be used to
facilitate subsequent analysis, further filtering steps should be
performed for the differential methylated genes. Two conditions had
to apply. First step is that the differential methylated CpGs with
β-value ≤0.2 or ≥0.8 in all samples should be deleted. The average
absolute β-values of difference between atherosclerosis and the
healthy groups should be ≥0.2. Finally, the differential methylated
genes were the simplified data, which could be used for the
following studies.
Clustering analysis
Clustering analysis is a Pattern Recognition
techniques (12,13). In order to establish that the
differential methylated genes extracted from the database could
distinguish atherosclerosis groups from the normal controls,
clustering analysis should be used to verify the effectiveness. In
the present study, the effective data visualization was presented
on the heatmap by Pheatmap package in R language 3.4 version,
setting the conditional parameter as the absolute percentage
methylation values (β-values) of the difference between the
atherosclerosis and the healthy groups as >0.5. The other
criterion was that the P-value should be <0.05 after t-test,
which represent a dataset with two dimensions, commonly genes and
samples (14). Ultimately, through
this heatmap, the distribution of the level of the differential
methylated CpGs in all samples were clearly found.
GO analysis
Due to the vast differential methylated data
generated from the microarray experiments, GoMiner's query gene
ontology (GO) database can be used to assess the function of
differentially expressed genes (15). The GO database (http:/www.geneontology.org/) is an open source tool
that covers three domains, which is widely used in functional
annotation and enrichment analysis (16,17).
Through the GO analysis, it is expected to discover the important
function related to the methylation changes and to find the
differential methylated genes corresponding to the related function
regions. In the present study, setting the threshold value of
P=0.01, we discarded the differential methylated genes with poor
P-value and carried out the enrichment on the rest of genes with
the category of biological process. Ultimately, the related
differential methylated genes were mapped to the same GO terms,
which could be used to check the function regions.
Remarkably, the differential methylated genes
mentioned above could be divided into two types of genes that,
respectively, were up-methylated and down-methylated. However, the
biological function of the two types of genes was the opposite.
When genes are transcribed into mRNA, the positively regulated
up-methylated genes will promote gene expression and the negatively
regulated, down-methylated genes will inhibit gene expression. In
consequence, in order to find out the respective biological
function of the two types of genes, the GO analysis was applied
respectively.
Pathway analysis
The difference is that the two methods focus on
different approaches and tools employed for the task. Generally
speaking, pathway is a process that proteins interacted with each
other to regulate cell function and metabolic activity. As is know,
the occurrence of a disease in biological pathway arises from the
expression change not only in the individual gene but also in the
interactions between related genes. Pathway analysis can help in
discovery of the most important biological pathway correlated to
the disease. In this study, the Kyoto Encyclopedia of Genes and
Genomes (KEGG) (http://www.geneme.jp/kegg/) was the common pathway
database (18). Using the stringent
filtering criteria of the pathways enriched by the differential
methylated genes with P<0.05, the related pathways were
extracted. Similarly, two types of pathways were presented,
respectively, enriched with the up-methylated and down-methylated
genes.
Results
Identification of differential
methylated genes between atherosclerosis samples and normal
controls
Analyzing the 15 atherosclerosis samples and 15
normal controls with the chip platform on Illumina Human Methlation
450 BeadChip, 427,909 methylated CpGs were obtained. Subsequently,
for the sake of the following analysis, there were two procedures
to filtrate the differential methylated genes. The first is the
preliminary screening with the criteria of the absolute β-values of
the difference between the atherosclerosis and the healthy groups
>0.5 and the P<0.05. Ultimately, a total of 58,903
differential methylated CpGs including 42,932 up-methylated CpGs
and 15,971 down-methylated CpGs were obtained. Details of these
genes are presented in Fig. 1.
