Introduction
Waardenburg syndrome (WS) is a rare genetic disorder
characterized by various degrees of deafness, abnormal pigmentation
and minor defects in structures arising from neural crest. WS was
first introduced by the ophthalmologist Petrus J Waardenburg in
1947 and was described in further detail in 1951 (1,2).
Overall, the syndrome affects 1 in 42,000 people in the population
(1). WS is caused by mutations of
several genes that affect the division and migration of neural
crest cells during embryonic development. Pathogenic genes
associated with WS include PAX3, MITF, SNAI2, EDN3, EDNRB and SOX10
(2,3). The clinical characteristics of the
syndrome include lateral displacement of the eye's medial canthi, a
hyperplastic broad high nasal root, hyperplasia of the medial
portions of the eyebrows, partial or total heterochromia iridum,
congenital deafness or partial (unilateral) deafness and
circumscribed albinism of the frontal head hair (white forelock)
(1). Currently, there are four
different types of WS according to various clinical
characteristics. WS type 1 is characterized by dystopia canthorum,
congenital sensorineural hearing loss, pigmentary disturbances of
iris and hair hypopigmentation (2).
WS type 1 and WS type 2 are distinguished by the presence or
absence of dystopia canthorum, respectively. WS type 3
(Klein-Waardenburg syndrome) is similar to type 1 with additional
musculoskeletal abnormalities. WS type 4 (Shah-Waardenburg syndrome
or Waardenburg-Hirschsprung disease) is characterized by the
presence of an aganglionic megacolon (3). The highly variable presentations of WS
make it difficult to reach a definitive diagnosis. Studies
reporting ocular manifestations are limited. Müllner-Eidenböck
et al (4) presented Turkish
family members with WS type 2 who presented with a fundus photo
with ipsilateral connections between the iris and fundus.
Cortés-González et al (5)
suggested that posterior microphthalmos may be associated with WS
type 2A. Previous studies have also reported that patients with WS
may suffer from additional open-angle glaucoma or branch retinal
vein occlusion (6,7). Furthermore, Meire et al
(8) reported a patient with WS who
presented with Marcus Gunn ptosis with jaw-winking. Despite
numerous findings connecting WS syndrome and intraocular
abnormalities, little is known regarding the abnormal configuration
of the eye and its comorbidities in WS. Therefore, the present
article focused on the external ocular manifestations in five
patients diagnosed with WS, and suggested potential treatments to
improve the ocular appearance of these patients.
Materials and methods
The present study followed the principles of the
Declaration of Helsinki and was approved by The Ethics Committee of
the Ninth People's Hospital, Shanghai Jiao Tong University School
of Medicine. Informed consent for publication of the images was
provided by the guardians of the patients. A total of five Chinese
patients with a clinical diagnosis of WS from the same age group,
examined in the Department of Ophthalmology, Ninth People's
Hospital, Shanghai Jiao Tong University School of Medicine
(Shanghai, China) between March 2014 and July 2018, were included
in the present study. The criteria for diagnosis of WS were based
on the Waardenburg Consortium, indicating that affected individuals
must demonstrate at least 2 major criteria, or 1 major criterion
plus 2 minor criteria (Table I)
(2,9,10).
 | Table IWaardenburg Consortium diagnostic
criteria for Type 1. |
Table I
Waardenburg Consortium diagnostic
criteria for Type 1.
| Major criteria | Minor criteria |
|---|
| Congenital
sensorineural hearing loss | Congenital
hypopigmentation of the skin |
| Pigmentary
disturbance of iris | Synophrys (eyebrows
that meet) |
| Pigmentary
disturbance of hair (white forelock) | Broad high nasal
root |
| Dystopia canthorum, W
index >1.95 | Hypoplasia of alae
nasi |
| Affected first degree
relative | Premature greying of
hair |
Data collected from patients included age, sex,
family history of WS, hair color, skin signs, hearing (confirmed by
audiology testing) and characteristic facial features (eyebrow
shape, nasal profile and nasal alae). No patients presented with
systemic diseases. Each patient underwent visual acuity, slit lamp,
fundus and external ocular examination. The collected data involved
color analysis of the iris and fundus images by anterior segment
and fundus photography, intraocular pressure, retinal nerve fiber
layer thickness and cup/disc ratio. External ocular examination
includes eyelid measurements of palpebral fissure width (FW),
interpupillary distance, inner and outer canthal distance, upper
eyelid margin reflex distance (the distance from the central
corneal light reflection to the upper lid margin distance; MRD1)
(11,12) and levator muscle function (LMF).
