Introduction
Cardiac remodelling is a series of complex molecular
and cellular mechanisms that lead to changes in cardiac structure,
function and phenotype (1). The most
common structural changes in pressure overload-induced cardiac
remodelling are left ventricular hypertrophy and interstitial
fibrosis (1). Pressure overload is a
cause of cardiac remodelling (2).
Pathological cardiac remodelling is associated with a series of
pathophysiological changes, involving oxidative stress (3), inflammation (4), apoptosis (5) and autophagy (6). Previous studies have indicated that
oxidative stress plays a vital role in the progression of cardiac
remodelling (7,8). NADPH oxidase (NOX) is one of the main
sources of reactive oxygen species (ROS) (7) and mice overexpressing NOX4 have
demonstrated an 8-fold increase in the production of ROS (8). Increased ROS not only mediates
cardiomyocyte hypertrophy and apoptosis, but also inactivates
nitric oxide (NO), which can induce or aggravate cardiac fibrosis
(9). Therefore, the inhibition of
oxidative stress may serve as a potential target for the treatment
of cardiac remodelling.
Carnosic acid (CA) is a phenolic terpenoid separated
from Rosmarinus officinalis that exerts multiple pharmacological
effects, including antioxidative stress (10), anti-inflammation (11) and anti-tumour effects (12). A previous study has revealed that CA
inhibits arsenic-induced hepatotoxicity by inhibiting oxidative
stress (10). Furthermore, CA
inhibits liver ischaemia/reperfusion injury by reducing ROS
(13). A previous study has
indicated that isoproterenol-induced cardiac oxidative stress
activates apoptosis-associated pathways directly or via ROS and
that pre-treatment with CA inhibits oxidative stress and apoptosis
in mice (14). Previous studies have
revealed that phosphorylated (p-) AKT and p-glycogen synthase
kinase 3 β (GSK3β) serve important roles in cardiac remodelling
(15,16). CA inhibits renal fibrosis and
oxidative stress by suppressing AKT-mediated NOX4(17). However, the effects of CA on pressure
overload-induced cardiac remodelling and the molecular mechanisms
underlying this remain unclear.
According to previous studies (15,16,17), CA
improves cardiac remodelling induced by pressure overload via the
AKT/GSK3β/NOX4 signalling pathway. Therefore, the present study
investigated the potential protective role of CA in cardiac
remodelling.
Materials and methods
Chemicals and reagents
CA (Fig. 1A; >98%
purity, as detected by high-performance liquid chromatography) was
acquired from Shanghai Winherb Medical S&T Development Co.,
Ltd., (cat. no. 3650--09-7). Total Superoxide Dismutase (SOD) Assay
Kit with WST-8 (cat. no. S0101), Lipid Peroxidation malondialdehyde
(MDA; cat. no. S0131) Assay kit and NADP+/NADPH Assay kit with
WST-8 (cat. no. S0179) were purchased from Beyotime Institute of
Biotechnology. The following antibodies were purchased from Abcam:
NOX2 (1:1,000; cat. no. ab80508), NOX4 (1:1,000; cat. no.
ab154244), collagen I (1:1,000; cat. no. ab34710), collagen III
(1:1,000; cat. no. ab7778) and connective tissue growth factor
(CTGF; 1:1,000; cat. no. ab209780). The following antibodies were
purchased from Cell Signalling Technology, Inc.: Bax (1:1,000; cat.
no. 2722), Bcl-2 (1:1,000; cat. no. 2870), cleaved (C-) caspase-3
(1:1,000; cat. no. 9661), p-AKT (1:1,000; cat. no. 4060), total
(t)-AKT (1:1,000; cat. no. 4691), p-GSK3β (1:1,000; cat. no. 9323),
T-GSK3β (1:1,000; cat. no. 9315) and GAPDH (1:1,000; cat. no.
2118). The following antibodies were obtained from Santa Cruz
Biotechnology, Inc: Atrial natriuretic peptide (ANP; 1:1,000; cat.
no. sc-20158), brain natriuretic peptide (BNP; 1:1,000; cat. no.
sc-271185) and β-myosin heavy chain (β-MHC; 1:1,000; cat. no.
sc-53090). These antibodies were normalized to GAPDH expression.
Secondary antibodies including IRDye 800CW Goat anti-Mouse (cat.
no. 926-32210) and IRDye 800CW Goat anti-Rabbit (cat. no.
926-32211), were obtained from LI-COR Biosciences (1:10,000).
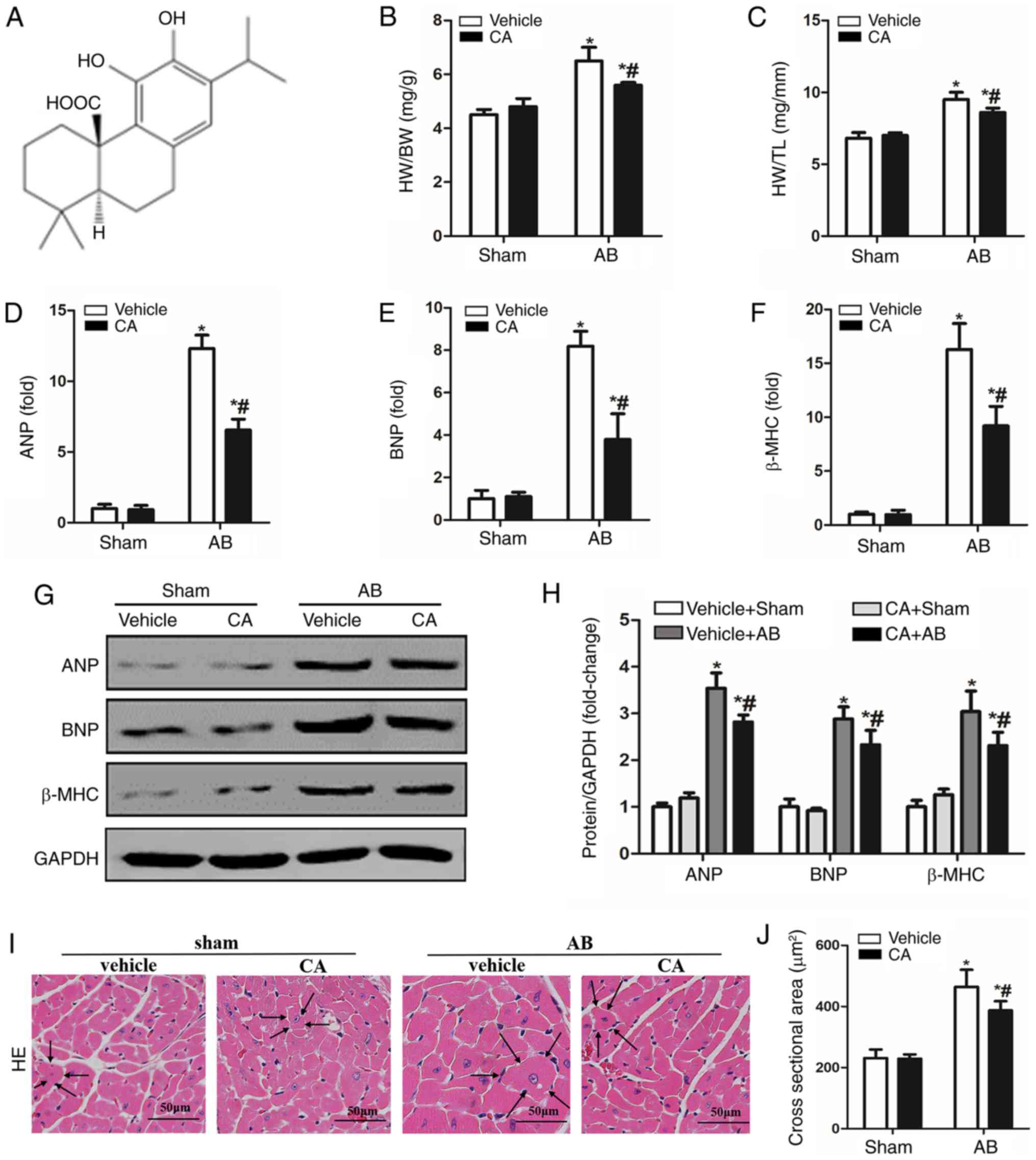 | Figure 1CA alleviated pressure
overload-induced cardiac hypertrophy in mice. (A) Chemical
structure of CA. Statistical results of (B) HW/BW and (C) HW/TL
(n=15). mRNA expressions of (D) ANP, (E) BNP and (F) β-MHC are
presented in each group. (G) Protein levels of ANP, BNP and β-MHC,
and (H) quantitative analysis is presented for each group. (I)
Haematoxylin and eosin staining (the surrounding arrows indicated
the size of each cardiomyocyte), and (J) statistical data of the
cardiomyocyte cross-sectional areas of each group (≥100
cells/heart). Scale bar, 50 µm. *P<0.05 vs. the
vehicle + sham group. #P<0.05 vs. the vehicle + AB
group. HW/BW, heart weight/body weight; ANP, atrial natriuretic
peptide; BNP, B-type natriuretic peptide; β-MHC, β-myosin heavy
chain; CA, carnosic acid; AB, aortic banding; HE, haematoxylin and
eosin. |
Animals and experimental design
All animal care and experiments were based on the
Guidelines for the Care and Use of Laboratory Animals published by
the United States National Institutes of Health (NIH Publication,
revised 2011) (18) and were
approved by the Animal Care and Use Committee of Wuhan University.