Subsequently, further filtering steps should be applied to
facilitate a more stringent analysis. The criterion that was
applied discarded the CpGs with β-values ≤0.2 or ≥0.8 in all
samples and the retained CpGs with the absolute β-values were the
difference between the atherosclerosis and the healthy groups of
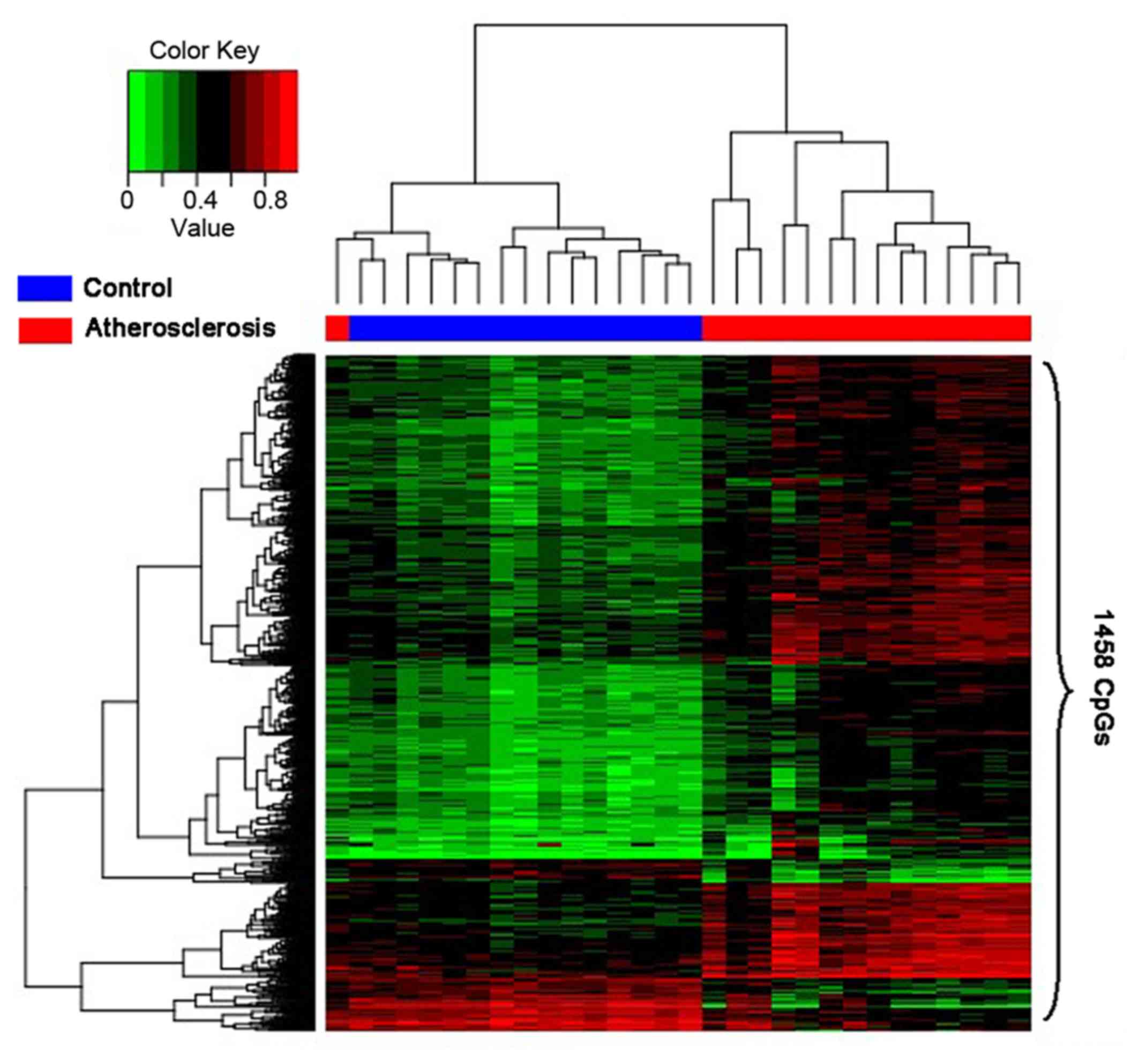
>0.2. Ultimately, 1,458 differently methylated CpGs covering 971
genes extracted by us were considered as the final results.