Surgical correction of ptosis was performed by one
surgeon (JL). The different plastic procedures (levator resection
or frontalis suspension) depended on the degree of ptosis and inner
canthus forms (13-15).
Patients with glaucoma were treated with anti-glaucoma medications
(Carteolol hydrochloride 1% twice a day) (16). For patients with iris heterochromia
only, follow-up lasted for 2-3 years, including visual acuity,
intraocular pressure and external ocular examinations.
Results
A total of five Chinese patients with WS (four male
and one female) were included in the present study. The mean age at
presentation was 3.8 years (median age, 3.5 years; age range, 1-8
years). All patients met the diagnostic criteria for WS type 1
(Table I). No patient had muscle
contractures (WS type 3) or Hirschsprung disease (WS type 4). As
listed in Table II, the general
manifestations included hearing loss (1/5), broad high nasal root
(1/5) and hypoplasia of alae nasi (1/5). Ophthalmological
evaluations demonstrated ptosis (1/5), strabismus (1/5), synophrys
(2/5), dystopia canthorum (W >1.95) (5/5), iris hypopigmentation
(5/5), high intraocular pressure (1/5) and choroidal
hypopigmentation (1/5). The ophthalmological manifestations are
presented in Table III.
| Table IIGeneral manifestations of Waardenburg
Syndrome in 5 patients. |
Table II
General manifestations of Waardenburg
Syndrome in 5 patients.
| | | | | Major criteria | Minor criteria |
|---|
| Case | Age, years | Sex | W index | Hearing loss | Disturbance of
iris | Disturbance of
hair | Dystopia
canthorum | Affected first degree
relative | Hypopigmentation of
the skin | Synophrys | Broad high nasal
root, hypoplasia of alae nasi | Premature greying of
hair |
|---|
| 1 | 3.5 | M | 1.97 | + | + | - | + | - | - | - | - | - |
| 2 | 8 | M | 2.89 | - | + | - | + | - | - | + | + | - |
| 3 | 1.5 | M | 1.99 | - | + | - | + | - | - | - | - | - |
| 4 | 1 | M | 2.35 | - | + | - | + | - | - | - | - | - |
| 5 | 5 | F | 2.23 | - | + | - | + | - | - | + | + | - |
| Table IIIOphthalmological manifestations of
Waardenburg Syndrome. |
Table III
Ophthalmological manifestations of
Waardenburg Syndrome.
| | | | | Iris
heterochromia | | | External ocular
examination |
|---|
| Cases | Eye | Visual acuity | Intraocular
pressure (mmHg) | Iris color | Sector or
diffuse | Optic nerve
anomalies | Choroidal
hypopigmentation | Strabismus | Ptosis | Heavy eyebrows,
medial portion | Telecanthus |
|---|
| 1 | OD | 20/80 | 12 | Pale blue | Diffuse | - | - | - | + | - | + |
| | OS | 20/50 | 11 | Pale blue | Diffuse | - | - | - | - | | |
| 2 | OD | 20/20 | 34 | Pale blue | Diffuse | C/D=0.6 | + | - | - | + | + |
| | OS | 20/20 | 30 | Normal | - | C/D=0.6 | - | + | - | | |
| 3 | OD | N/A | 16 | Pale blue | Diffuse | - | - | - | - | - | + |
| | OS | N/A | 16 | Normal | - | - | - | - | - | | |
| 4 | OD | N/A | 11 | Normal | - | - | - | - | - | - | + |
| | OS | N/A | 11 | Pale blue | Diffuse | - | - | - | - | | |
| 5 | OD | 20/20 | 15 | Pale blue | Diffuse | - | - | - | - | + | + |
| | OS | 20/20 | 16 | Normal | - | - | - | - | - | | |
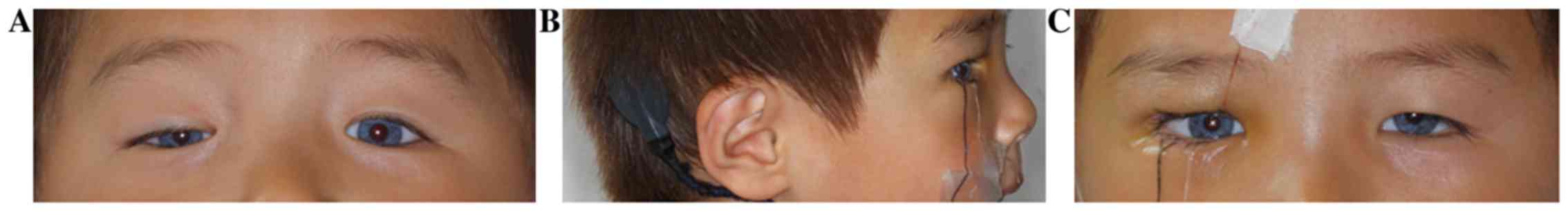
Case 1
A 3-year and 6-month-old male child presented with
ptosis in his right eye (Fig. 1).