Male C57/B6 mice (age, 8-10 weeks; weight, 23.5-27.5 g) were
purchased from the Institute of Laboratory Animal Science, Chinese
Academy of Medical Sciences. The mice had free access to food and
water and were housed under a specific-pathogen-free environment
and a controlled temperature (20-25˚C) and humidity (50±5%) with a
12 h light/dark cycle. A total of 60 animals were randomly divided
into four groups: i) Vehicle + sham (n=15); ii) CA + sham (n=15);
iii) vehicle + aortic banding (AB; n=15); and iv) AB + CA (n=15).
Mice were subjected to AB surgery to establish a model of cardiac
fibrosis and surgery was performed based on previously described
methods (19). Via intraperitoneal
injection, in total 3% 80 mg/kg pentobarbital was used to
anaesthetize animals. After anaesthesia, hair over the thoracic
surgery area was sheared. Following local disinfection with iodine,
fluoride and alcohol, mice were fixed on the operating table. An
insert was made into the trachea and the skin was cut along the
second and third intercostal level. The muscles and soft tissues
were separated in turn, revealing the thoracic aorta. The aorta was
then freed from connective tissue and a surgical suture was wrapped
around the vessel. The thread was tightened against a 27-guauge
needle placed on the freed portion of the aorta. Following
ligation, the needle was removed. For the sham-operated groups,
after the aorta was freed, surgical thread was wound around the
vessel, but no ligation was performed. The two AB groups received
the same surgical procedure. After 2 days of AB, 50 mg/kg CA was
administered orally once a day for 12 days. A period of four weeks
following the AB operation, echocardiography and haemodynamic
parameters were analysed and mice were sacrificed via cervical
dislocation. The heart weight was then measured, along with tibia
length.
Echocardiography measurement and
invasive hemodynamic pressure-volume analysis
A MyLabTM 30CV (Esaote SpA) and a 10-MHz linear
array ultrasound transducer (Esaote SpA) were used to detect
cardiac function, as reported previously (20). A total of 1.5% isoflurane (21) was used to anaesthetize mice on a 37˚C
temperature-controlled warming pad. Corneal reflex, pain reflex,
respiration and muscle tension were monitored in mice to ensure
they were anesthetized. When the mouse was appeared calm, was in
the supine position, did not feel pain, did not exhibit righting
reflex and had relaxed limbs the mouse was determined to have
entered the anesthesia maintenance period. The following parameters
were measured: Ejection fraction (EF), left ventricle end-diastolic
diameter (LVEDd), left ventricle end-systolic diameter (LVESd) and
the percentage fraction of shortening, all of which were calculated
as described previously (21). The
present study first measured the LVEDd and LVESd. The LVFS was
calculated as [(LVEDd-LVESd)/LVEDd]*100%, and LVEF was
calculated based on the Teichhotz formula (22).
A 1.4-F Millar microtip pressure transducer was used
for invasive haemodynamic pressure-volume detection. Mice were then
anesthetised with 2% isoflurane on a 37˚C temperature-controlled
warming pad. A pressure-volume conductance system was used to
detect heart rate and the derivative of pressure over time (dp/dt).
PVAN data analysis software (Millar) was used to analyse the
end-systolic pressure (ESP), end-diastolic pressure (EDP), the
maximal rate of pressure development (dp/dtmax) and the minimal
rate of pressure decay (dp/dtmin).
Histological analysis
After 12 days of treatment with CA, extracted hearts
were rapidly soaked in 10% KCl solution to keep the heart at
diastole. Hearts were then fixed with 4% formalin for 2-3 days at
room temperature, dehydrated and embedded in paraffin, then cut to
4-5 µm sections. To assess the cross-sectional area of
cardiomyocytes, haematoxylin and eosin (HE) staining was performed
under room temperature for 1 h. To evaluate collagen deposition,
picrosirius red (PSR) staining and Masson staining were performed
on the cardiac sections under room temperature for 3 h (23). A light microscope was used to
visualize the cardiac sections at magnifications of x400 and x200.
The cross-sectional area of the cardiomyocytes and the degree of
collagen deposition were analysed using the Image-Pro Plus v6.0
quantitative digital image analysis system (Media Cybernetics,
Inc.). ≥100 myocardial cells in each group were analysed (24).
Immunohistochemistry staining
Immunohistochemistry for 4-hydroxynonenal (4-HNE)
and α-smooth muscle actin (α-SMA) was conducted to evaluate the
level of oxidative stress and the activation of myofibroblasts,
respectively. The hearts of each group were fixed with 4% neutral
formaldehyde solution for 2-3 days under room temperature and then
dewaxed to hydrate. Antigen repair was performed using the citric
acid method (Citrate Antigen Retrieval Solution; P0081; Beyotime
Institute of Biotechnology) according to the manufacturer's
protocol. The sections blocked with 8% goat serum for 30 min at
37˚C. Cardiac sections (4-5 µm) were incubated with 4-HNE (1:100;
cat. no. ab46545; Abcam) and α-SMA (1:100; cat. no. ab5694; Abcam)
antibodies at 37˚C for 2 h. After rinsing, EnVisionTM+/HRP
(K400011-2, Dako; Agilent Technologies, Inc.) was added to cardiac
sections for 1 h at 37˚C and samples were processed using a
peroxide-based substrate diaminobenzidine kit (Gene Tech
Biotechnology Co., Ltd.) and haematoxylin for staining nuclei for 1
sec at room temperature. Neutral resin adhesive was used for
sealing. Finally, heart sections were photographed using a light
microscope (magnification, x200 and x400) as previously described
(23).
Determination of oxidative stress
To assess the SOD1 concentration, NADPH oxidase and
the lipid peroxidation product MDA of the left ventricle were
assessed. Commercial kits purchased from Beyotime Institute of
Biotechnology were used to determine oxidative stress and
performance based on the manufacturer's protocol.
Western blotting and reverse
transcription-quantitative PCR (RT-qPCR)
Protein extraction and SDS-PAGE of the cardiac
tissues were performed according to previous study (23). Left ventricular tissues were
homogenized in Radio Immunoprecipitation Assay lysis buffer (cat.
no. P0013C; Beyotime Institute of Biotechnology). The concentration
of the protein samples was detected using a BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). Sample buffer and water were
added to homogenate the protein samples. Equal quantities of
protein were placed into 10% SDS-PAGE gel for electrophoresis. The
target protein was then transferred to PVDF membranes using
membrane transfer apparatus and blocked in Tri-buffered saline
containing 5% skim milk powder for 60 min at room temperature, and
incubated with primary antibodies at 4˚C overnight. Subsequently,
the membranes were incubated with IRDye 800CW conjugated goat
anti-rabbit IgG (cat. no. P/N 925-32211; 1:1,250; LI-COR
Biosciences) for 1 h at room temperature. The PVDF membranes were
scanned and analysed using an Odyssey Infrared Imaging System
(LI-COR Biosciences). Each sample was normalized to GAPDH.