Hierarchical clustering analysis on
the differential methylated genes
The purpose of extracting the differential
methylated genes was to confirm the related GO terms or biological
pathways based on these genes, which could help us to make certain
the mechanism of the occurrence of atherosclerosis and discover the
approach to diagnose or treat the disease. Therefore, it is
imperative that the reliability of the differential methylated
genes should be verified. The results of the analysis will be
displayed by the heatmap. In consequence, the heatmap (Fig. 2) was able to make clear that the
disease samples had been separated from the controls.
GO analysis on the differential
methylated genes
The purpose of performing GO analysis on the
differential methylated genes is to establish the important
function related to the methylation changes and find the
differential methylated genes corresponding to the related function
regions. By setting the threshold value at P=0.01 and the threshold
value at FDR=0.01, the differential up and down-methylated genes,
respectively, enriched the GO terms according to the classification
of biological process. The classification results are presented in
Tables I and II, which clearly illustrate the relevant
function enrichment.
 | Table ILast 30 GO terms with P-value enriched
with the up-methyalted genes. |
Table I
Last 30 GO terms with P-value enriched
with the up-methyalted genes.
| GO terms | P-value | FDR |
|---|
| Cell adhesion | 4.47E-07 | 2.83E-07 |
| Positive regulation
of GTPase activity | 2.24E-05 | 1.40E-05 |
| Anterior/posterior
pattern specification | 4.37E-05 | 2.75E-05 |
| Positive regulation
of bone mineralization | 1.12E-04 | 6.94E-05 |
| Positive regulation
of endothelial cell migration | 1.52E-04 | 9.43E-05 |
| Negative regulation
of osteoblast differentiation | 2.50E-04 | 1.56E-04 |
| Glossopharyngeal
nerve morphogenesis | 3.30E-04 | 2.06E-04 |
| Positive regulation
of transcription, DNA-templated | 3.81E-04 | 2.39E-04 |
| Peptidyl-tyrosine
phosphorylation | 4.21E-04 | 2.64E-04 |
| Thymus
development | 5.04E-04 | 3.15E-04 |
| Positive regulation
of phosphatidylinositol 3-kinase signaling organization | 5.17E-04 | 3.24E-04 |
| Transforming growth
factor beta receptor signaling pathway | 7.00E-04 | 4.37E-04 |
| Regulation of
epithelial cell migration activity | 7.97E-04 | 4.97E-04 |
| Negative regulation
of transcription from RNA polymerase II promoter | 9.97E-04 | 6.22E-04 |
| Cartilage
development | 0.001039 | 6.51E-04 |
| Cellular response
to transforming growth factor beta stimulus | 0.001243 | 7.87E-04 |
| Positive regulation
of catenin import into nucleus | 0.001394 | 8.66E-04 |
| Embryonic skeletal
system development | 0.00172 | 0.001081 |
| Central nervous
system development | 0.002427 | 0.001510 |
| Extracellular
matrix organization | 0.002647 | 0.001651 |
| Negative regulation
of transcription, DNA-templated | 0.002654 | 0.001654 |
| Positive regulation
of epithelial to mesenchymal transition | 0.002868 | 0.001791 |
| Stress fiber
assembly | 0.00346 | 0.002158 |
| Negative regulation
of sodium ion transmembrane transporter activity | 0.004041 | 0.002519 |
| Lymph vessel
development | 0.004041 | 0.002519 |
| Positive regulation
of MAP kinase activity | 0.004157 | 0.002599 |
| Integrin-mediated
signaling pathway | 0.004187 | 0.002618 |
| Nervous system
development | 0.004236 | 0.002652 |
| Ephrin receptor
signaling pathway | 0.004477 | 0.002786 |
| Axon guidance | 0.004509 | 0.002815 |
| Table IIGO terms with P-value enriched with
the down-methylated genes. |
Table II
GO terms with P-value enriched with
the down-methylated genes.