Prior to examination, the patient had a history of congenital
bilateral hearing loss and was treated by cochlear implantation for
2 years. The patient exhibited complete iris hypopigmentation (pale
blue eyes) bilaterally. He did not have any neurological defect or
skin abnormality, and had no family history of WS. The uncorrected
distance visual acuity (UDVA) was 20/80 in the right eye and was
20/50 in the left eye. Cycloplegic refraction for the right eye
[oculus dexter (OD)] was: +3.25 diopters sphere (DS)/-0.75 diopters
cylinder (DC) x160˚, with a corrected distance visual acuity (CDVA)
of 20/50. Cycloplegic refraction for the left eye [oculus sinister
(OS)] was: +2.75 DS/-0.75DC x150˚, with CDVA of 20/30. The FW was
17 mm in the right eye and was 18 mm in the left eye. The MRD1 was
0 in the right eye and 3 mm in the left eye. The LMF was 2 mm in
the right eye and 9 mm in the left eye. For the development of
vision, ptosis of the patient was surgically corrected by levator
resection. The patient underwent timely treatment of amblyopia in
order to improve visual function (17). Current treatment for amblyopia mainly
includes optical correction, patching, pharmacological treatment,
optical treatment, Bangerter filters and surgery (17). After 3 years of postoperative
amblyopia therapy the patient's visual function was well developed.
The UDVA was 20/30 in the right eye and 20/20 in the left eye.
Cycloplegic refraction was OD: +2.75 DS/-0.75 DC x160˚, with CDVA
of 20/20, and OS: +1.50 DS/-1.25 DC x160˚, with CDVA of 20/20. This
patient underwent levator resection in hospital, and him and his
parents underwent genetic testing. Complete sequence analysis of
PAX3, MITF and SOX10 genes was performed. The results demonstrated
