Introduction
Doxorubicin (DOX), which is a member of the
anthracycline family, has been widely used as a chemotherapeutic
agent for solid tumors owing to its effective therapeutic outcomes
(1). However, the administration of
DOX is limited since anthracycline therapy has been indicated to be
closely associated with dose-dependent cardiotoxicity (1). Although the exact molecular mechanism
of cardiotoxicity induced by DOX treatment has not fully
elucidated, DOX has been reported to promote mitochondrial
dysfunction and alter the mitochondrial membrane by eliciting lipid
peroxidation, generating free radicals, increasing the myocardial
levels of sodium and calcium and inducing autophagy/mitophagy,
thereby resulting in apoptotic and non-apoptotic cell death in
cardiomyocytes (2-5).
DOX-induced toxicity has also been demonstrated to be primarily
attributed to the generation of reactive oxygen species
(ROS)/reactive nitrogen species (RNS) as a result of drug redox
recycling (6). Therefore, decreasing
or attenuating DOX-induced ROS accumulation has become a
therapeutic strategy to reduce the risk of heart failure (6).
Leucine-rich pentatricopeptide repeat-containing
(LRPPRC), which is also known as LRP130, is a well-known
mitochondria-associated protein that has been indicated to serve
critical roles in mitochondria by maintaining mitochondrial
membrane potential and function (7).
In the mitochondrial matrix, LRPPRC has been reported to bind
stem-loop-interacting RNA-binding protein and subsequently regulate
mRNA stability and polyadenylation and the coordination of
translation (7-9).
Mutations in LRPPRC have been indicated to result in cytochrome
c oxidase deficiency, decreased mitochondrial mRNA levels
and reduced mitochondrial translation in the liver and brain
(7,10). Several reports have revealed that
LRPPRC regulates mRNA transport from the cytoplasm to mitochondria
(11) and RNA transport from the
nucleus to mitochondria (12);
however, LRPPRC has been indicated to be primarily localized in the
mitochondria (13). Knockdown of
LRPPRC in cells (13) and knockout
of Lrpprc in mice (8) has
been indicated to reduce mitochondrial membrane potential and
homeostasis, and DNA mass and function. Therefore, LRPPRC regulates
physiological processes both in vitro and in vivo via
regulating mitochondrial function.
Numerous studies have been performed to investigate
the effects and the mechanisms by which DOX targets mitochondrial
energy metabolism and ROS production. Wang et al (14) reported that fibroblast growth factor
21, which is a well-known regulator of glucose and lipid
metabolism, exerted cardioprotective effects against DOX-induced
toxicity via the NAD-dependent protein deacetylase sirtuin-1/liver
kinase B1/AMP-activated protein kinase pathway in H9C2 cells. Liu
et al also reported that pterostilbene, which is a natural
analogue of resveratrol and an antioxidant, exerted
cardioprotective effects against DOX-induced cardiotoxicity in H9C2
cells and injury in mouse cardiomyocytes by reducing oxidative
stress, including ROS accumulation, which indicated that oxidative
stress may be the principal cause of cardiotoxicity following DOX
exposure (15). It has also been
reported that salsolinol, a plant-based isoquinoline alkaloid,
ameliorated cardiomyocyte function, promoting mitochondrial
respiratory and energy metabolism (16). However, little is known of the effect
of LRPPRC on oxidative stress induced by DOX exposure. The present
study aimed to investigate the effects of LRPPRC on H9C2
cardiomyocytes under DOX-induced oxidative stress, and the
regulatory roles of LRPPRC in mitochondrial function.
Materials and methods
Cell culture and treatment
H9C2 myoblast cells derived from the rat myocardium
were obtained from the Institute of Biochemistry and Cell Biology,
Chinese Academy of Sciences. Cells were maintained in DMEM
supplemented with 10% FBS and 10 ml/l 100X antibiotic-antimycotic
solution containing 10,000 units of penicillin and 10 mg/ml
streptomycin at 37 in a 5% CO2 humidified incubator. All
reagents were purchased from Thermo Fisher Scientific, Inc.
For cell treatment, 30% inhibitory concentration
(IC30, 4.75 µM) or 50% inhibitory concentration
(IC50, 7.16 µM) of DOX was added into the culture medium
for 24, 48, 72 and 96 h at 37˚C and cells were used for subsequent
analysis.
To induce oxidative stress, 200 mM
H2O2 was added into the culture medium for 24
h at 37˚C and cells were used for subsequent analysis.
Cell Counting Kit-8 (CCK-8) assay
H9C2 cells were collected and resuspended in
serum-free DMEM at a final concentration of 1x106
cells/ml. A total of 6x103 cells/well were seeded into a
96-well plate and cultured overnight. The cells were subsequently
treated with various concentrations of DOX (1.0, 2.5, 5.0, 7.5,
10.0, 12.5, 15.0 and 17.5 µM) for 24 h at 37˚C and CCK-8 solution
(Sigma-Aldrich; Merck KGaA) was added to the cells and incubated
for between 30 min and 2 h at 37˚C in the dark according to the
manufacturer's protocol. The absorbance at 450 nm was measured
using a microplate reader (Synergy 2 Multi-Mode Microplate Reader;
BioTek Instruments, Inc.) to determine cell viability.
Reverse transcription-quantitative PCR
(RT-qPCR)
A total of 1x106 H9C2 cells were employed
for RNA extraction using TRIzol® (Invitrogen; Thermo
Fisher Scientific, Inc.) following the manufacturer's instructions.