| GO terms | P-value | FDR |
|---|
| Anterior/posterior
pattern specification | 7.46E-06 | 4.68E-06 |
| Transcription from
RNA polymerase II promoter | 3.18E-04 | 1.99E-04 |
| Positive regulation
of transcription from RNA polymerase II promoter | 0.001554 | 0.000974 |
| Negative regulation
of transcription from RNA polymerase II promoter | 0.002829 | 0.001774 |
| Cell
development | 0.008129 | 0.005063 |
| Thymus
development | 0.00935 | 0.005842 |
| Regulation of
vasculogenesis | 0.010149 | 0.006375 |
Pathway analysis on the differential
methylated genes
The pathway analysis was performed with the
up-methylated and down-methylated genes independently, which could
be expected to discover the related function corresponding to the
development of this disease. In consequence, the results are
presented in Table III. However,
different from the GO analysis there were no down-methylated genes
mapped to the related biological pathways and only 50 pathways
presented with up-methylated genes. For easy observation, only
those in the last 30 pathways with P-value enriched with the
differential methylated genes were chosen, as they were considered
the most relevant biological pathways.
| Table IIIThe 30 pathways with P-value enriched
with the up-methylated genes. |
Table III
The 30 pathways with P-value enriched
with the up-methylated genes.
| Pathways | P-value |
|---|
| Focal adhesion | 3.60E-05 |
| PI3K-Akt signaling
pathway | 2.72E-04 |
| Ras signaling
pathway | 9.84E-04 |
| Regulation of actin
cytoskeleton | 0.001012 |
| HIF-1 signaling
pathway | 0.003851 |
| Glutamatergic
synapse | 0.0044 |
| Rap1 signaling
pathway | 0.005313 |
| cAMP signaling
pathway | 0.006573 |
| Pathways in
cancer | 0.008167 |
| Cholinergic
synapse | 0.009791 |
| Endocytosis | 0.010149 |
| Axon guidance | 0.010285 |
| Mucin type O-Glycan
biosynthesis | 0.01085 |
| Adrenergic
signaling in cardiomyocytes | 0.012417 |
| Adherens
junction | 0.012676 |
| ErbB signaling
pathway | 0.014367 |
| ECM-receptor
interaction | 0.014367 |
| Proteoglycans in
cancer | 0.015585 |
| Hippo signaling
pathway | 0.016108 |
| Tight junction | 0.017989 |
| Fatty acid
metabolism | 0.018393 |
| Osteoclast
differentiation | 0.030227 |
| TGF-beta signaling
pathway | 0.031804 |
| cGMP-PKG signaling
pathway | 0.032235 |
| Insulin
secretion | 0.033815 |
| Choline metabolism
in cancer | 0.034316 |
| Prostate
cancer | 0.040362 |
| Fatty acid
degradation | 0.043614 |
| Oxytocin signaling
pathway | 0.047152 |
| Morphine
addiction | 0.047703 |
Discussion
As a chronic inflammatory disease, atherosclerosis
is involved in activation of adaptive and innate immunity (19). Hypertension, smoking and diabetes
could all become the incentives for this disease (20). The pathogenesis of the disease has
been widely researched by many scholars, including formation of
aggregating homocysteinylated lipoproteins with microorganisms
(21), reactive oxygen species
(22), and microRNA-155(23). However, the pathogenesis has not been
fully elucidated yet. Moreover, because of the irreversible damage
on tissues and organs, the treatment on atherosclerosis is not
ideal. Therefore, in order to clarify the occurrence and
development and reduce the incidence of this disease, finding the
biomarkers for the early detection and prevention has become
critical in dealing with the occurrence of atherosclerosis.
Epigenetics is emerging as an attractive candidate for the update
and future therapy perspectives in atherosclerosis (24). Epigenetics may not only help
understanding the molecular mechanisms of atherosclerosis as
genetic predisposition explains only part of cardiovascular disease
risk, but also play an important role in early diagnosis of some
diseases, such as cancer and adult-onset disease (25). Therefore, as the most studied
epigenetic modification, DNA methylation was investigated in
atherosclerosis by us.
In the present study, 971 differential DNA
methylated genes (1,458 CpGs), including up-methylated and
down-methylated genes were identified with the stringent filtering
criteria. Then, after enriching the up-methylated and
down-methylated genes into the GO terms and biological pathways
respectively, we sought out several critical GO terms and pathways
with lower P-value, which could be considered as the important
function to reveal the occurrence and development of this
disease.