that no pathogenic mutations were detected.
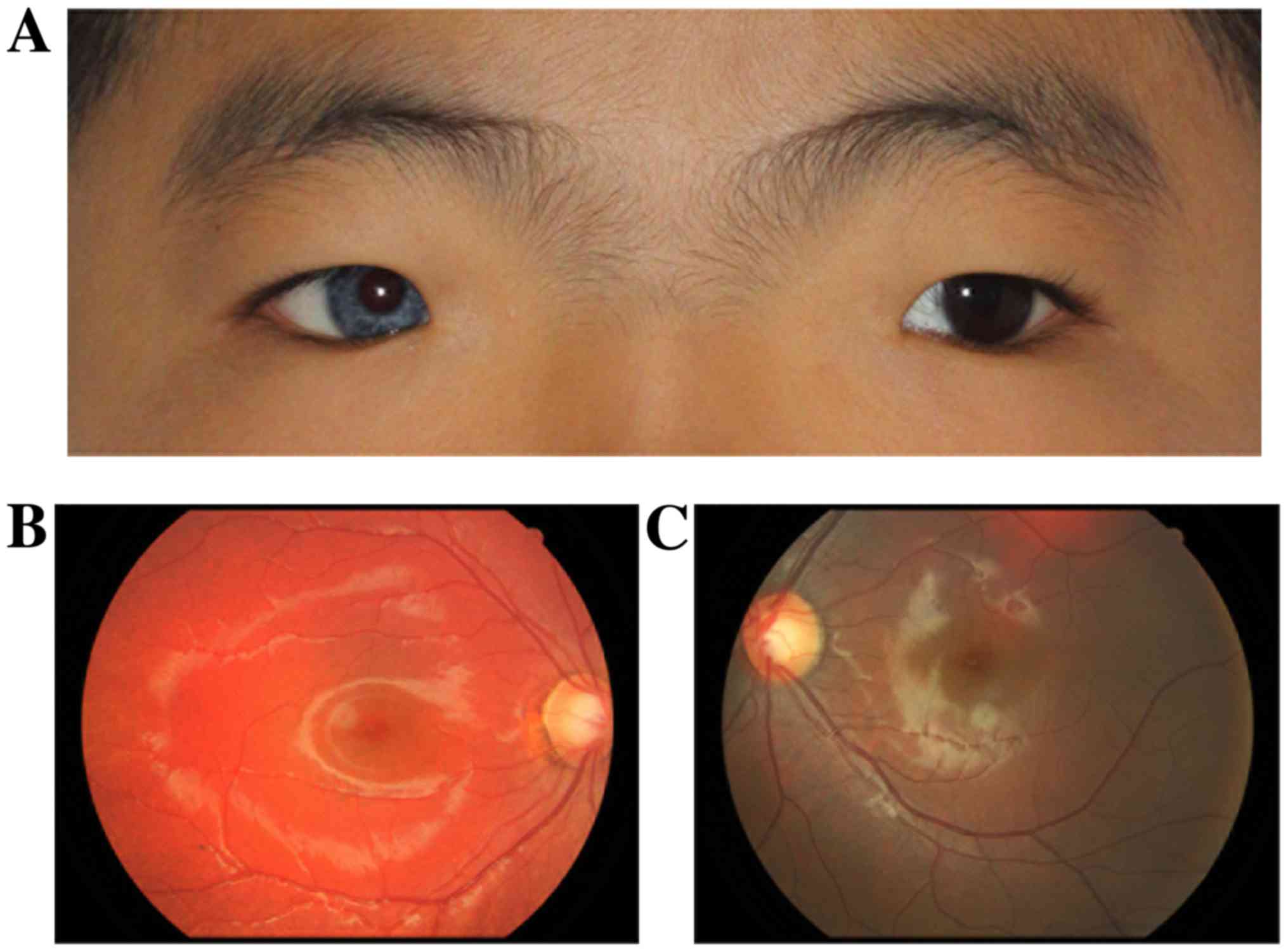
Case 2
An 8-year-old male child presented with strabismus
in his left eye (Fig. 2).
Ophthalmological examination demonstrated complete iris
hypopigmentation (pale blue eye) unilaterally. There was an
increased difference between the medial canthus of the eyes and
broad nasal root. The inner canthal distance was 55 mm. The FW was
19 mm, the MRD1 was 3 mm and the LMF was 10 mm in both eyes. No
hearing loss, neurological defects or skin abnormalities were
reported in this case. The binocular vision was 20/20 and there was
a post-equatorial hypopigmented choroid with scattered pigmented
spots in the right eye compared with the left one. Examination
showed enlarged cup to disc ratio (C/D) in both eyes [oculus
uterque [(OU) (OU C/D=0.6)] and high intraocular pressure (OD=35
mmHg, OS=33 mmHg), but RNFL thickness was within the normal range,
suggesting early onset of elevated intraocular pressure. The type
of glaucoma was open angle glaucoma. The peripheral anterior
chamber depth was >1/3 of the peripheral corneal thickness in
slit-lamp examination and no peripheral anterior synechia was seen.
The patient was treated with anti-glaucoma eye drops (Carteolol
hydrochloride 1% twice a day) and the intraocular pressure was well
controlled at the 3-year follow-up. There was a post-equatorial
hypopigmented choroid with scattered pigmented spots in the right
fundus photo. The enlarged inner canthal distance did not affect
the patient's visual function, and no plastic surgery was
needed.
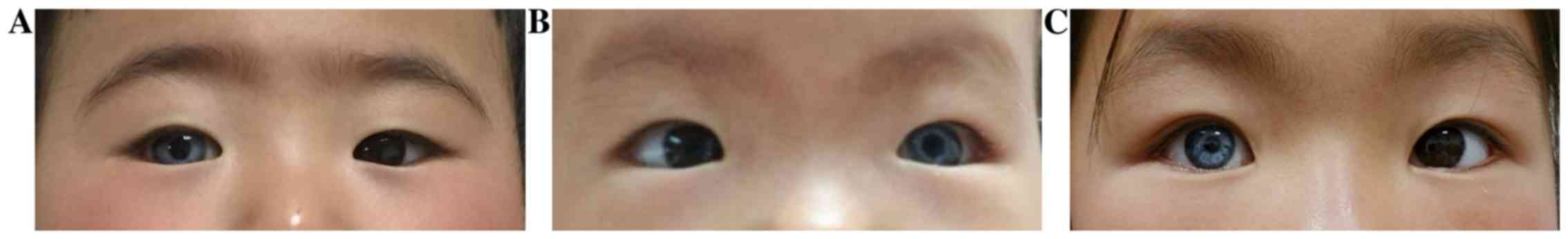
Cases 3-5
Cases 3-5 presented with the initial complaint of
abnormal color of the iris (Fig. 3).