Complementary DNA was obtained via reverse transcription using the
RevertAid First Strand cDNA Synthesis kit (Fermentas; Thermo Fisher
Scientific, Inc.). The reactions were performed according to the
following temperature protocol: 65˚C for 5 min, ice bath for 2 min,
37˚C for 45 min and 85˚C for 10 min. qPCR was performed using the
SYBR Green Real-Time PCR Master Mixes (Fermentas; Thermo Fisher
Scientific, Inc.) according to the following thermocycling
conditions: 95˚C for 2 min; 35 cycles of 95˚C for 30 sec and 60˚C
for 1 min. The primer sequences used are as follows: LRPPRC
forward, 5'-CTGCACTGTGCTCTTCAAGC-3' and reverse,
5'-GACTGCACACTACCGAAGCA-3'; Bcl-2 forward,
5'-CGACTTTGCAGAGATGTCCA-3' and reverse, 5'-ATGCCGGTTCAGGTACTCAG-3';
Bax forward, 5'-CGAGCTGATCAGAACCATCA-3' and reverse,
5'-CTCAGCCCATCTTCTTCCAG-3'; β-actin forward,
5'-AGCCATGTACGTAGCCATCC-3' and reverse, 5'-CTCTCAGCTGTGGTGGTGAA-3';
cytochrome c oxidase subunit (COX) 1 forward,
5'-GGAGCAGTATTCGCCATCAT-3' and reverse, 5'-CGACGAGGTATCCCTGCTAA-3';
COX 3 forward, 5'-GAACATACCAAGGCCACCAC-3' and reverse,
5'-TAATTCCTGTTGGGGGTCAG-3'; NADH dehydrogenase subunit 1 forward,
5'-CTCCCTATTCGGAGCCCTAC-3' and reverse, 5'-GGAGCTCGATTTGTTTCTGC-3';
and cytochrome b forward, 5'-GTCGGCGAAGAAAAATGTGT-3' and
reverse, 5'-AAGCTGCTCACAGAGGGGTA-3'. β-actin was employed as an
internal standard. Gene expression was calculated using the
2-ΔΔCq method (17). Each
experiment was repeated three times.
MitoTracker Green and MitoTracker Red
staining
A total of 1x106 H9C2 cells were loaded
with 100 nM green-fluorescing MitoTracker Green (MitoGreen, YEASEN
Technology Company, Shanghai) for 30 min at 37˚C to measure
mitochondrial content, and 500 nM MitoTracker Red (MitoRed, YEASEN
Technology Company, Shanghai) for 30 min at 37˚C. Images were taken
using a X71 (U-RFL-T) fluorescence microscope (Olympus
Corporation). All data were obtained from experiments with at least
three replicates.
Western blot analysis
H9C2 cells were washed 3 times with ice-cold PBS and
lysed in RIPA total protein lysis buffer (Guangzhou RiboBio Co.,
Ltd.) using the SoniConvert® sonicator (DocSense,
Chengdu, China) according to the manufacturer's instructions for 3
sec(s) at room temperature. Concentration of protein in the lysate
was measured using BCA kit (Sigma-Aldrich; Merck KGaA) and protein
was mixed with 5X SDS loading buffer (Beyotime Institute of
Biotechnology) and incubated for 10 min at 100˚C to denature.
Subsequently, 30 µg of each proteins was separated on a 6-15%
gradient SDS-PAGE gels and transferred on to a PVDF membrane. Then
membrane was blocked in blocking buffer containing 5% BSA (Beyotime
Institute of Biotechnology) in PBS at room temperature for 1 h.
Western blotting was performed using the primary antibodies
anti-β-actin (1:5,000; cat. no. ab8226), mouse monoclonal
anti-LRPPRC (1:500; cat. no. ab21864), rabbit polyclonal anti-Bcl-2
(1:2,000; cat. no. ab196495) and rabbit monoclonal anti-Bax
(1:1,000; cat. no. ab32503; all from Abcam). Membranes were
incubated with primary antibodies at room temperature for 1 h.
Horseradish peroxidase-conjugated anti-rabbit (cat. no. ab79080) or
anti-mouse IgG secondary antibodies (cat. no. ab47827; both
1:20,000; both from Abcam) were then incubated with the membrane at
room temperature for a further 1 h. Signals were detected using ECL
reagent (Thermo Fisher Scientific, Inc.) and Image J software
(version 2.0; National Institutes of Health) was used for
densitometry. β-actin was used as the internal reference.
Fluorescence immunostaining
H9C2 (1x105) cells attached onto 18 mm
coverslips were fixed with 4% paraformaldehyde for 10 min at room
temperature and permeabilized with 0.5% Triton X-100
(Sigma-Aldrich; Merck KGaA) for 10 min at room temperature. To
block unspecific staining, the cells were incubated with 5% normal
goat serum (Thermo Fisher Scientific, Inc.) diluted in 1X PBS at
room temperature for 30 min. Staining was performed using primary
LRPPRC antibodies (1:200; cat. no. ab21864; Abcam) at room
temperature for 2 h in the dark, followed by four washes with
PBS-Tween 20 (0.5%) and incubation with rabbit
Cy5®-conjugated secondary antibody (cat. no. ab6563,
1:2,000 in 0.5% normal goat serum) for 1 h at room temperature.
Nuclei were subsequently stained with 5 µg/ml DAPI (Sigma-Aldrich;
Merck KGaA) at room temperature for 10 min. Images were captured
with a X71 (U-RFL-T) fluorescence microscope (Olympus Corporation)
at magnification, x100.
LRPPRC knockdown and
overexpression
Knockdown of LRPPRC was achieved via transient
transfection of H9C2 cells with small interfering RNA (siRNA)
duplexes (cat. no. for si and NC sequence RSH045567; Guangzhou
FulenGen Co., Ltd.) targeting LRPPRC mRNA, according to the
manufacturer's instructions. A total of 1x106 cells in 2
ml serum-free DMEM were seeded per well of 6-well plates.
Transfection complexes were formed via mixing 5 µl
Lipofectamine® 2000 reagent (Thermo Fisher Scientific,
Inc.) with the siRNA oligomer (50 nM) in 0.5 ml OptiMEM medium
(Thermo Fisher Scientific, Inc.) at room temperature for 20 min.
The complexes were subsequently added to the cells, which were
cultured for 48 h at 37˚C and collected for subsequent analysis.
Control cells were transfected with negative control siRNA.
For LRPPRC overexpression, the coding sequence of
rat LRPPRC (accession no. NM_001008519.1) was provided by Guangzhou
FulenGen Co. Ltd. and was inserted into the mammalian expressing
vector pSG5L-Flag-HA (Addgene, Inc.). Overexpression of LRPPRC was
achieved via transient transfection as aforementioned. Notably, for
each transfection in 6-well plate, 1.6 µg of plasmid was used.