Through the enrichment analysis with up-methylated
genes, there were GO terms and some pathways that attracted our
attention, including the GO terms with cell adhesion and the
pathway with Focal adhesion, PI3K-Akt signaling pathway, Ras
signaling pathway and HIF-1 signaling pathway. Previously, there
has been considerable research focused on the relationship between
cell adhesion and atherosclerosis. Additionally, there have been
research on the treatment of atherosclerosis on mice indicating
that bilirubin can prevent atherosclerotic plaque formation by
inhibiting monocyte migration through the disruption of endothelial
vascular cell adhesion (26). This
discovery demonstrated that the cell adhesion played an important
part in the development of the atherosclerosis. The other point
that deserves attention is the PI3K-Akt signaling pathway.
Similarly, this pathway has been focused on by many scholars. Among
them, Zhai et al (27) found
that the selective inhibition of PI3K-Akt signaling pathway would
markedly affect atherosclerotic plaque inflammation. In addition,
by experiments on mice, quercetin and myricitrin were confirmed to
inhibit atherosclerosis via PI3K-Akt signaling pathway (28). Research on the cell adhesion and
PI3K-Akt signaling pathway confirmed that it can reveal the
development of this disease.
Literature has documented that targeting Ras homolog
gene family member A (RhoA) inhibits the proliferation and
migration of vascular smooth muscle cells (VSMCs) identified as
major cellular events in hypertension-induced vascular remodeling,
closely involved in the progression of atherosclerosis, implicating
targeting Ras signaling is a promising strategy for treating
atherosclerosis (29,30). In addition, previous studies have
demonstrated that HIF-1, as one of the main regulators of cellular
responses in a low-oxygen environment, is recognized to have vital
roles in the development of atherosclerosis via cell-specific
responses, reacting on endothelial cells, VSMCs and macrophages and
emphasized to behave as a potential therapeutic target on the
atherosclerosis development (31,32).
Feng et al (33) concluded
that HIF1α triggers atherosclerosis initiation by inducing
excessive endothelial cell proliferation and inflammation via
inducing production of glycolysis enzymes. Altogether, these
enrichment pathways hold regulatory implications for
atherosclerosis initiation and development, which provides some
valuable insight or feasible means for treating atherosclerosis in
further clinical studies.
Furthermore, the GO terms and pathways enriched by
the down-methylated genes deserve attention. After careful
analysis, the GO term transcription from RNA polymerase II promoter
with the relatively low P-value became a focus of our attention,
which may have great relevance with the occurrence of
atherosclerosis. Specifically, human cytomegalovirus (HCMV) has
been thought to be the most possible etiological factor of
atherosclerosis (34). During late
HCMV infection with atherosclerosis, this virus facilitates viral
transcription by regulating the elongation rate of RNA polymerase
II (35). Therefore, it can explain
the close relationship between RNA polymerase II and the
development of this disease.
Factors such as the location and degree of
methylation (that is, a specific residue can bind to different
numbers of methyl groups) can influence whether gene expression is
promoted or inhibited. Uncovering the underlying mechanism of gene
methylation affecting targeted genes is of significance for our
work and further study.
Increasing evidence has identified that DNA
methylation is involved in multiple processes and diseases,
including atherosclerosis (36-38).
As shown in the supplement Table SII of up-methylated genes,
namely, HOXA3, PIK3R2, FOXD3, MAP3K8, ITPKB, IGF1R, and FOXC2. It
is reported that methylation sites located in HOXA3 are identified
to relate with cholesterol efflux capacity (39). In addition, Balakrishnan et al
(40) demonstrated that promoter DNA
methylation status of PIK3R2, FOXD3, MAP3K8, ITPKB, IGF1R, and
FOXC2 were validated by bisulfite DNA sequencing involved in
activation of insulin signaling and angiogenesis. Furthermore, as
presented in supplement Table SIII of down-methylated genes,
namely, HOXD4, SMAD3, HDAC4. It is determined that HOXD4 and HOXA2
are found to be most pronouncedly hypomethylated in carotid
atherosclerotic plaques in comparison with their methylation
patterns in intact tissues of interior mammary arteries and
saphenous veins (41). HOXD4 is also
confirmed to be hypomethylated in the independent dataset of the
right coronary arteries in advanced atherosclerotic plaques
compared with the other tissues of vascularture (42). Riffo-Campos et al (43) identified that SMAD3 is recognized to
be a hub regulator correlated with metal-related differentially
methylated region and atherosclerosis; moreover, HDAC4 is discerned
to be associated with effectors of atherosclerosis.