The children in case 3 (a 1-year and 5-month-old boy) and case 4 (a
1-year-old boy) were too young to perform a visual examination; the
eye examination therefore only assessed eyesight, which was
followed up regularly. The MRD1 was 3 mm and the LMF was 10 mm in
both eyes in case 3. In case 4, the MRD1 was 4 mm and the LMF was
10 mm in both eyes. Furthermore, during examination of case 5 (A
5-year-old girl), the patient was diagnosed with a slight
photophobia, although no intraocular defect was found except for
iris morphology. The visual acuity was 20/20 in both eyes. The MRD1
was 4 mm and the LMF was 11 mm in both eyes. For patients with
symptoms of iris heterochromia only, no special treatment was
required and colored contact lenses could be worn.
Discussion
WS is characterized by a variety of typical clinical
manifestations caused by pigmentation disorders. Pathogenic genes
related to WS include PAX3, MITF, SNAI2, EDN3, EDNRB and SOX10
(2,3). In the present study, the patient in
case 1 underwent levator resection due to ptosis in hospital;
therefore, the patient and their parents underwent genetic testing.
Complete sequence analysis of the PAX3, MITF and SOX10 genes was
performed. The results demonstrated that no pathogenic mutations
were detected. The other four children were examined at the
outpatient clinic and diagnosed with WS type 1. At the time of the
study, there was no special treatment, and their parents declined
genetic testing.
The clinical manifestations of pigment synthesis
disorders include premature whitening of the hair, hypopigmentation
of the skin and heterochromia of the iris, accompanied by various
degrees of sensorineural hearing loss. Patients may also have other
systematic signs, such as dystopia canthorum, dysplastic skeletal
muscles of the extremities, Hirschsprung disease and abnormalities
of the intestinal nervous system (18). According to Farrer's 1992 WS
diagnostic criteria (9), all cases
in the present study were classified as WS type 1. Certain signs,
including ptosis, synophrys, telecanthus, iris hypopigmentation,
choroidal hypopigmentation, high intraocular pressure and
strabismus were identified. For WS, the ocular abnormal
manifestations are diverse. Subsequently, the eyes of patients with
WS must be examined carefully to rule out eye problems. If there
are any problems, proper intervention and treatment are
necessary.
Ptosis, which refers to abnormal drooping of the
upper lid due to partial or total loss of levator function
(11), is one of the most common
congenital abnormalities in children, although it is a rare
characteristic in WS. Clinical features include continuous
limitations in the visual field and, and in some severe cases,
amblyopia. Patients may need to raise their eyebrows or lift their
eyelid with a finger (19). The
specific facial features, including increased forehead lines,
raised eyebrows and looking upwards, not only affect appearance but
also affect the patient's psychological development to varying
degrees. Surgery is an effective method of correcting ptosis.
Ptosis has been reported in the literature regarding WS, but it has
not been described in detail, nor have specific treatments been
described. Chen et al (3) and
Tamayo et al (20) reported
cases of ptosis in WS; however, they did not discuss treatments.
Meire et al (8) hypothesized
that the occurrence of Marcus Gunn ptosis in a patient with WS may
suggest a lesion in the sympathetic pathway.
The correct time for surgery in WS-associated ptosis
is not well established. Since the patient can prevent the
interference of the upper eyelid when looking up or looking down,
less amblyopia, strabismus or refractive errors occur. Therefore,
for mild to moderate ptosis, surgery is considered to be better at
3-6 years of age. For severe cases, early intervention is suggested
to protect their visual development and prevent amblyopia. In case
1 of the present study, the levator muscle strength of the patient
was 2 mm, which was a severe ptosis that met the surgical
correction criteria. The operation method was levator resection.