Cell cycle analysis
To analyze the distribution of the cell cycle phases
via quantification of the DNA content, H9C2 cells
(1x106/well) grown in 6-well plates were washed with
ice-cold 1X PBS and fixed overnight at 4˚C with ice-cold 70%
ethanol. Subsequently, fixed cells were washed 3 times with
ice-cold 1X PBS, collected via centrifugation at 1,000 x g at 4˚C
for 5 min and incubated with a final concentration of 100 µg/ml
RNase A and 10 µg/ml propidium iodide (PI; Beyotime Institute of
Biotechnology) for 15 min in the dark at room temperature. The
cells were analyzed using a three laser Navios flow cytometer
(Beckman Coulter, Inc.) and data was analyzed using FlowJo (FlowJo
LLC; v9.7.4).
Apoptosis analysis
To detect apoptotic cell death, annexin V-FITC/PI
double staining was performed using Annexin V FITC apoptosis
detection kit (Becton, Dickinson and Company) following the
manufacturer's instructions. H9C2 cells (1x106) were
washed with ice-cold 1X PBS and pelleted by centrifugation at 400 x
g at 4˚C for 10 min. Following removing of the supernatant, the
pellet was resuspended in 1X binding buffer from the Annexin V FITC
apoptosis detection kit and stained with 5 µl FITC-labeled annexin
V at 4˚C for 15 min in the dark. Subsequently, 10 µl PI was added
to the mixture and incubated at 4˚C for 5 min in the dark. The
stained cells were analyzed via flow cytometry using a three lasers
Navios flow cytometer within 1 h following staining (Beckman
Coulter, Inc.) and data was analyzed using FlowJo.
ATP measurement
To measure total ATP content, 5x105 H9C2
cells were suspended and lysed in a buffer containing 0.22 M
sucrose, 0.12 M mannitol, 40 mM tricine, pH 7.5 and 1 mM EDTA at
4˚C for 10 min. The total lysate was analyzed using ATP
Bioluminescence Assay kit (Sigma-Aldrich; Merck KGaA) according to
the manufacturer's instructions and quantitatively measured using
the Optocomp I BG-1 luminometer (GEM Biomedical, Inc.).
ROS staining
Subsequently, 2',7'-Dichlorofluorescin diacetate
(DCFH-DA; Sigma-Aldrich; Merck KGaA) was used for ROS staining
following 4.75 µM of DOX exposure at 37˚C for 24 h. To scavenge
induced ROS, 10 µM of N-acetyl-L-cysteine (NAC) was added to the
culture for 24 h treatment. Briefly, 1x106 H9C2
cells/well cultured in a 6-well plate were incubated with 10 µM
DCFH-DA in DMEM medium at 37˚C for 20 min. The plates were washed
three times with ice-cold 1X PBS to remove excess DCFH-DA dye.
Fluorescence was visualized immediately at 485 nm (excitation) and
530 nm (emission) using an inverted fluorescence microscope
(Olympus Corporation) at magnification, x100.
Statistical analysis
All data were analyzed using SPSS v13.0 (SPSS,
Inc.). Data are presented as the mean ± standard deviation. The
statistical significances of the differences between groups were
assessed using unpaired Student's t-test or one-way ANOVA followed
by Bonferroni's post hoc analysis. P<0.05 was considered to
indicate a statistically significant difference. All experiments
were repeated three times independently.
Results
Low-dose DOX upregulates the
expression of LRPPRC
Prior to assessing the effect of DOX exposure on the
expression levels of LRPPRC, the cytotoxic effect of DOX on H9C2
cells was measured using the CCK-8 assay. As illustrated in
Fig. 1A, the IC30
(4.75±0.31 µM) and IC50 (7.16±0.52 µM) of DOX were
employed for subsequent analysis. Following 0, 24, 48, 72 and 96 h
of DOX exposure at IC30 or IC50, the mRNA and
protein levels of LRPPRC were detected. The levels of Bcl-2 and
Bax, which have been indicated to be regulated by LRPPRC (13), and therefore may be associated with
mitochondrial homeostasis, were also examined. Following 24-96 h of
DOX exposure at the IC30, LRPPRC mRNA levels were
significantly upregulated compared with control cells. By contrast,
DOX at the IC50 did not affect LRPPRC, Bcl-2 and Bax
mRNA levels, indicating that a low dose of DOX may regulate LRPPRC,
and thus modulate mitochondrial function (Fig. 1B). After 48-h treatment under
IC30 of DOX, Bcl-2 mRNA was upregulated significantly.
Subsequently, western blotting was performed to detect the effect
of DOX at IC30 on the protein levels of LRPPRC, Bcl-2
and Bax. In consistence with the alterations at the mRNA level, an
increase in the LRPPRC protein expression level was observed
following DOX exposure. Notably, the protein levels of Bcl-2 and
Bax were increased with no evident increase observed at their mRNA
levels, which may be attributed to their possible stabilization by
LRPPRC at the protein level (Fig.
1C). A significant increase in Bcl-2 mRNA and protein levels
was observed at 48-h exposure, which suggested that Bcl-2 was
potentially involved in LRPPRC regulation. The upregulation of
LRPPRC following DOX exposure was additionally verified via
immunofluorescence staining. As anticipated, the expression of
LRPPRC was increased following 24-96 h of IC30 of DOX
exposure compared with control cells (Fig. 1D).