In conclusion, compared with the traditional method,
the study of atherosclerosis with DNA methylation used related GO
terms and pathways related to atherosclerosis. After the analysis,
the GO terms and pathways termed cell adhesion, PI3K-Akt signaling
pathway and transcription from RNA polymerase II promoter were
considered to be the most related function regions with the
development of the disease. As a discovery-based study, our
findings may provide a potential biomarker for the early detection
and prevention. Although the analysis results require clinical data
of large samples as support, it still provides a new research
direction for studying atherosclerosis.
Some limitations are present in the current work.
Data from bioinformatics analysis was used in the whole study. Pure
bioinformatics analysis is not prior to integrated bioinformatics
and experimental verification, which is a limitation for our
present study. This work focused on uncovering GO function and
pathways involved in aberrant DNA methylation, thereinto a mass of
methylated genes were included. It is difficult to realize a small
range of gene level detection or methylation level or status
detection, using RT-PCR, MSP and Bisulfite sequencing. Additional
tools for pathway analysis are required. Reactome and IPA were used
to perform pathway analysis and found less pathways were
enriched.
In the present study, we preliminarily chose the
KEGG, as a common pathway enrichment analysis method. Further
research is necessary, with more datasets or our clinical sample
collection, combined with more systematic analytical means.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WC and TS conceived the study and drafted the
manuscript. RL and HW acquired the data. XC and ZC analyzed the
data and revised the manuscript. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hasanov Z, Ruckdeschel T, Kapel S, Mogler
C, Appak S, Spegg C and Augustin HG: Role of Endosialin during
atherosclerosis progression. Atherosclerosis. 241(e14)2015.
View Article : Google Scholar
|
|
2
|
Salic K, Wielinga PY, Verschuren L,
Gjorstrup P, Kleemann R and Kooistra T: Intervention with
anti-inflammatory RVE1 attenuates atherosclerosis without
decreasing plasma cholesterol and adds to the anti-atherogenic
effect of atorvastatin. Atherosclerosis. 235(e267)2014. View Article : Google Scholar
|
|
3
|
Campbell LA and Rosenfeld ME: Infection
and atherosclerosis development. Arch Med Res. 46:339–350.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ross R: The pathogenesis of
atherosclerosis: A perspective for the 1990s. Nature. 362:801–809.
1993.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Campbell JH, Efendy JL, Smith NJ and
Campbell GR: Molecular basis by which garlic suppresses
atherosclerosis. J Nutr. 131:1006–1009. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Portales-Casamar E, Lussier AA, Jones MJ,
MacIsaac JL, Edgar RD, Mah SM, Barhdadi A, Provost S,
Lemieux-Perreault LP, Cynader MS, et al: DNA methylation signature
of human fetal alcohol spectrum disorder. Epigenetics Chromatin.
9(25)2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Razin A and Riggs AD: DNA methylation and
gene function. Science. 210:604–610. 1980.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Esteller M and Herman JG: Cancer as an
epigenetic disease: DNA methylation and chromatin alterations in
human tumours. J Pathol. 196:1–7. 2002.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Masliah E, Dumaop W, Galasko D and
Desplats P: Distinctive patterns of DNA methylation associated with
Parkinson disease: Identification of concordant epigenetic changes
in brain and peripheral blood leukocytes. Epigenetics. 8:1030–1038.
2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gopalakrishnan S, Van Emburgh BO and
Robertson KD: DNA methylation in development and human disease.