Following the operation, the ptosis of the patient was well
corrected. After 3 years of postoperative amblyopia training, the
patient's visual function was well developed. The UDVA was 20/30 in
the right eye and 20/20 in the left eye. Cycloplegic refraction was
OD: +2.75 DS/-0.75 DC x160˚ with CDVA of 20/20, and OS: +1.50
DS/-1.25 DC x160˚ with CDVA of 20/20. No amblyopia was observed in
this patient. Previous studies have reported that children with
amblyopia due to astigmatic anisometropia or deprivation could
benefit from early surgical correction (21,22).
Kadoi et al (7) reported a case of WS with hypopigmented
fundi, branch retinal vein occlusion and high intraocular pressure.
Nork et al (23) and Gupta
and Aggarwal (6) reported cases of
WS with bilateral glaucoma. Although glaucoma has not been
considered as an associated characteristic of WS, a possible
mechanism could be that ocular melanocytes may be derived from the
neural crest and that a defect in pigmentation may therefore lead
to developmental abnormalities in these structures. As discussed in
the literature, the iridocorneal angle structures are largely of
neural crest origin, and the predisposition to glaucoma should be
investigated with a larger number of WS cases, which may support
the association of glaucoma with WS (6). In the case 2 of the present study, the
patient had high intraocular pressure and enlarged cup to disc
ratio. This patient was treated with anti-glaucoma eye drops and
follow-up observation was needed regularly. This finding suggested
that examination of intraocular pressure, optic disc ratio and RNFL
measurement for patients with WS may be necessary.
The characteristic ocular abnormality of WS is
abnormal pigmentation of the iris. Iris hypopigmentation occurred
in all 5 patients from the present study. Changes in iris
pigmentation in WS included the following: Complete heterochromia
(different colors in both eyes), partial heterochromia (segmented
blue) and stunted bright blue iris (24). In the present study, all 5 patients
presented with complete heterochromia and no partial heterochromia
was observed. Patients with heterochromia of the iris in WS may
need to use sunglasses to protect the intraocular structures of the
eye against excess radiation from the sun's rays. Colored contact
lenses can be used for cosmetic reasons.
In the present study, several abnormal
characteristics of the eyes were reported, including synophrys,
epicanthal folds, telecanthus, ptosis and strabismus. Not only the
external abnormalities, but also the intraocular defects of
patients with WS should be carefully examined in clinic.
Abnormalities in the external eye, such as ptosis, epicanthal folds
or telecanthus, can be corrected by ocular plastic surgery, which
is beneficial to the physical development of child.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81870688 and 81970834) and
the Science and Technology Commission of Shanghai (grant no.
17DZ2260100).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and HP obtained the data and wrote the
manuscript. JW, QY and ML analyzed the datasets. BM and LJ designed
the study and critically revised the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
the Ninth People's Hospital, Shanghai Jiao Tong University School
of Medicine (Shanghai, China).
Patient consent for publication
Informed consent for publication of the images was
provided by the guardians of the patients
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Waardenburg PJ: A new syndrome combining
developmental anomalies of the eyelids, eyebrows and nose root with
pigmentary defects of the iris and head hair and with congenital
deafness. Am J Hum Genet. 3:195–253. 1951.PubMed/NCBI
|
|
2
|
Read AP and Newton VE: Waardenburg
syndrome. J Med Genet. 34:656–665. 1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chen H, Jiang L, Xie Z, Mei L, He C, Hu Z,
Xia K and Feng Y: Novel mutations of PAX3, MITF, and SOX10 genes in
Chinese patients with type I or type II Waardenburg syndrome.
Biochem Biophys Res Commun. 397:70–74. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Müllner-Eidenböck A, Moser E, Frisch H and
Read AP: Waardenburg syndrome type 2 in a Turkish family:
Implications for the importance of the pattern of fundus
pigmentation. Br J Ophthalmol. 85:1384–1386. 2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cortés-González V, Zenteno JC,
Guzmán-Sánchez M, Giordano-Herrera V, Guadarrama-Vallejo D,
Ruíz-Quintero N and Villanueva-Mendoza C: Tietz/Waardenburg type 2A
syndrome associated with posterior microphthalmos in two unrelated
patients with novel MITF gene mutations. Am J Med Genet A.
170:3294–3297. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gupta V and Aggarwal HC: Open angle
glaucoma as a manifestation of Waardenburg's syndrome. Indian J
Ophthalmol. 48:49–50. 2000.PubMed/NCBI
|
|
7
|
Kadoi C, Hayasaka S and Yamamoto S: Branch
retinal vein occlusion in a patient with Waardenburg syndrome.