For reverse transcription-quantitative PCR analysis,
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was
used to extract total mRNA from left ventricular tissues at room
temperature. A cDNA synthesis kit (cat. no. 04896866001; Roche
Applied Science) and oligo (dT) primers were subsequently used to
synthesize the first strand cDNA. RT and qPCR were performed as
reported previously (25), 20 µl
reactions according to the standard protocol of the manufacturer
and ran the cycle 95˚C for 5 min, 45 cycles (of 95˚C for 10 sec,
60˚C for 10 sec and 72˚C for 10 sec), 95˚C for 5 sec, 60˚C for 1
min, 97˚C for 0.11 sec and 40˚C for 10 min. The mRNA expression
level normalized against GAPDH mRNA levels. The primers used for
qPCR are presented in Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Species | Sequences
(5'-3') |
|---|
| ANP | Mouse | F:
ACCTGCTAGACCACCTGGAG |
| | | R:
CCTTGGCTGTTATCTTCGGTACCGG |
| BNP | Mouse | F:
GAGGTCACTCCTATCCTCTGG |
| | | R:
GCCATTTCCTCCGACTTTTCTC |
| β-MHC | Mouse | F:
CCGAGTCCCAGGTCAACAA |
| | | R:
CTTCACGGGCACCCTTGGA |
| Collagen I | Mouse | F:
TGGTACATCAGCCCGAAC |
| | | R:
GTCAGCTGGATAGCGACA |
| Collagen III | Mouse | F:
GTCAGCTGGATAGCGACA |
| | | R:
GAAGCACAGGAGCAGGTGTAGA |
| CTGF | Mouse | F:
GACATGCCGCCTGGAGAAAC |
| | | R:
AGCCCAGGATGCCCTTTAGT |
| NOX2 | Mouse | F:
GACCATTGCAAGTGAACACCC |
| | | R:
AAATGAAGTGGACTCCACGCG |
| NOX4 | Mouse | F:
GACCATTGCAAGTGAACACCC |
| | | R:
AAATGAAGTGGACTCCACGCG |
| GAPDH | Mouse | F:
TCATCAACGGGAAGCCCATC |
| | | R:
CTCGTGGTTCACACCCATCA |
TUNEL Staining
An apoptosis detection kit was used for staining
cardiac tissue slides based on the manufacturer’s protocol. The
hearts of each group were fixed with 4% neutral formaldehyde
solution for 2-3 days under room temperature and then dewaxed to
hydrate. After xylene dewaxing and ethanol dehydration, cardiac
tissue sections were incubated in 20 μg/ml proteinase K for 20 min
and fluorescein-labelled dUTP for 1 h at 37 ℃. DAPI Hematoxylin was
used to stain cardiomyocyte nuclei for 1 second under room
temperature. Neutral resin adhesive used for sealing. Fluorescence
microscopy (200X) was used to view the slides and ≥100 myocardial
cells in each group were analysed. Image-Pro Plus 6.0 was used to
calculate the results (24).
Statistical analysis
Each experiment was repeated three times. Data are
presented as mean ± SEM, and were analysed using SPSS v16.0
software (SPSS, Inc.). Two-way ANOVA followed by a Tukey post hoc
test was used for data analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
CA alleviates pressure
overload-induced cardiac hypertrophy in mice
To evaluate the protective role of CA in pressure
overload-induced cardiac hypertrophy, the present study established
a mouse model of cardiac hypertrophy with or without CA for 12
days. The vehicle + AB group demonstrated increased heart
weight/body weight (HW/BW) and HW/tibia length (HW/TL) ratios
compared with the vehicle + sham and CA + sham groups. Treatment
with CA in mice subjected to AB surgery significantly decreased
cardiac hypertrophy, as indicated by decreased HW/BW and HW/TL
ratios (Fig. 1B and C). Furthermore, hypertrophic markers
including ANP, BNP and β-MHC were significantly increased in the
vehicle + AB group compared with the vehicle + sham group but were
decreased following CA treatment in the CA + AB group (Fig. 1D-F). The present results also
indicated that CA significantly decreased the protein expression of
ANP, BNP and β-MHC induced by AB (Fig.
1G and H). Furthermore, HE
staining indicated that the enlarged cardiomyocyte area induced by
pressure overload was alleviated by CA treatment (Fig. 1I and J). The present results suggested that CA
alleviated pressure overload-induced cardiac hypertrophy in
mice.
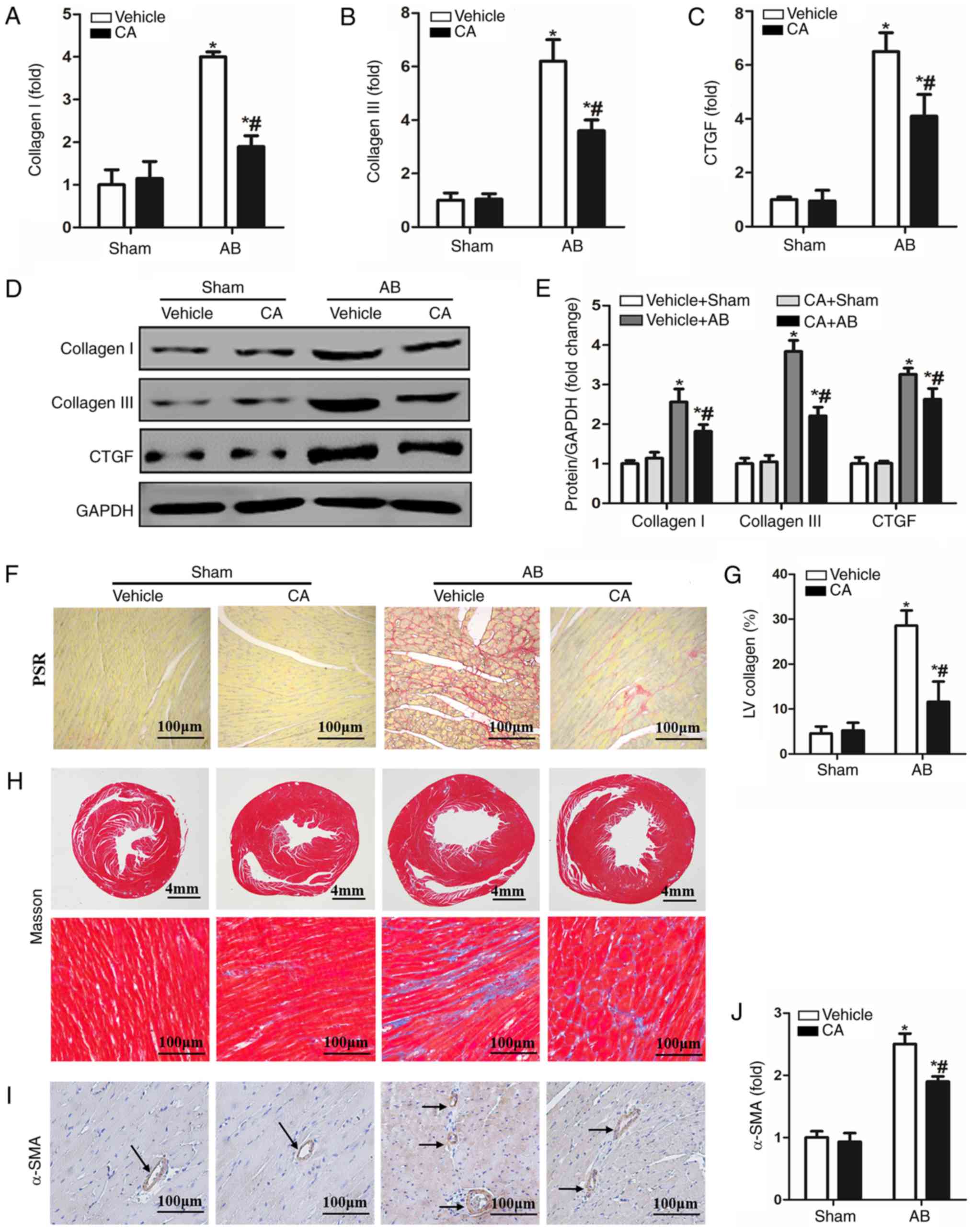
CA attenuates pressure
overload-induced cardiac fibrosis in mice
Cardiac fibrosis is a characteristic of cardiac
remodelling, which ultimately leads to the conversion of cardiac
hypertrophy to heart failure (2).
