Introduction
Ketone bodies, which are mainly generated from fatty
acids, are important alternative energy sources during fasting
(1,2). Mitochondrial
3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS2; Nomenclature
Committee of the International Union of Biochemistry and Molecular
Biology classification no. EC 2.3.3.10) catalyzes the condensation
of acetoacetyl-CoA and acetyl-CoA to form
3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), which is the
rate-limiting step of ketone body synthesis (3,4).
HMGCS2 deficiency (Online Mendelian Inheritance in
Man reference no. 605911) is a metabolic disorder caused by
mutations in the HMGCS2 gene, which consists of 10 exons
located on chromosome 1p12-13(5).
To date, nearly 30 cases that have been reported in the literature
describe an autosomal recessive inheritance pattern (6). Most patients present with symptomatic
hypoketotic hypoglycemia and hepatomegaly after a period of
prolonged fasting or intercurrent illness (6). Metabolic acidosis is often noted
during the acute phase (7-12).
During the metabolic crisis, the serum concentration of free fatty
acids (FFAs) is relatively high when compared with that of the
total ketone bodies (TKBs) (6). The
clinical presentation is similar to that observed with fatty acid
β-oxidation defects, but the urinary organic acids and blood
acylcarnitine profiles are non-specific in cases of HMGCS2
deficiency. Urinary 4-hydroxy-6-methyl-2-pyrone (4HMP) is a
possible specific marker of the disorder (10). While the use of liver samples for an
enzyme assay or immunoblotting aids in diagnosis (13), obtaining these samples is an
invasive procedure. The diagnosis of HMGCS2 deficiency is therefore
usually based on genetic analysis.
Genetic analyses were performed on several suspected
cases referred to Department of Pediatrics, Graduate School of
Medicine, Gifu-University (Gifu, Japan) from 2005. From these
cases, four patients carried six rare non-synonymous variants of
HMGCS2, among which five of the variants have, to the best
of our knowledge, not been reported previously. To confirm their
pathogenicity in vitro, the variant enzymes were expressed
in 293T cells and the Escherichia coli strain BL21(DE3).
Materials and methods
Cases
The patients in all four cases were born to
non-consanguineous Japanese parents with no family history of
inherited metabolic disease. All of the patients were born
uneventfully following an uncomplicated term pregnancy. Only
patient 3 was a light-for-date infant; the others had normal birth
weights. After birth, physical growth and neurological development
until the onset of disease were normal except for in patient 4.
Clinical features and the results of first blood examinations
showing metabolic decompensation are listed in Table I. Before enrolling the four patients
into this study, written informed consent was obtained from the
parents of each patient by their attending pediatrician. This study
was approved by the Ethical Committee of the Graduate School of
Medicine, Gifu University, Japan (approval no. 29-503).
 | Table ILaboratory findings from a single
blood sample taken during hypoglycemic crisis in each of the four
patients with HMGCS2 variants. |
Table I
Laboratory findings from a single
blood sample taken during hypoglycemic crisis in each of the four
patients with HMGCS2 variants.
| |
Laboratory
findings | Mutation in
HMGCS2 |
|---|
| Patient | Sex | Age of onset
months | pH | BE mM | Glucose mM | TKBs mM | FFAs mM | FFAs/TKBs | Glucose x TKBs | AST IU/l | ALT IU/l | C0 µM | C2 µM | C2/C0 | Allele 1 | Allele 2 |
|---|
| 1 | F | 8 | 7.02 | -26.8 | 0.78 | 0.502 | 3.32 | 6.6 | 0.39 | 358 | 173 | 20.6 | 56.1 | 2.72 | p.S392L | p.R500H |
| 2 | M | 6 | 6.95 | -28.9 | 1.17 | 0.4 | 2.5 | 6.3 | 0.47 | 241 | 140 | 8.03 | 75.8 | 9.44 | p.G219E | p.R500C |
| 3 | M | 7 | 7.13 | -24.4 | 1.83 | 0.161 | 2.20 | 13.7 | 0.29 | 230 | 156 | 8.75 | 53.4 | 6.10 | p.M235T | p.S392L |
| 4 | F | 7 | 6.92 | -29.7 | 1.11 | | 2.39 | | | 193 | 154 | 10.9 | 23.0 | 2.12 | p.M235T | p.V253A |
On the day of symptom onset, in July 2013 (the exact
day remained anonymous to protect the patient's identity), patient
1 was 8 months old. She took breast milk in the early morning.