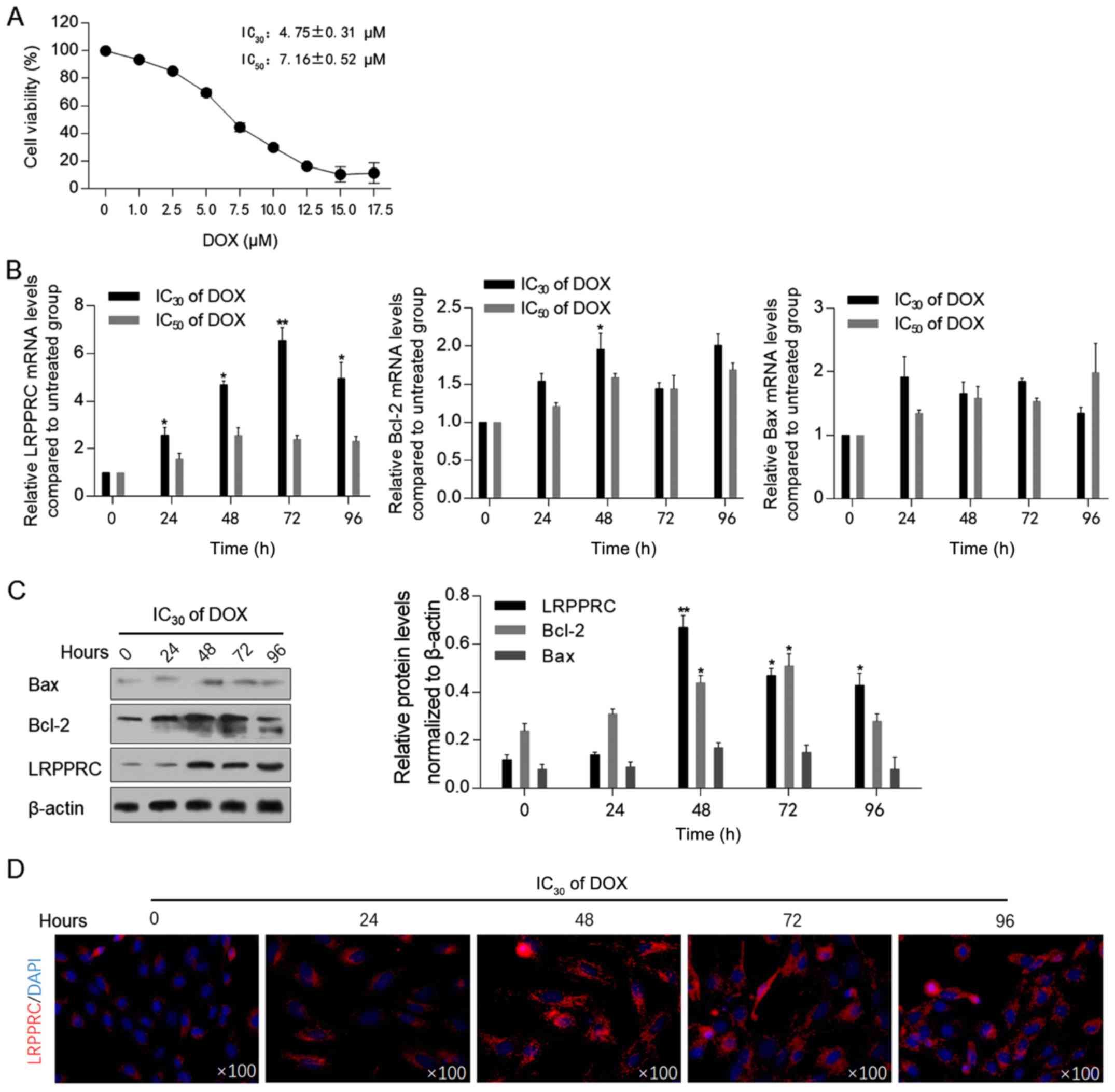 | Figure 1DOX treatment upregulates LRPPRC at
the transcriptional level. (A) Cell Counting Kit-8 assay was
performed to detect DOX cytotoxicity in H9C2 cells. (B) Following
DOX treatment at IC30 or IC50 for 0, 24, 48,
72 and 96 h, the mRNA levels of LRPPRC, Bcl-2 and Bax were detected
via reverse transcription-quantitative PCR. (C) Protein levels of
LRPPRC, Bcl-2 and Bax were detected via western blot (left panel),
and quantitatively analyzed via ImageJ software (right panel). (D)
Via immunofluorescence staining, the expression and localization of
LRPPRC (red) and nucleus (blue) were visualized.
*P<0.05 and **P<0.01 vs. 0 h DOX
exposure group. LRPPRC, leucine-rich pentatricopeptide
repeat-containing; DOX, doxorubicin. |
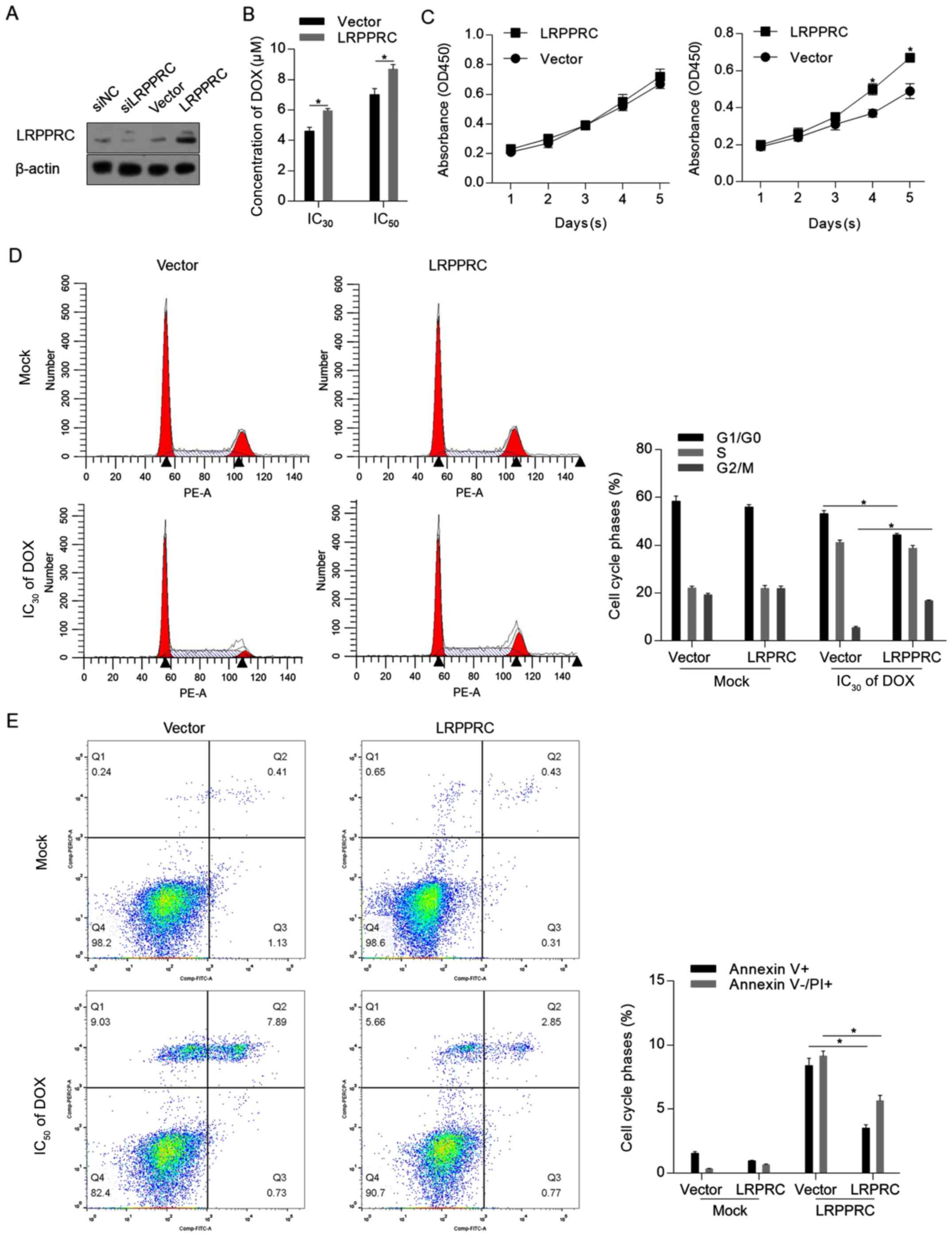
LRPPRC overexpression exerts
protective effects against DOX treatment
To assess the effect of LRPPRC under normal
conditions and DOX treatment, LRPPRC expression level was firstly
modulated via knockdown or overexpression. As demonstrated in
Fig. 2A, the LRPPRC expression
vector substantially increased the LRPPRC protein level. As
knockdown of LRPPRC slightly affected the protein level compared
with control cells, which may be attributed to the low endogenous
level of LRPPRC, overexpression of LRPPRC was employed for
subsequent functional analysis. The CCK-8 assay indicated that
overexpression of LRPPRC sensitized H9C2 cells to DOX (Fig. 2B). Under normal conditions,
overexpression of LRPPRC did not affect cell proliferation, while
cell proliferation was induced by LRPPRC overexpression following
DOX exposure compared with control cells (Fig. 2C). The protective effect of LRPPRC