Therefore, the present study investigated the effect of CA on
cardiac fibrosis. PSR staining and Masson staining were performed
to evaluate cardiac fibrosis 4 weeks after AB surgery. The results
indicated enhanced mRNA (Fig. 2A-C)
and protein (Fig. 2D and E) levels of fibrotic markers, including
collagen I, collagen III and CTGF, which was consistent with
significant perivascular and cardiac fibrosis (Fig. 2F-H), compared with the vehicle + sham
and CA + sham groups. CA treatment in AB mice led to a significant
reduction in cardiac fibrosis and the mRNA and protein expression
of certain fibrotic markers. Furthermore, the expression of α-SMA
was increased in the vehicle + AB group and decreased in the CA +
AB group (Fig. 2I and J). The present results indicated that CA
alleviated pressure overload-induced cardiac fibrosis in mice.
CA improves pressure overload-induced
cardiac dysfunction in mice
Echocardiography and pressure-volume loop
measurements were performed to assess cardiac function in mice with
or without AB surgery. The result revealed no significant
difference in heart rate between each group (Fig. 3A), indicating that CA exerted no
clear effect on heart rate. It was also revealed that
echocardiographic parameters, including LVEDd and LVESd, were
increased (Fig. 3B and C) and the levels of LVEF were decreased in
the vehicle + AB group compared with the vehicle + sham and CA +
sham groups (Fig. 3D). The
haemodynamic parameters, ESP and EDP, were elevated (Fig. 3E and F) and the dp/dt max and dp/dt min,
indicating the systolic and diastolic function respectively, were
decreased in the vehicle + AB group compared with the vehicle +
sham and CA + sham groups (Fig. 3G
and H). The present results
suggested that all the impaired parameters could be improved and
harmful parameters could be restored after CA treatment following
AB surgery.
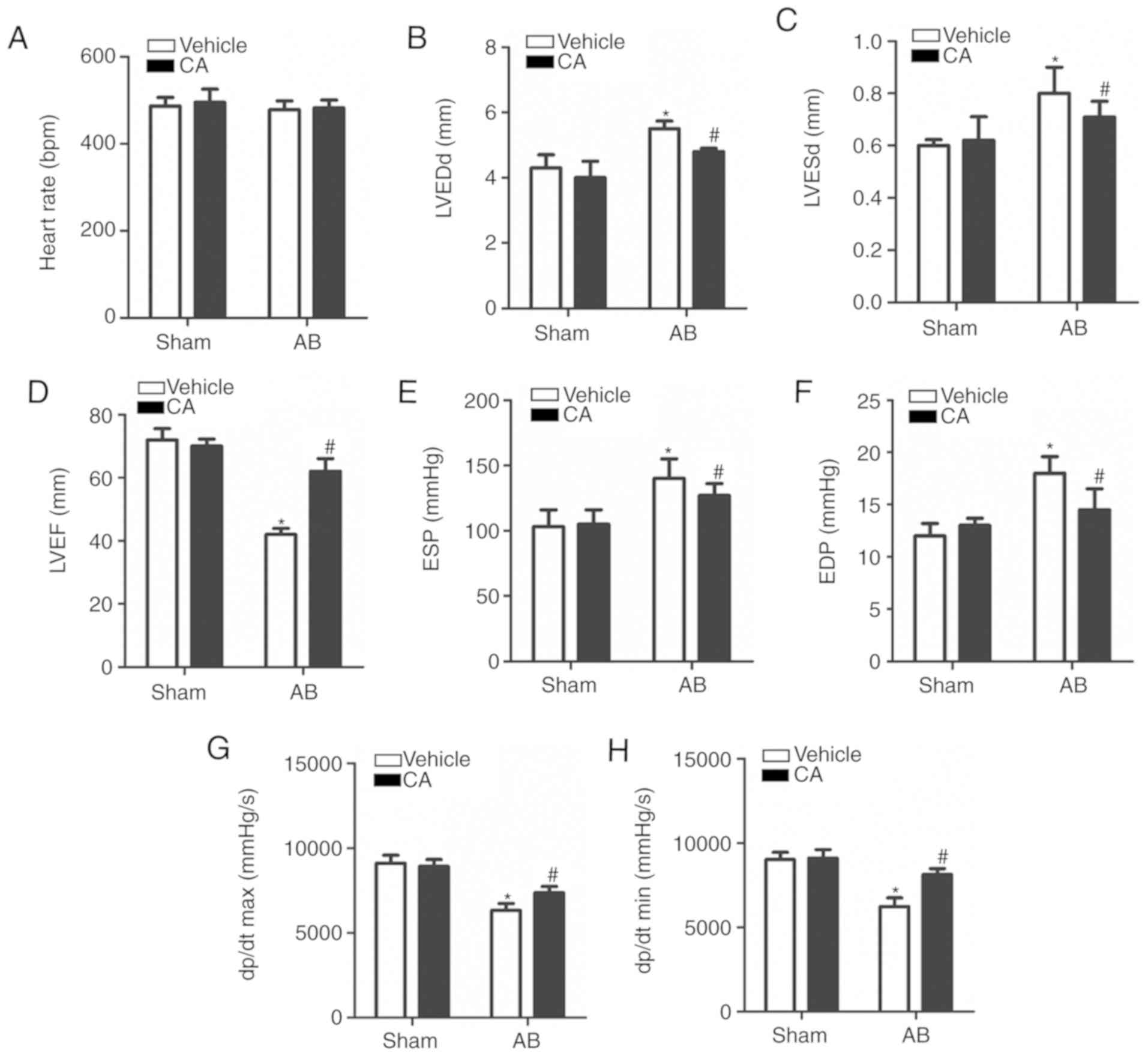 | Figure 3CA improved pressure overload-induced
cardiac dysfunction in mice. (A) Heart rates of the mice in each
group (n=15). Echocardiographic parameters, including (B) LVEDd,
(C) LVESd and (D) LVEF in each group. Haemodynamic parameters
including (E) ESP, (F) EDP, (G) dp/dt max and (H) dp/dt min were
presented for each group. *P<0.05 vs. the vehicle +
sham group. #P<0.05 vs. the vehicle + AB group.