Mutat Res. 647:30–38. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wu P, Farrell WE, Haworth KE, Emes RD,
Kitchen MO, Glossop JR, Hanna FW and Fryer AA: Maternal genome-wide
DNA methylation profiling in gestational diabetes shows distinctive
disease-associated changes relative to matched healthy pregnancies.
Epigenetics. 13:122–128. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chen CY and Ye F: Particle swarm
optimization algorithm and its application to clustering analysis.
In: IEEE International Conference on Networking, Sensing and
Control. IEEE, Taiwan. pp789–794. 2004. View Article : Google Scholar
|
|
13
|
Diday E and Simon JC: Clustering analysis.
Commun Cybern. 10:47–94. 1976.
|
|
14
|
Deu-Pons J, Schroeder MP and Lopez-Bigas
N: Heatmap: An interactive heatmap viewer for the web.
Bioinformatics. 30:1757–1758. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kestler HA, Müller A, Kraus JM, Buchholz
M, Gress TM, Liu H, Kane DW, Zeeberg BR and Weinstein JN:
VennMaster: Area-proportional Euler diagrams for functional GO
analysis of microarrays. BMC Bioinformatics. 9(67)2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Du Z, Zhou X, Ling Y, Zhang Z and Su Z:
agriGO: A GO analysis toolkit for the agricultural community.
Nucleic Acids Res. 38:64–70. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ogata H, Goto S, Sato K, Fujibucji W, Bono
H and Kanehisa M: KEGG: Kyoto encyclopedia of genes and genomes.
Nucleic Acids Res. 27:29–34. 1999.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kim HJ: Role of nucleotide-binding and
oligomerization domain 2 protein (NOD2) in the development of
atherosclerosis. Korean J Physiol Pharmacol. 19:479–484.
2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bernhard D, Pfister G, Huck CW, Kind M,
Salvenmoser W, Bonn GK and Wick G: Disruption of vascular
endothelial homeostasis by tobacco smoke: Impact on
atherosclerosis. FASEB J. 17:2302–2304. 2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
McCully KS: Homocysteine and the
pathogenesis of atherosclerosis. Expert Rev Clin Pharmacol.
8:211–219. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Goncharov NV, Avdonin PV, Nadeev AD,
Zharkikh IL and Jenkins RO: Reactive oxygen species in pathogenesis
of atherosclerosis. Curr Pharm Des. 21:1134–1146. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ma X, Ma C and Zheng X: MicroRNA-155 in
the pathogenesis of atherosclerosis: A conflicting role? Heart Lung
Circ. 22:811–818. 2013. View Article : Google Scholar
|
|
24
|
Khyzha N, Alizada A, Wilson MD and Fish
JE: Epigenetics of atherosclerosis: Emerging mechanisms and
methods. Trends Mol Med. 23:332–347. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bustos FJ, Ampuero E, Jury N, Aguilar R,
Falahi F, Toledo J, Ahumada J, Lata J, Cubillos P, Henríquez B, et
al: Epigenetic editing of the Dlg4/PSD95 gene improves cognition in
aged and Alzheimer's disease mice. Brain. 140:3252–3268.
2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kawamoto R, Ninomiya D, Hasegawa Y, Kasai
Y, Kusunoki T, Ohtsuka N, Kumagi T and Abe M: Mildly elevated serum
bilirubin levels are negatively associated with carotid
atherosclerosis among elderly persons. PLoS One.
9(e114281)2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhai C, Cheng J, Mujahid H, Wang H, Kong
J, Yin Y, Li J, Zhang Y, Ji X and Chen W: Selective inhibition of
PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage
and vulnerability of atherosclerotic plaque. PLoS One.
9(e90563)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lu XL, Zhao CH, Yao XL and Zhang H:
Quercetin attenuates high fructose feeding-induced atherosclerosis
by suppressing inflammation and apoptosis via ROS-regulated
PI3K/AKT signaling pathway. Biomed Pharmacother. 85:658–671.