overexpression was additionally verified by detecting the
distribution of the cell cycle phases and the apoptotic and
non-apoptotic cell death. As illustrated in Fig. 2D and E, although no detectable effects were
observed on proliferation and cell death under normal conditions,
overexpression of LRPPRC significantly reversed the DOX-induced
G1/G0 arrest, increased the cell population
in the G2/M phase and significantly reversed Dox-induced
apoptotic and non-apoptotic cell death. Taken together, these
results indicated that LRPPRC overexpression exerts protective
effects against DOX-induced cytotoxicity of H9C2 cardiomyocytes,
while slightly affecting the cells under normal conditions.
LRPPRC overexpression maintains
mitochondrial function potentially via scavenging ROS induced by
DOX
Considering the critical regulatory roles of LRPPRC
on mitochondrial function (7),
cellular ATP synthesis was detected following DOX exposure. The
results indicated that LRPPRC overexpression significantly
increased ATP synthesis both under normal conditions and following
DOX exposure (Fig. 3A).
Subsequently, mitochondrial function was examined via detecting the
mitochondrial mass and transcriptional activity. As illustrated in
Fig. 3B and C, the DOX treatment decreased mitochondrial
mass and transcriptional activity compared with Mock/vector group,
and overexpression of LRPPRC reversed the decrease in mitochondrial
mass and transcriptional activity.. Moreover, ROS accumulation was
decreased in LRPPRC-overexpressing cells after DOX treatment, which
additionally indicated that LRPPRC may exert protective effects via
scavenging ROS (Fig. 3D). Following
DOX treatment at IC50 for 24 h, DOX-induced cell death