LVEDd, left ventricle end-diastolic diameter; LVESd, left ventricle
end-systolic diameter; LVEF, left ventricle ejection fraction; ESP,
end-systolic pressure; EDP, end-diastolic pressure; dp/dt max,
maximal rate of pressure development; dp/dt min, minimal rate of
pressure decay; CA, carnosic acid; AB, aortic banding. |
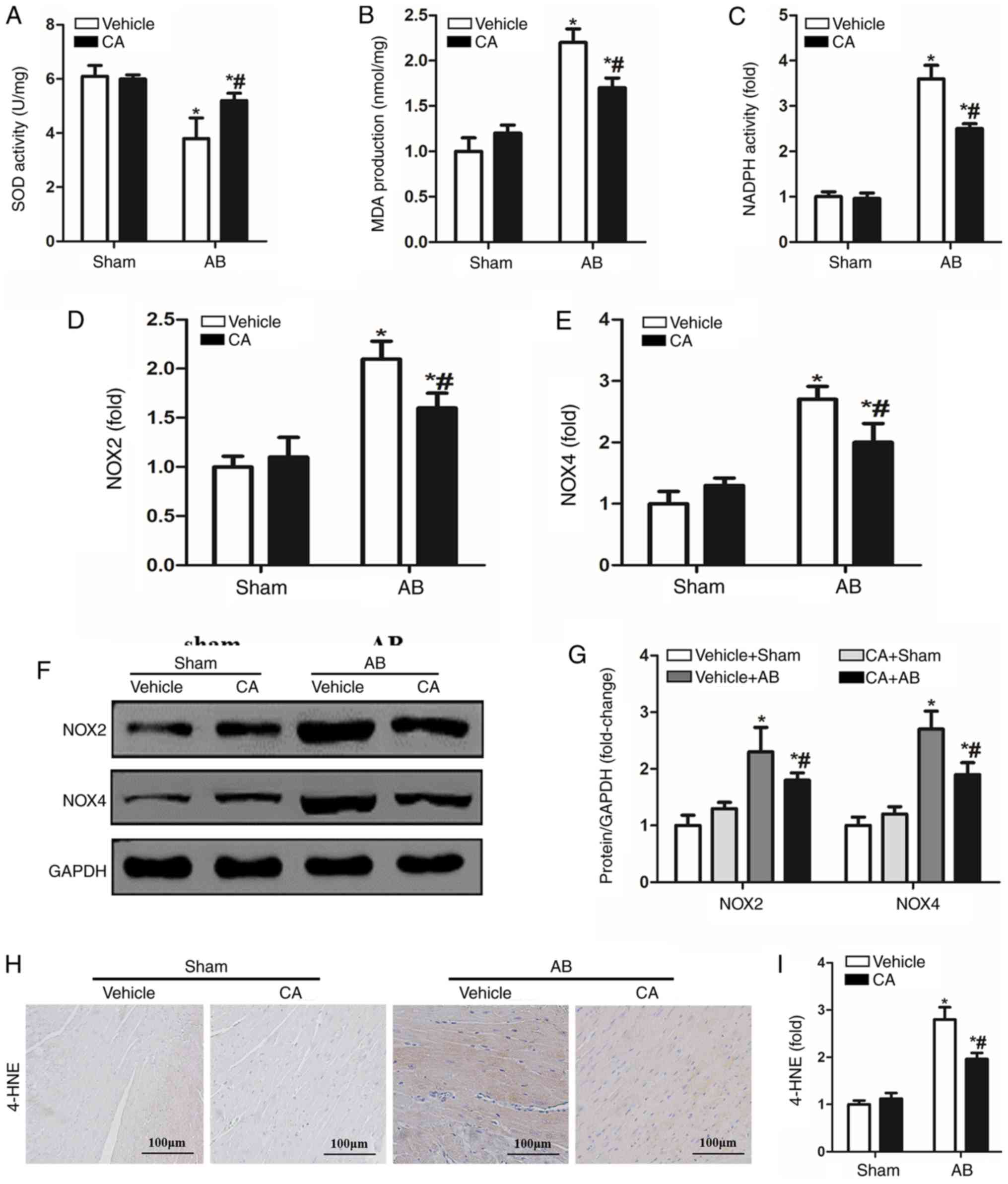
CA suppresses pressure
overload-induced oxidative stress in mice
Oxidative stress is involved in the pathophysiology
of cardiac remodelling and a previous study has demonstrated that
CA possesses strong antioxidant abilities (14). Therefore, the present study
investigated whether CA treatment alleviated pressure
overload-induced cardiac remodelling by inhibiting oxidative
stress. SOD activity was increased by CA treatment in the pressure
overload-induced hearts of mice compared with the vehicle + AB
group (Fig. 4A). However, MDA
production and NADPH activity induced by AB surgery were
significantly decreased by CA treatment when compared with the
vehicle + AB group (Fig. 4B and
C). The present study also
investigated the expression of NOX induced by pressure overload and
identified that the expression of NOX2 and NOX4 were increased at
both mRNA (Fig. 4D and E) and protein levels (Fig. 4F and G) compared with the vehicle + sham and CA +
sham groups. Additionally, CA treatment significantly reduced the
expression of NOX2 and NOX4 at the mRNA and protein levels.
Furthermore, immunohistochemical staining indicated that the
expression of 4-HNE, a product of oxidative stress, was increased
following AB surgery and reversed by CA treatment (Fig. 4H and I). The present results suggested that CA
may suppress cardiac fibrosis by inhibiting NADPH oxidase mediated
oxidative stress.
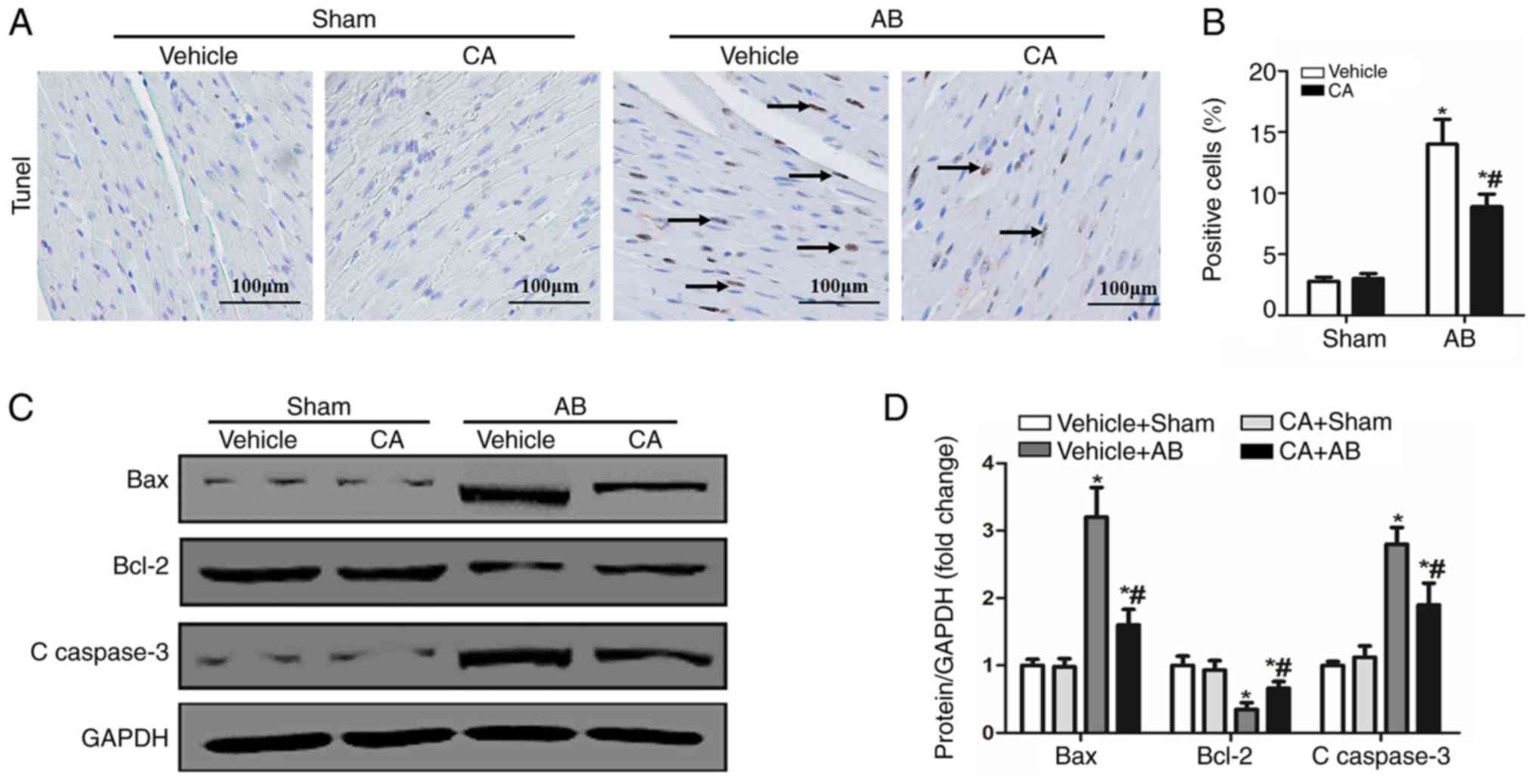
CA inhibits apoptosis in mice induced
by pressure overload
Previous studies have demonstrated that oxidative
stress can activate apoptosis via different pathways and cause
cardiac injury (14,26). CA has the ability to inhibit
oxidative stress and apoptosis (11). Therefore, the present study performed
TUNEL staining and detected the expression of apoptosis-associated
proteins to evaluate the cardioprotective effect of CA following
pressure overload in mice. The present results identified that the
apoptosis ratio was increased in the vehicle + AB group compared
with the vehicle + sham and CA + sham groups, and decreased after
CA treatment (Fig. 5A and B). Furthermore, the expression of certain
pro-apoptotic proteins, including Bax and C-caspase-3 were
increased in the vehicle + AB group compared with the vehicle +
sham and CA + sham groups, and decreased after CA treatment
(Fig. 5C and D). The expression of the anti-apoptotic
protein, Bcl-2, decreased in the vehicle + AB group compared with
the vehicle + sham and CA + sham groups, and increased after CA
treatment (Fig. 5C and D). However, there were no differences
between the vehicle + sham group and the CA + sham group.