2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cui C, Wang X, Shang XM, Li L, Ma Y, Zhao
GY, Song YX, Geng XB, Zhao BQ, Tian MR, et al: lncRNA 430945
promotes the proliferation and migration of vascular smooth muscle
cells via the ROR2/RhoA signaling pathway in atherosclerosis. Mol
Med Rep. 19:4663–4672. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yu MH, Lin MC, Huang CN, Chan KC and Wang
CJ: Acarbose inhibits the proliferation and migration of vascular
smooth muscle cells via targeting Ras signaling. Vascul Pharmacol.
103-105:8–15. 2018. View Article : Google Scholar
|
|
31
|
Jain T, Nikolopoulou EA, Xu Q and Qu A:
Hypoxia inducible factor as a therapeutic target for
atherosclerosis. Pharmacol Ther. 183:22–33. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Tanaka T and Eckardt KU: HIF activation
against CVD in CKD: Novel treatment opportunities. Semin Nephrol.
38:267–276. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Feng S, Bowden N, Fragiadaki M, Souilhol
C, Hsiao S, Mahmoud M, Allen S, Pirri D, Ayllon BT, Akhtar S, et
al: Mechanical activation of hypoxia-inducible factor 1α drives
endothelial dysfunction at atheroprone sites. Arterioscler Thromb
Vasc Biol. 37:2087–2101. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chen R, Xiong S, Yang Y, Fu W, Wang Y and
Ge J: The relationship between human cytomegalovirus infection and
atherosclerosis development. Mol Cell Biochem. 249:91–96.
2003.PubMed/NCBI
|
|
35
|
Perng YC, Campbell JA, Lenschow DJ and Yu
D: Human cytomegalovirus pUL79 is an elongation factor of RNA
polymerase II for viral gene transcription. PLoS Pathog.
10(e1004350)2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tabaei S and Tabaee SS: DNA methylation
abnormalities in atherosclerosis. Artif Cell Nanomed Biotechnol.
47:2031–2041. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jiang D, Sun M, You L, Lu K, Gao L, Hu C,
Wu S, Chang G, Tao H and Zhang D: DNA methylation and
hydroxymethylation are associated with the degree of coronary
atherosclerosis in elderly patients with coronary heart disease.
Life Sci. 224:241–248. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Aavik E, Babu M and Ylä-Herttula S: DNA
methylation processes in atheosclerotic plaque. Atherosclerosis.
281:168–179. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Sayols-Baixeras S, Hernáez A, Subirana I,
Lluis-Ganella C, Muñoz D, Fitó M, Marrugat J and Elosua R: DNA
methylation and high-density lipoprotein functionality - brief
report: The REGICOR study (Registre Gironi del Cor). Arterioscler
Thromb Vasc Biol. 37:567–569. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Balakrishnan A, Guruprasad KP,
Satyamoorthy K and Joshi MB: Interleukin-6 determines protein
stabilization of DNA methyltransferases and alters DNA promoter
methylation of genes associated with insulin signaling and
angiogenesis. Lab Invest. 98:1143–1158. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Nazarenko MS, Markov AV, Lebedev IN,
Sleptsov AA, Frolov AV, Barbash OL and Puzyrev VP: DNA methylation
profiling of the vascular tissues in the setting of
atherosclerosis. Mol Biol (Mosk). 47:398–404. 2013.PubMed/NCBI View Article : Google Scholar : (In Russian).
|
|
42
|
Nazarenko MS, Markov AV, Lebedev IN,
Freidin MB, Sleptcov AA, Koroleva IA, Frolov AV, Popov VA,
Barbarash OL and Puzyrev VP: A comparison of genome-wide DNA
methylation patterns between different vascular tissues from
patients with coronary heart disease. PLoS One.
10(e0122601)2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Riffo-Campos AL, Fuentes-Trillo A, Tang
WY, Soriano Z, De Marco G, Rentero-Garrido P, Adam-Felici V,
Lendinez- Tortajada V, Francesconi K and Goessler W: In silico
epigenetics of metal exposure and subclinical atherosclerosis in
middle aged men: Pilot results from the Aragon Workers Health
Study. Philos Trans R Soc Lond B Biol Sci. 373(1748)2018.PubMed/NCBI View Article : Google Scholar
|