was significantly attenuated by overexpression of LRPPRC or ROS
scavenger NAC, indicating that LRPPRC potentially regulates ROS
accumulation (Fig. 3E).
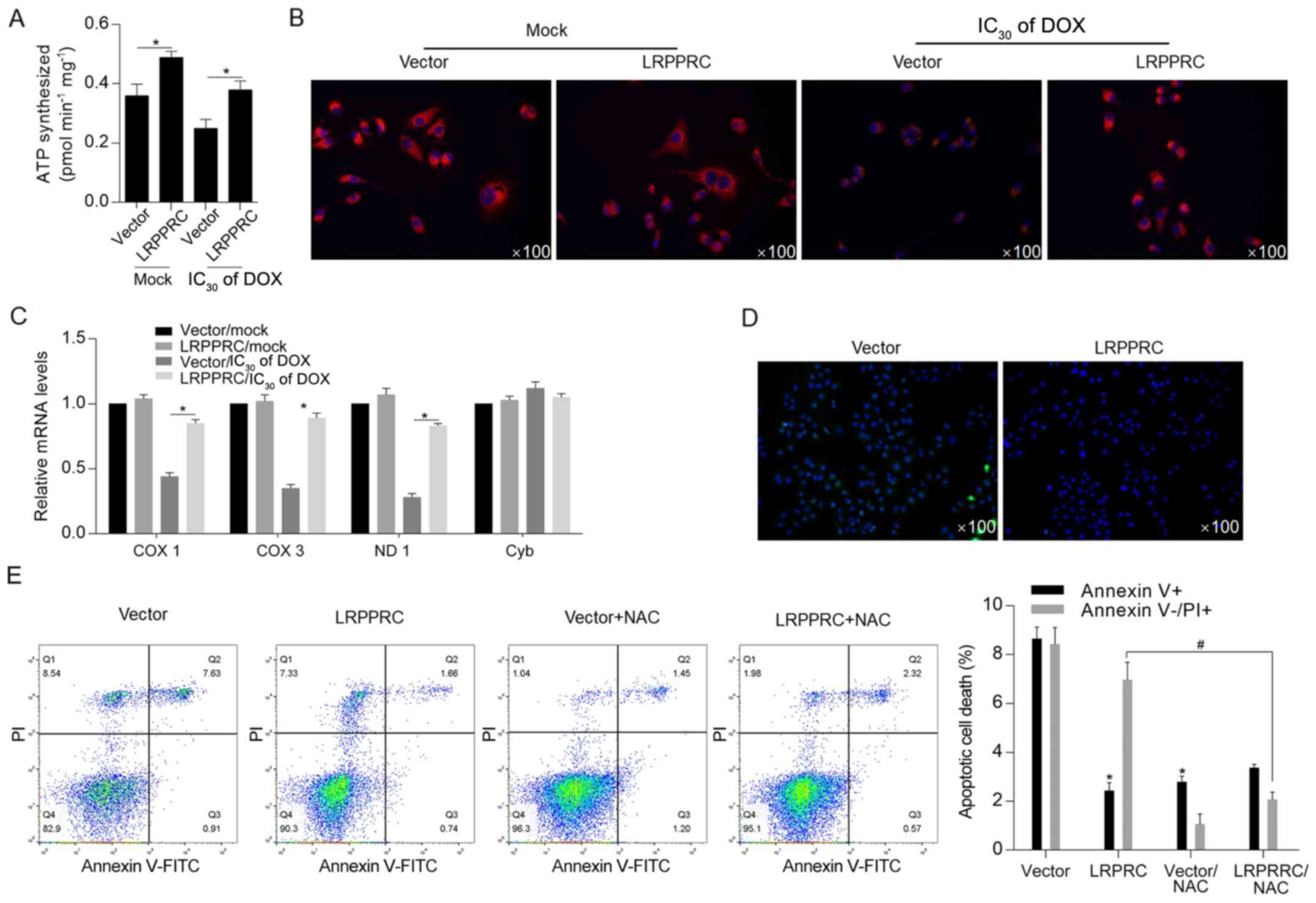 | Figure 3DOX-induced LRPPRC exerts protective
effects against DOX exposure potentially via scavenging ROS. (A)
Following DOX exposure, the effect of LRPPRC overexpression on ATP
synthesis was examined. (B) Mitochondrial mass was measured using
MitoTracker Red staining. (C) To evaluate the effect of LRPPRC on
mitochondrial transcriptional activity, the expression levels of
COX 1, COX 3, ND1 and Cyb were detected via reverse
transcription-quantitative PCR. (D) ROS accumulation was detected
following DOX exposure at IC30 for 24 h. (E) Annexin
V-FITC/PI double staining followed by flow cytometric analysis was
performed to detect apoptotic and non-apoptotic cell death.
*P<0.05 vs. vector group; #P<0.05 vs.
LRPPRC group. LRPPRC, leucine-rich pentatricopeptide
repeat-containing; DOX, doxorubicin; PI, propidium iodide; COX,
cytochrome c oxidase subunit; ND1, NADH dehydrogenase
subunit 1; Cyb, cytochrome b; NAC, N-acetyl-L-cysteine; ROS,
reactive oxygen species. |
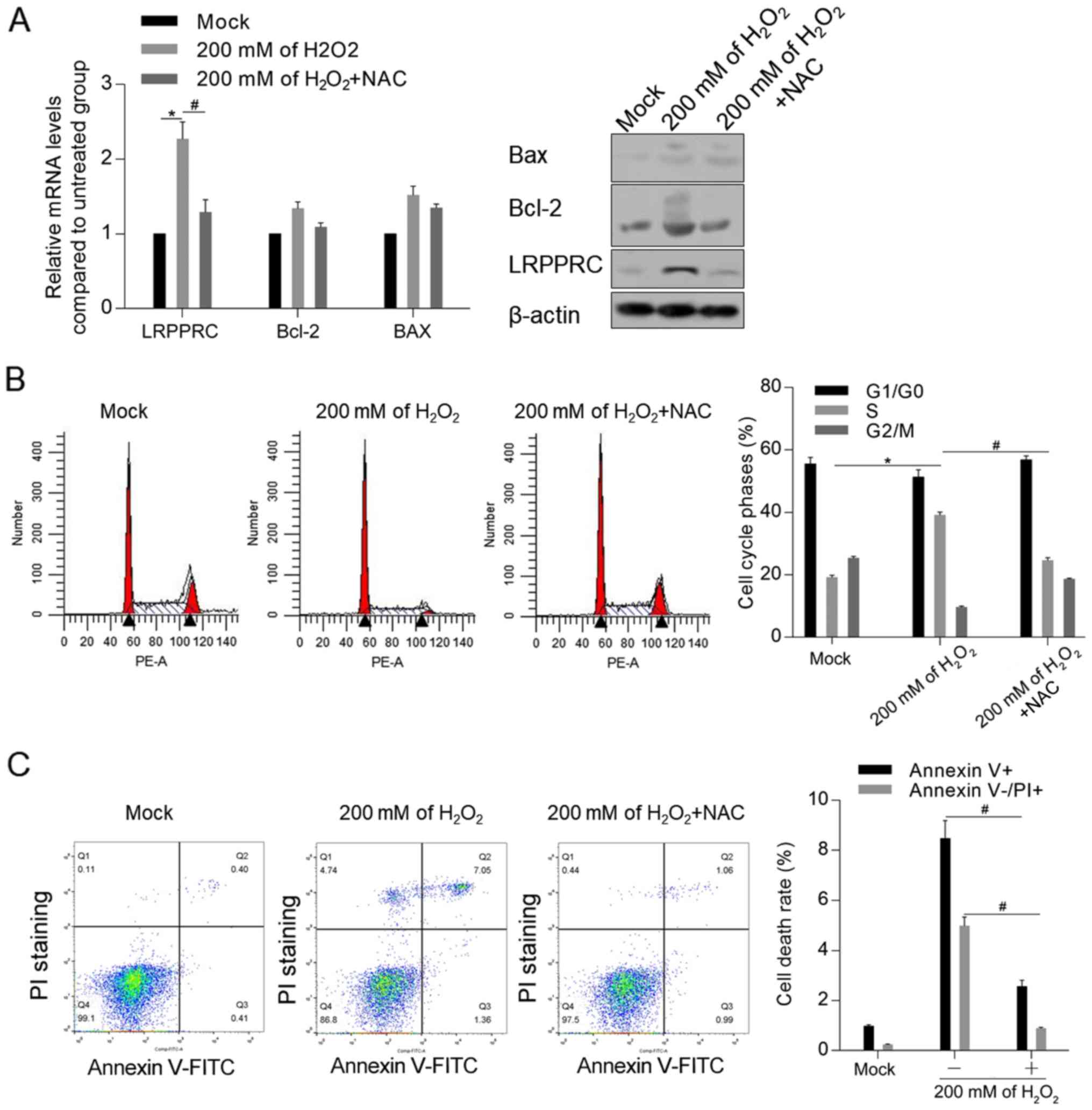
Accumulated ROS is essential for
LRPPRC upregulation
To investigate the mechanism of LRPPRC induction by
DOX, it was examined whether accumulated ROS induced by
H2O2 was associated with the upregulation of
LRPPRC. As demonstrated in Fig. 4A,
treatment with 200 mM H2O2 for 24 h
significantly upregulated the mRNA and protein level of LRPPRC
compared with control cells, which was reversed by co-treatment
with ROS scavenger NAC. Moreover, NAC attenuated the
H2O2-induced cell cycle arrest and apoptosis
(Fig. 4B and C).