Therefore, the present results suggested that CA inhibited pressure
overload induced-cardiomyocyte apoptosis.
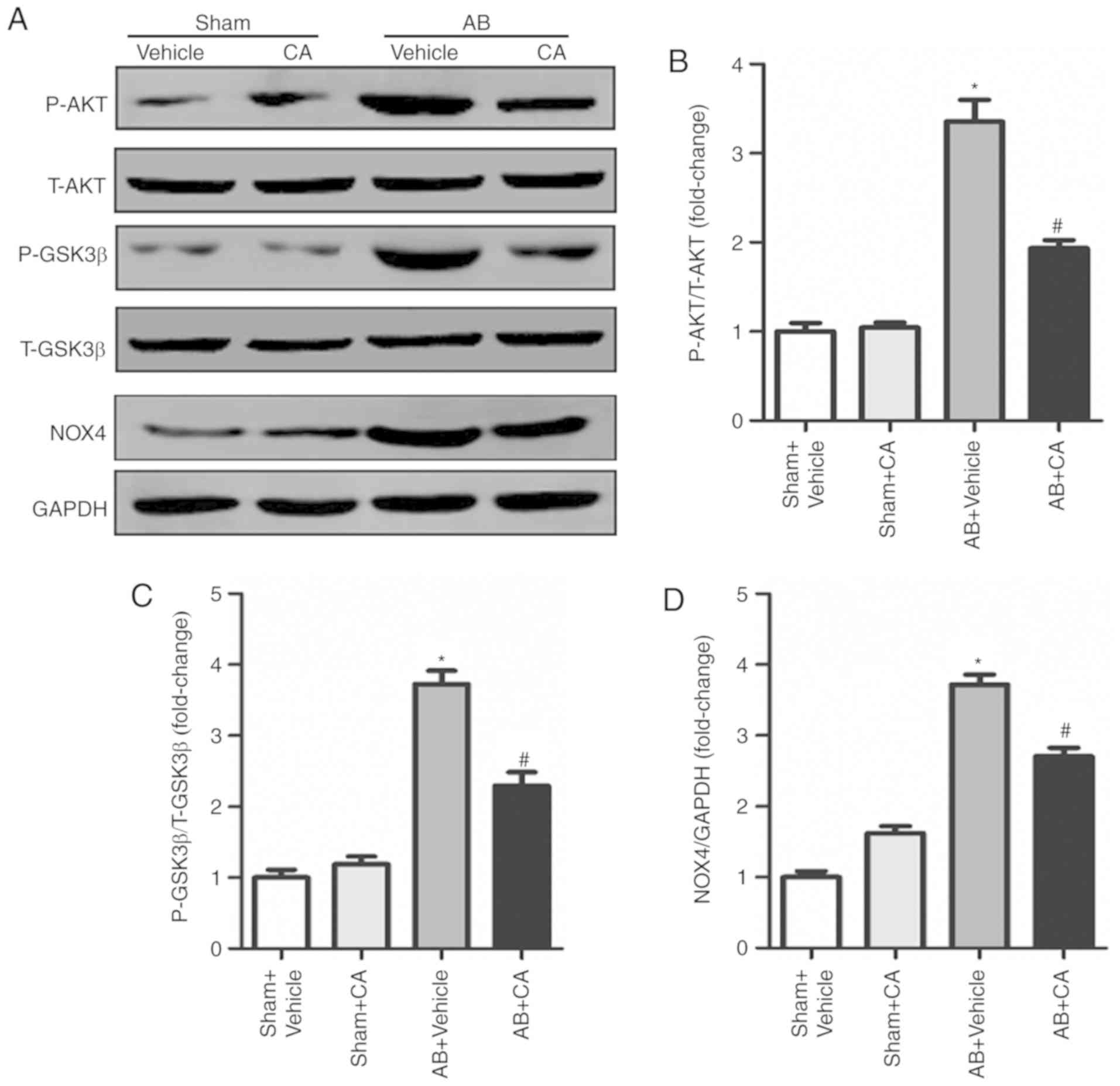
CA protects against cardiac
remodelling by regulating the AKT/GSK3β/NOX4 signalling
pathway
To assess the underlying mechanism of the effect of
CA acting on cardiac remodelling, the present study investigated
the AKT/GSK3β/NOX4 signalling pathway. The present results
suggested that CA treatment significantly reduced the expression of
p-AKT, p-GSK3β and NOX4 induced by AB (Fig. 6A-D). The present results indicated
that CA may exert its protective effect against cardiac remodelling
by suppressing the AKT/GSK3β/NOX4 signalling pathway.
Discussion
The present study performed AB surgery to establish
a mouse cardiac remodelling model. The present results suggested CA
treatment not only protected against pressure overload-induced
cardiac hypertrophy, but also attenuated cardiac fibrosis.
Furthermore, CA treatment inhibited oxidative stress and apoptosis
following AB surgery. Taken together, the present results suggested
that CA may be a potential candidate for cardiac remodelling
induced by pressure-overload (Fig.
7).
Heart failure is a clinical syndrome of cardiac
circulation disorder caused by ventricular systolic or diastolic
insufficiency and has become a serious social and public health
problem (27). Pathological cardiac
remodelling is induced by various stimuli, including inflammation,
biomechanical stress and pressure overload. In pressure overload,
cardiomyocytes develop hypertrophy with a series of changes, such
as abnormal gene transcription and protein synthesis and apoptosis.
With the prolongation of stimulation, these changes result in
increasing extracellular matrix and cardiac fibrosis (2). Cardiac fibrosis is the adaptive
response of the heart to various forms of injury during which
cardiac fibroblasts transform into myofibroblasts (2).
Previous studies have reported that oxidative stress
is implicated in the pathophysiology of cardiac remodelling, which
impairs heart pumping capacity and ultimately contributes to heart
failure (28,29). NOX is the main source of ROS that
induces oxidative stress in the heart and NOX2 and NOX4 are
primarily associated with the heart (30). Under pathological conditions,
activated NOX2 reduces oxygen molecules into oxygen ions via NADPH,
thus leading to heart injury (31).
Liu et al (32), demonstrated
that NOX2 promoted the synthesis of collagen I and III in neonatal
rat cardiac fibroblasts, leading to cardiac fibrosis. Furthermore,
NOX2 enhanced angiotensin II induced ROS generation and
cardiomyocyte hypertrophy (33).
NOX4 produces hydrogen peroxide via a disproportionation reaction
with oxygen ions, thereby regulating ROS production (34). A previous study revealed that NOX4
upregulated transverse aortic constriction-induced cardiomyocyte
hypertrophy and left ventricular dysfunction (35). A further study demonstrated that NOX4
contributed to angiotensin-II-induced adult mouse cardiac
fibroblast proliferation and migration (36). The present results indicated that the
production of MDA and the activities of SOD and NADPH were
significantly increased following AB surgery. In addition, the
present results identified that the mRNA and protein expression of
NOX2 and NOX4 were significantly increased following AB surgery. As
a lipid peroxide formed by the lipid peroxidation reaction between
ROS and biomacromolecules, 4-HNE exerts a destructive effect on
cardiac tissue by interfering with adducts formed by organelles
such as mitochondria (37). In the
present study, the expression of 4-HNE was significantly increased
in the vehicle + AB group. The present results indicated that the
inhibition of oxidative stress may improve cardiac hypertrophy and
fibrosis.