Ophthalmologica. 210:354–357. 1996.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Meire F, Standaert L, De Laey JJ and Zeng
LH: Waardenburg syndrome, Hirschsprung megacolon, and Marcus Gunn
ptosis. Am J Med Genet. 27:683–686. 1987.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Farrer LA, Grundfast KM, Amos J, Arnos KS,
Asher JH Jr, Beighton P, Diehl SR, Fex J, Foy C, Friedman TB, et
al: Waardenburg syndrome (WS) type I is caused by defects at
multiple loci, one of which is near ALPP on chromosome 2: First
report of the WS consortium. Am J Hum Genet. 50:902–913.
1992.PubMed/NCBI
|
|
10
|
Shields CL, Nickerson SJ, Al-Dahmash S and
Shields JA: Waardenburg syndrome: Iris and choroidal
hypopigmentation: Findings on anterior and posterior segment
imaging. JAMA Ophthalmol. 131:1167–1173. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li J, Lin M, Zhou H, Jia R and Fan X:
Double-eyelid blepharoplasty incorporating blepharoptosis surgery
for ‘latent’ aponeurotic ptosis. J Plast Reconstr Aesthet Surg.
64:993–999. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Quaranta-Leoni FM, Sposato S, Leonardi A,
Iacoviello L and Costanzo S: Timing of surgical correction for the
treatment of unilateral congenital ptosis: Effects on cosmetic and
functional results. Orbit. 36:382–387. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Clauser L, Tieghi R and Galie M: Palpebral
ptosis: Clinical classification, differential diagnosis, and
surgical guidelines: An overview. J Craniofac Surg. 17:246–254.
2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Gazzola R, Piozzi E, Vaienti L and Wilhelm
Baruffaldi Preis F: Therapeutic algorithm for congenital ptosis
repair with levator resection and frontalis suspension: Results and
literature review. Semin Ophthalmol. 33:454–460. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Dave TV, Sharma P, Nayak A, Moharana R and
Naik MN: Outcomes of frontalis sling versus levator resection in
patients with monocular elevation deficiency associated ptosis.
Ophthalmic Plast Reconstr Surg. 35:251–255. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chrisp P and Sorkin EM: Ocular carteolol.
A review of its pharmacological properties, and therapeutic use in
glaucoma and ocular hypertension. Drugs Aging. 2:58–77.
1992.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wallace DK, Repka MX, Lee KA, Melia M,
Christiansen SP, Morse CL and Sprunger DT: American Academy of
Pediatric Ophthalmology/Strabismus Preferred Practice Pattern
Pediatric Ophthalmology Panel: Amblyopia preferred practice
pattern®. Ophthalmology. 125:P105–P142. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Liu XZ, Newton VE and Read AP: Waardenburg
syndrome type II: Phenotypic findings and diagnostic criteria. Am J
Med Genet. 55:95–100. 1995.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cahill KV, Bradley EA, Meyer DR, Custer
PL, Holck DE, Marcet MM and Mawn LA: Functional indications for
upper eyelid ptosis and blepharoplasty surgery: A report by the
American Academy of Ophthalmology. Ophthalmology. 118:2510–2517.
2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tamayo ML, Gelvez N, Rodriguez M, Florez
S, Varon C, Medina D and Bernal JE: Screening program for
Waardenburg syndrome in Colombia: Clinical definition and
phenotypic variability. Am J Med Genet A. 146A:1026–1031.
2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Akal A, Göncü T, Boyaci N and Yilmaz ÖF:
Anisometropic amblyopia in a case of type 2 Waardenburg syndrome.
BMJ Case Rep. 2013(bcr2013201140)2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
SooHoo JR, Davies BW, Allard FD and
Durairaj VD: Congenital ptosis. Surv Ophthalmol. 59:483–492.
2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Nork TM, Shihab ZM, Young RS and Price J:
Pigment distribution in Waardenburg's syndrome: A new hypothesis.
Graefes Arch Clin Exp Ophthalmol. 224:487–492. 1986.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ohno N, Kiyosawa M, Mori H, Wang WF,
Takase H and Mochizuki M: Clinical findings in Japanese patients
with Waardenburg syndrome type 2. Jpn J Ophthalmol. 47:77–84.
2003.PubMed/NCBI View Article : Google Scholar
|