Discussion
The present study investigated the effect of DOX on
the expression of LRPPRC, which is a mitochondria-associated
regulator, and subsequently evaluated the regulatory effects of
DOX-induced LRPPRC on cultured H9C2 cardiomyocytes. DOX has been
indicated to exhibit well-known side effects, including the
induction of cardiotoxicity, by altering the mitochondrial
membrane, which comprises lipid peroxidation, generation of free
radicals, the increase in the myocardial levels of sodium and
calcium and the induction of autophagy/mitophagy, thereby resulting
in apoptotic and non-apoptotic cell death in cardiomyocytes
(2-5).
The results of the present study demonstrated that DOX induced
transcriptional upregulation of LRPPRC, which was followed by
stabilization of Bcl-2 and Bax at the protein level. Antioxidants
have been indicated to exert cardioprotective effects against DOX
exposure via scavenging ROS (18,19). To
the best of our knowledge, the current study indicated for the
first time that the expression of LRPPRC, which exerted
cardioprotective effects against DOX-induced injury, was increased
following DOX exposure.
The results of the present study demonstrated that
overexpressed LRPPRC exerted protective effects against DOX
exposure. LRPPRC reversed the DOX-mediated inhibition of
proliferation and cell cycle arrest and improved mitochondrial
function. Under normal conditions, overexpressed LRPPRC increased
ATP synthesis, mitochondrial DNA mass and transcriptional levels
without affecting proliferation, which is consistent with previous
findings (20,21). However, overexpressed LRPPRC did not
affect cell proliferation and cell cycle distribution under normal
conditions, indicating that LRPPRC primarily reversed DOX-induced
oxidative stress. To further evaluate the cardioprotective effect
of LRPPRC, DOX was administered to cells overexpressing LRPPRC. As
anticipated, DOX exposure at a relatively high concentration
(IC50 of DOX) induced cell death, which was
significantly reversed following overexpression of LRPPRC.
Consistently, overexpressed LRPPRC did not affect cell death under
normal conditions. However, overexpressed LRPPRC significantly
inhibited both apoptotic and non-apoptotic cell death induced by
DOX treatment, with apoptotic cell death being inhibited to a
greater extent. Interestingly, LRPPRC-knockdown did not efficiently
reduce LRPPRC levels, which may be attributed to the low levels of
endogenous LRPPRC in H9C2 cells.
Mitochondria have been indicated to serve essential
roles cardiac cell homeostasis and preventing cells from
injury-induced death (22).
Dysregulated mitochondrial homeostasis has been reported to induce
mitochondrial dysfunction, including a decrease in ATP synthesis,
the accumulation of mitochondrial and cytoplasmic ROS and
alterations in redox balance in cardiomyocytes (23-25).
ROS are considered to be the major mediators of DOX-induced
cardiotoxicity (26,27). Excess ROS induced by DOX have been
demonstrated to result in irreversible mitochondrial damage and
exacerbate cardiac diseases (22).
In the present study, DOX-induced LRPPRC and overexpressed LRPPRC
were revealed to maintain mitochondrial homeostasis and function,
as they increased ATP synthesis, mitochondrial mass and
transcriptional activity compared with the mock group.
Additionally, overexpression of LRPPRC in H9C2 cells reversed
DOX-induced ROS accumulation and subsequent apoptotic and
non-apoptotic cell death. It was also revealed that accumulated ROS
induced by H2O2 exposure increased LRPPRC
protein levels, indicating that ROS may directly regulate LRPPRC.
Notably, considering the low endogenous ROS level in the absence of
H2O2, the effect of
H2O2 was specifically detected with or
without NAC treatment. Therefore, it was hypothesized that the
ROS-LRPPRC axis may form a feedback loop, and it is worth
investigating whether accumulated ROS regulates LRPPRC protein
level in subsequent studies.
The current study aimed to evaluate the
cardioprotective effects of LRPPRC and its function in energy
metabolism in H9C2 rat cardiomyocytes. However, certain limitations
exist in the present study. Firstly, the cardioprotective effects
of LRPPRC were evaluated by introducing exogenous LRPPRC instead of
using DOX-induced LRPPRC. Additional studies with DOX pretreatment
are required to determine whether DOX-induced LRPPRC exhibits
similar effects to those of exogenous LRPPRC. Secondly, although
several mitochondrial functions were investigated in the current
study (ATP synthesis, mitochondrial transcriptional activity and
mass), mitochondria-associated autophagy and mitophagy in H9C2
cells under DOX treatment still require additional investigation.
Taken together, these findings may provide evidence of the
cardioprotective effects of LRPPRC against DOX treatment.
Acknowledgements
The authors would like to thank Mrs. Yun Bai (Third
Military Medical University, Chongqing, China) for language
editing.
Funding
The present study was funded by Starting Scientific
Foundation of Zunyi Medical University (grant no.
SS20180601ZF).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
QT and DL designed the investigation strategy. WX,
XK and JZ participated in data collection, performed the
statistical analysis and wrote the manuscript. WX and XK performed
cell experiments. YX performed part of the molecular experiments,
including transfection and RT-qPCR. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Menna P, Salvatorelli E and Minotti G:
Doxorubicin degradation in cardiomyocytes. J Pharmacol Exp Ther.
322:408–419. 2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Umansky SR, Shapiro JP, Cuenco GM, Foehr
MW, Bathurst IC and Tomei LD: Prevention of rat neonatal
cardiomyocyte apoptosis induced by simulated in vitro ischemia and
reperfusion. Cell Death Differ. 4:608–616. 1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Olson HM, Young DM, Prieur DJ, LeRoy AF
and Reagan RL: Electrolyte and morphologic alterations of
myocardium in adriamycin-treated rabbits. Am J Pathol. 77:439–454.