The accumulation of ROS components such as
superoxide anions, hydroxyl free radicals and hydrogen peroxide,
can damage the integrity and function of the cell membrane and lead
to cell apoptosis (38). A previous
study has suggested that increased ROS can induce cardiomyocyte
apoptosis by activating certain apoptotic signals, including
caspase-3(39). Additionally,
angiotensin II-mediated NADPH oxidase-induced ROS mediate
cardiomyocyte apoptosis via the mitochondrial apoptotic pathway
(26). The present results
demonstrated that the number of TUNEL-positive cells was
significantly increased upon pressure overload. Furthermore, the
expression of certain pro-apoptotic proteins, including Bax and
C-caspase-3, were increased, while the expression of the
anti-apoptotic protein, Bcl-2, was decreased following AB surgery.
The present results indicated that ROS-mediated oxidative stress
serves a key role in cell apoptosis and that the inhibition of
apoptosis may serve a protective role in the heart.
CA is the major component extracted from rosemary
plants and has anti-cancer, anti-inflammatory and anti-obesity
effects (40). A previous study
demonstrated that CA suppressed oxidative stress by inhibiting the
expression of NOX4 in transforming growth factor β-stimulated
fibroblasts and unilateral ureteral obstruction-operated kidneys
(41). In addition, CA alleviated
bile duct ligation induced the expression of α-SMA and collagen I
in the liver and served an anti-fibrotic role in the livers of rats
(42). Previous studies have
indicated that CA serves an antioxidant role in different diseases,
including arsenic-induced hepatotoxicity (10) and isoproterenol-induced myocardial
stress (14). Furthermore, it has
been demonstrated that CA attenuated amyloid-β peptide-induced
human neuroblastoma cell apoptosis (43) and isoproterenol-induced cardiomyocyte
apoptosis (14). However, several
studies have determined that CA induced apoptosis in various cancer
models, possibly due to different doses and models affecting the
pharmacological effects of CA (44,45). The
present results indicated that CA treatment inhibited pressure
overload-induced cardiac hypertrophy and fibrosis, and reduced
oxidative stress and apoptosis, as well as improving cardiac
function.
AKT is a serine-threonine kinase that serves an
important role in cardiac growth and metabolism (46). Activated AKT stimulates downstream
GSK3β and participates in the pathophysiology of cardiac
hypertrophy (15,16). p-Akt increases the size of cardiac
myocytes and promotes the development of heart failure (47). Furthermore, NOX4 serves an important
role in the differentiation of myofibroblasts into different
phenotypes (48,49). A previous study indicated that CA
alleviated AKT-mediated renal fibrosis by reducing the expression
of NOX4(17). The present results
indicated that CA significantly decreased the expression of p-AKT,
p-GSK3β and NOX4 induced by AB. Therefore, the role of CA in
cardiac remodelling may rely on the AKT/GSK3K3β/NOX4 signalling
pathway.
In conclusion, the present results indicated that CA
significantly attenuated pressured overload-induced cardiac
hypertrophy and fibrosis, and that the potential mechanism may be
attributed to the inhibition of oxidative stress and apoptosis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YJW and FC conceived the present study and designed
the experiments. HJX and JJC performed the experiments. YJW and FC
wrote and revised the manuscript; XY, JX and JW analysed
experimental results. All authors read and approved the final
manuscript
Ethics approval and consent to
participate
All animal care and experiments were based on the
Guidelines for the Care and Use of Laboratory Animals published by
the United States National Institutes of Health (NIH Publication,
revised 2011) and were approved by the Animal Care and Use
Committee of Wuhan University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jaiswal A, Nguyen VQ, Carry BJ and le
Jemtel TH: Pharmacologic and endovascular reversal of left
ventricular remodeling. J Card Fail. 22:829–839. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu
C, Zhu JX, Yang Z, Deng W and Tang QZ: Mechanisms contributing to
cardiac remodelling. Clin Sci (Lond). 131:2319–2345.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang H, Sun X, Lin MS, Ferrario CM, Van
Remmen H and Groban L: G protein-coupled estrogen receptor (GPER)
deficiency induces cardiac remodeling through oxidative stress.
Transl Res. 199:39–51. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Al-Darraji A, Haydar D, Chelvarajan L,
Tripathi H, Levitan B, Gao E, Venditto VJ, Gensel JC, Feola DJ and
Abdel-Latif A: Azithromycin therapy reduces cardiac inflammation
and mitigates adverse cardiac remodeling after myocardial
infarction: Potential therapeutic targets in ischemic heart
disease. PLoS One. 13(e0200474)2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Eid RA, Alkhateeb MA, Al-Shraim M, Eleawa
SM, Shatoor AS, El-Kott AF, Zaki MSA, Shatoor KA, Bin-Jaliah I and
Al-Hashem FH: Ghrelin prevents cardiac cell apoptosis during
cardiac remodelling post experimentally induced myocardial
infarction in rats via activation of Raf-MEK1/2-ERK1/2 signalling.
Arch Physiol Biochem. 125:93–103. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sciarretta S, Yee D, Nagarajan N, Bianchi
F, Saito T, Valenti V, Tong M, Del Re DP, Vecchione C, Schirone L,
et al: Trehalose-Induced Activation of Autophagy Improves Cardiac
Remodeling After Myocardial Infarction. J Am Coll Cardiol.
71:1999–2010. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang M, Perino A, Ghigo A, Hirsch E and
Shah AM: NADPH oxidases in heart failure: Poachers or gamekeepers?
Antioxid Redox Signal. 18:1024–1041. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhao QD, Viswanadhapalli S, Williams P,
Shi Q, Tan C, Yi X, Bhandari B and Abboud HE: NADPH oxidase 4
induces cardiac fibrosis and hypertrophy through activating
Akt/mTOR and NFκB signaling pathways. Circulation. 131:643–655.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhang Y, Tocchetti CG, Krieg T and Moens
AL: Oxidative and nitrosative stress in the maintenance of
myocardial function. Free Radic Biol Med. 53:1531–1540.
2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Das S, Joardar S, Manna P, Dua TK,
Bhattacharjee N, Khanra R, Bhowmick S, Kalita J, Saha A, Ray S, et
al: Carnosic acid, a natural diterpene, attenuates arsenic-induced
hepatotoxicity via reducing oxidative stress, MAPK activation, and
apoptotic cell death pathway. Oxid Med Cell Longev.
2018(1421438)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Liu M, Zhou X, Zhou L, Liu Z, Yuan J,
Cheng J, Zhao J, Wu L, Li H, Qiu H, et al: Carnosic acid inhibits
inflammation response and joint destruction on osteoclasts,
fibroblast-like synoviocytes, and collagen-induced arthritis rats.
J Cell Physiol. 233:6291–6303. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Manoharan S, Vasanthaselvan M, Silvan S,
Baskaran N, Kumar Singh A and Vinoth Kumar V: Carnosic acid: A
potent chemopreventive agent against oral carcinogenesis. Chem Biol
Interact. 188:616–622. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li H, Sun JJ, Chen GY, Wang WW, Xie ZT,
Tang GF and Wei SD: Carnosic acid nanoparticles suppress liver
ischemia/reperfusion injury by inhibition of ROS, Caspases and
NF-κB signaling pathway in mice. Biomed Pharmacother. 82:237–246.
2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sahu BD, Putcha UK, Kuncha M, Rachamalla
SS and Sistla R: Carnosic acid promotes myocardial antioxidant
response and prevents isoproterenol-induced myocardial oxidative
stress and apoptosis in mice. Mol Cell Biochem. 394:163–176.
2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bénard L, Oh JG, Cacheux M, Lee A,
Nonnenmacher M, Matasic DS, Kohlbrenner E, Kho C, Pavoine C, Hajjar
RJ and Hulot JS: Cardiac stim1 silencing impairs adaptive
hypertrophy and promotes heart failure through inactivation of
mTORC2/Akt signaling. Circulation. 133:1458–1471. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li J, Kritzer MD, Michel JJ, Le A, Thakur
H, Gayanilo M, Passariello CL, Negro A, Danial JB, Oskouei B, et
al: Anchored p90 ribosomal S6 kinase 3 is required for cardiac
myocyte hypertrophy. Circ Res. 112:128–139. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jung KJ, Min KJ, Park JW, Park KM and Kwon
TK: Carnosic acid attenuates unilateral ureteral
obstruction-induced kidney fibrosis via inhibition of Akt-mediated
Nox4 expression. Free Radic Biol Med. 97:50–57. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. National
Academies Press, Washington, DC, 2011.
|
|
19
|
Jiang DS, Liu Y, Zhou H, Zhang Y, Zhang
XD, Zhang XF, Chen K, Gao L, Peng J, Gong H, et al: Interferon
regulatory factor 7 functions as a novel negative regulator of
pathological cardiac hypertrophy. Hypertension. 63:713–722.
2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ma ZG, Yuan YP, Xu SC, Wei WY, Xu CR,
Zhang X, Wu QQ, Liao HH, Ni J and Tang QZ: CTRP3 attenuates cardiac
dysfunction, inflammation, oxidative stress and cell death in
diabetic cardiomyopathy in rats. Diabetologia. 60:1126–1137.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gao S, Ho D, Vatner DE and Vatner SF:
Echocardiography in mice. Curr Protoc Mouse Biol. 1:71–83.
2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Troy BL, Pombo J and Rackley CE:
Measurement of left ventricular wall thickness and mass by
echocardiography. Circulation. 45:602–611. 1972.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wu QQ, Xiao Y, Jiang XH, Yuan Y, Yang Z,
Chang W, Bian ZY and Tang QZ: Evodiamine attenuates TGF-β1-induced
fibroblast activation and endothelial to mesenchymal transition.
Mol Cell Biochem. 430:81–90. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang N, Wei WY, Yang Z, Che Y, Jin YG,
Liao HH, Wang SS, Deng W and Tang QZ: Nobiletin, a polymethoxy
flavonoid, protects against cardiac hypertrophy induced by
pressure-overload via inhibition of NAPDH oxidases and endoplasmic
reticulum stress. Cell Physiol Biochem. 42:1313–1325.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ibarra-Lara L, Hong E, Soria-Castro E,
Torres-Narváez JC, Pérez-Severiano F, Del Valle-Mondragón L,
Cervantes-Pérez LG, Ramírez-Ortega M, Pastelín-Hernández GS and
Sánchez-Mendoza A: Clofibrate PPARα activation reduces oxidative
stress and improves ultrastructure and ventricular hemodynamics in
no-flow myocardial ischemia. J Cardiovasc Pharmacol. 60:323–334.
2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Qin F, Patel R, Yan C and Liu W: NADPH
oxidase is involved in angiotensin II-induced apoptosis in H9C2
cardiac muscle cells: Effects of apocynin. Free Radic Biol Med.
40:236–246. 2006.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yang CJ and Yang J, Fan ZX, Zhang J, Liu
XW and Yang J: Diagnostic value of soluble ST2 in heart failure: A
meta-analysis. Bachu Med J. 1:52–59. 2018.
|
|
28
|
Liu JJ, Lu Y, Ping NN, Li X, Lin YX and Li
CF: Apocynin ameliorates pressure overload-induced cardiac
remodeling by inhibiting oxidative stress and apoptosis. Physiol
Res. 66:741–752. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li W, Wu X, Li M, Wang Z, Li B, Qu X and
Chen S: Cardamonin alleviates pressure overload-induced cardiac
remodeling and dysfunction through inhibition of oxidative stress.
J Cardiovasc Pharmacol. 68:441–451. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bryk D, Olejarz W and Zapolska-Downar D:
The role of oxidative stress and NADPH oxidase in the pathogenesis
of atherosclerosis. Postepy Hig Med Dosw. 71:57–68. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cangemi R, Calvieri C, Bucci T, Carnevale
R, Casciaro M, Rossi E, Calabrese CM, Taliani G, Grieco S, Falcone
M, et al: SIXTUS study group: Is NOX2 upregulation implicated in
myocardial injury in patients with pneumonia? Antioxid Redox
Signal. 20:2949–2954. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Liu Y and Zhang J: Nox2 contributes to
cardiac fibrosis in diabetic cardiomyopathy in a transforming
growth factor-β dependent manner. Int J Clin Exp Pathol.
8:10908–10914. 2015.PubMed/NCBI
|
|
33
|
Hingtgen SD, Tian X, Yang J, Dunlay SM,
Peek AS, Wu Y, Sharma RV, Engelhardt JF and Davisson RL:
Nox2-containing NADPH oxidase and Akt activation play a key role in
angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics.
26:180–191. 2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Siu KL, Lotz C, Ping P and Cai H: Netrin-1
abrogates ischemia/reperfusion-induced cardiac mitochondrial
dysfunction via nitric oxide-dependent attenuation of NOX4
activation and recoupling of NOS. J Mol Cell Cardiol. 78:174–185.
2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Matsushima S, Kuroda J, Zhai P, Liu T,
Ikeda S, Nagarajan N, Oka S, Yokota T, Kinugawa S, Hsu CP, et al:
Tyrosine kinase FYN negatively regulates NOX4 in cardiac
remodeling. J Clin Invest. 126:3403–3416. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Somanna NK, Valente AJ, Krenz M, Fay WP,
Delafontaine P and Chandrasekar B: The Nox1/4 Dual inhibitor
GKT137831 or Nox4 knockdown inhibits angiotensin-II-induced adult
mouse cardiac fibroblast proliferation and migration. AT1
physically associates with Nox4. J Cell Physiol. 231:1130–1141.
2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Deshpande M, Mali VR, Pan G, Xu J, Yang
XP, Thandavarayan RA and Palaniyandi SS: Increased
4-hydroxy-2-nonenal-induced proteasome dysfunction is correlated
with cardiac damage in streptozotocin-injected rats with
isoproterenol infusion. Cell Biochem Funct. 34:334–342.
2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kumar D and Jugdutt BI: Apoptosis and
oxidants in the heart. J Lab Clin Med. 142:288–297. 2003.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sahu BD, Rentam KK, Putcha UK, Kuncha M,
Vegi GM and Sistla R: Carnosic acid attenuates renal injury in an
experimental model of rat cisplatin-induced nephrotoxicity. Food
Chem Toxicol. 49:3090–3097. 2011.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Jung KJ, Min KJ, Park JW, Park KM and Kwon
TK: Carnosic acid attenuates unilateral ureteral
obstruction-induced kidney fibrosis via inhibition of Akt-mediated
Nox4 expression. Free Radic Biol Med. 97:50–57. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhang S, Wang Z, Zhu J, Xu T, Zhao Y, Zhao
H, Tang F, Li Z, Zhou J, Gao D, et al: Carnosic acid alleviates
BDL-induced liver fibrosis through miR-29b-3p-mediated inhibition
of the high-mobility group Box 1/Toll-Like receptor 4 signaling
pathway in rats. Front Pharmacol. 8(976)2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Meng P, Yoshida H, Tanji K, Matsumiya T,
Xing F, Hayakari R, Wang L, Tsuruga K, Tanaka H, Mimura J, et al:
Carnosic acid attenuates apoptosis induced by amyloid-β 1-42 or
1-43 in SH-SY5Y human neuroblastoma cells. Neurosci Res. 94:1–9.
2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Shi B, Wang LF, Meng WS, Chen L and Meng
ZL: Carnosic acid and fisetin combination therapy enhances
inhibition of lung cancer through apoptosis induction. Int J Oncol.
50:2123–2135. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Han NN, Zhou Q, Huang Q and Liu KJ:
Carnosic acid cooperates with tamoxifen to induce apoptosis
associated with Caspase-3 activation in breast cancer cells in
vitro and in vivo. Biomed Pharmacother. 89:827–837. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Abeyrathna P and Su Y: The critical role
of Akt in cardiovascular function. Vascul Pharmacol. 74:38–48.
2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Chaanine AH and Hajjar RJ: AKT signalling
in the failing heart. Eur J Heart Fail. 13:825–829. 2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Barnes JL and Gorin Y: Myofibroblast
differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney
Int. 79:944–956. 2011.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Hecker L, Vittal R, Jones T, Jagirdar R,
Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ and Thannickal
VJ: NADPH oxidase-4 mediates myofibroblast activation and
fibrogenic responses to lung injury. Nat Med. 15:1077–1081.
2009.PubMed/NCBI View Article : Google Scholar
|