1974.PubMed/NCBI
|
|
4
|
Myers CE, Gianni L, Simone CB, Klecker R
and Greene R: Oxidative destruction of erythrocyte ghost membranes
catalyzed by the doxorubicin-iron complex. Biochemistry.
21:1707–1712. 1982.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Olson RD and Mushlin PS: Doxorubicin
cardiotoxicity: Analysis of prevailing hypotheses. FASEB J.
4:3076–3086. 1990.PubMed/NCBI
|
|
6
|
Piasek A, Bartoszek A and Namiesnik J:
Phytochemicals that counteract the cardiotoxic side effects of
cancer chemotherapy. Postepy Hig Med Dosw (Online). 63:142–158.
2009.PubMed/NCBI(In Polish).
|
|
7
|
Sasarman F, Brunel-Guitton C, Antonicka H,
Wai T and Shoubridge EA: LSFC Consortium. LRPPRC and SLIRP interact
in a ribonucleoprotein complex that regulates posttranscriptional
gene expression in mitochondria. Mol Biol Cell. 21:1315–1323.
2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ruzzenente B, Metodiev MD, Wredenberg A,
Bratic A, Park CB, Cámara Y, Milenkovic D, Zickermann V, Wibom R,
Hultenby K, et al: LRPPRC is necessary for polyadenylation and
coordination of translation of mitochondrial mRNAs. EMBO J.
31:443–456. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chujo T, Ohira T, Sakaguchi Y, Goshima N,
Nomura N, Nagao A and Suzuki T: LRPPRC/SLIRP suppresses
PNPase-mediated mRNA decay and promotes polyadenylation in human
mitochondria. Nucleic Acids Res. 40:8033–8047. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Xu F, Morin C, Mitchell G, Ackerley C and
Robinson BH: The role of the LRPPRC (leucine-rich pentatricopeptide
repeat cassette) gene in cytochrome oxidase assembly: Mutation
causes lowered levels of COX (cytochrome c oxidase) I and COX III
mRNA. Biochem J. 382:331–336. 2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mili S and Pinol-Roma S: LRP130, a
pentatricopeptide motif protein with a noncanonical RNA-binding
domain, is bound in vivo to mitochondrial and nuclear RNAs. Mol
Cell Biol. 23:4972–4982. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Topisirovic I, Siddiqui N, Orolicki S,
Skrabanek LA, Tremblay M, Hoang T and Borden KLB: Stability of
eukaryotic translation initiation factor 4E mRNA is regulated by
HuR, and this activity is dysregulated in cancer. Mol Cell Biol.
29:1152–1162. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sterky FH, Ruzzenente B, Gustafsson CM,
Samuelsson T and Larsson NG: LRPPRC is a mitochondrial matrix
protein that is conserved in metazoans. Biochem Biophys Res Commun.
398:759–764. 2010.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang S, Wang Y, Zhang Z, Liu Q and Gu J:
Cardioprotective effects of fibroblast growth factor 21 against
doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell
Death Dis. 8(e3018)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liu D, Ma Z, Xu L, Zhang X, Qiao S and
Yuan J: PGC1α activation by pterostilbene ameliorates acute
doxorubicin cardiotoxicity by reducing oxidative stress via
enhancing AMPK and SIRT1 cascades. Aging (Albany NY).
11:10061–10073. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wen J, Zhang L, Liu H, Wang J, Li J, Yang
Y, Wang Y, Cai H, Li R and Zhao Y: Salsolinol attenuates
doxorubicin-induced chronic heart failure in rats and improves
mitochondrial function in H9c2 cardiomyocytes. Front Pharmacol.
10(1135)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Errami M, Galindo CL, Tassa AT, Dimaio JM,
Hill JA and Garner HR: Doxycycline attenuates isoproterenol- and
transverse aortic banding-induced cardiac hypertrophy in mice. J
Pharmacol Exp Ther. 324:1196–1203. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hori Y, Kunihiro S, Sato S, Yoshioka K,
Hara Y, Kanai K, Hoshi F, Itoh N and Higuchi S: Doxycycline
attenuates isoproterenol-induced myocardial fibrosis and matrix
metalloproteinase activity in rats. Biol Pharm Bull. 32:1678–1682.
2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mourier A, Ruzzenente B, Brandt T,
Kühlbrandt W and Larsson NG: Loss of LRPPRC causes ATP synthase
deficiency. Hum Mol Genet. 23:2580–2592. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cuillerier A, Honarmand S, Cadete V, Ruiz
M, Forest A, Deschênes S, Beauchamp C, LSFC Consortium; Charron G,
Rioux JD, et al: Loss of hepatic LRPPRC alters mitochondrial
bioenergetics, regulation of permeability transition and
trans-membrane ROS diffusion. Hum Mol Genet. 26:3186–3201.
2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sack MN, Fyhrquist FY, Saijonmaa OJ,
Fuster V and Kovacic JC: Basic biology of oxidative stress and the
cardiovascular system: Part 1 of a 3-part series. J Am Coll
Cardiol. 70:196–211. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L,
Reiter RJ, Qiao S and Yuan J: AMPK/PGC1a activation by melatonin
attenuates acute doxorubicin cardiotoxicity via alleviating
mitochondrial oxidative damage and apoptosis. Free Radic Biol Med.
129:59–72. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ma Z, Xin Z, Di W, Yan X, Li X, Reiter RJ
and Yang Y: Melatonin and mitochondrial function during
ischemia/reperfusion injury. Cell Mol Life Sci. 74:3989–3998.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Galley HF, McCormick B, Wilson KL, Lowes
DA, Colvin L and Torsney C: Melatonin limits paclitaxel-induced
mitochondrial dysfunction in vitro and protects against
paclitaxel-induced neuropathic pain in the rat. J Pineal Res.
63(e12444)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Takemura G and Fujiwara H:
Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms
to management. Prog Cardiovasc Dis. 49:330–352. 2007.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Octavia Y, Tocchetti CG, Gabrielson KL,
Janssens S, Crijns HJ and Moens AL: Doxorubicin-induced
cardiomyopathy: From molecular mechanisms to therapeutic
strategies. J Mol Cell Cardiol. 52:1213–1225. 2012.PubMed/NCBI View Article : Google Scholar
|