Afterward, she could not obtain sufficient nutrition orally and
gradually became lethargic, before she began vomiting at 6 PM. At
Nagoya City University Hospital (looked after by TI; Nagoya,
Japan), she presented with tachypnea, tachycardia and hepatomegaly,
prompting the first blood test. In spite of the immediate
correction of hypoglycemia with intravenous 20% glucose infusion,
metabolic acidosis progressed. Continuous hemodiafiltration was
performed for 12 h to improve severe acidosis. Urinary organic acid
analysis showed increased excretion of dicarboxylic acid. In total,
5 µl serum was used for acylcarnitine analysis using NeoSMAAT kit
(cat. nos. 509254, 509261 and 509278; https://www.sekisuimedical.jp/business/diagnostics/others/neosmaat/;
Sekisui Medical Co., Ltd.) and the LCMS-8040 instrument (Shimadzu
Corporation). Her metabolic state was stabilized within 2 days from
the onset and she recovered with no neurological complications.
On the day of symptom onset, in November 2014,
patient 2 was 6 months old. He had a low-grade fever and showed
loss of appetite. The next day, he began having diarrhea and
vomited three times in the evening. After showing impaired
consciousness 2 days later, the first blood test was performed.
Ultrasonography revealed an enlarged and fatty liver. He was
transferred to Fujita Health University Hospital (Toyoake, Japan),
where continuous hemodiafiltration was performed for 18 h in a
similar procedure to that performed on patient 1. In total, 6 µl
serum was used for acylcarnitine analysis using MS2
Screening Neo II kit (https://www.siemens-healthineers.com/jp/newborn-mass-screening/ms2-screening-series;
Siemens Healthineers) and API 3200™ LC-MS/MS System (SCIEX). He
recovered without neurological complications, similar to patient
1.
On the day of symptom onset, in October 2015,
patient 3 was 7 months old. He appeared drowsy in the afternoon and
then gradually developed tachypnea and loss of appetite. The next
morning, the first blood examination was performed and hepatomegaly
was identified at Kitasato University Hospital (looked after by KA;
Sagamihara, Japan). Two dried blood spots were used for
acylcarnitine analysis using MS2 Screening Neo II kit
(https://www.siemens-healthineers.com/jp/newborn-mass-screening/ms2-screening-series;
Siemens Healthineers) and API 3200™ LC-MS/MS System (SCIEX). A
computed tomography scan revealed an enlarged and fatty liver.
Hypoglycemia was immediately corrected by glucose infusion.
Metabolic acidosis was corrected by continuous bicarbonate infusion
(about 2 mEq/kg/h) for 5 h. He recovered without neurological
complications.
As a newborn, patient 4 was fed with both breast
milk and infant formula and showed normal physical growth until 3
months of age. Thereafter, she was fed with only breast milk and
her physical growth and development slowed down. On the day of
symptom onset, in November 2016, she was 7 months old. She vomited
twice around noon and then gradually developed tachypnea and
impaired consciousness. She was hospitalized at Kurume University
Hospital (Kurume, Japan) early the next morning because of severe
hypoglycemia and metabolic acidosis. An infusion of glucose and
bicarbonate was started immediately. The patient's enlarged liver
showed homogeneous hyperechogenicity compared with the kidney,
which suggested a fatty liver. After 11 h, her metabolic status and
consciousness had almost fully recovered. In total, 6 µl serum was
used for acylcarnitine analysis using Quattro Premier MS/MS (cat.
no. 7200000594; Waters Coporation). Labeled carnitine standards set
B-op (cat. no. NSK-B-OP-1; Otsuka Pharmaceutical Co., Ltd.) was
used as internal standards.
In all 4 cases, urinary organic acids were analyzed
using GCMS-QP2010 Plus (Shimadzu Corporation). Urine samples
containing 0.1 mg creatinine were used for this analysis. In
Patient 4, QP5050 GC/MS (Shimadzu Corporation) was also used for
confirmation. Urine sample containing 0.2 mg creatinine was used
for this analysis. Urine samples were prepared according to the
previous report (14). The
mass/charge ratios (m/z) used to detect 4-HMP were 73, 99,
127, 139, 155, 170, 183 and 198(10). The details of blood acylcarnitine
analyses were summarized in Table
SI.
On first evaluation, the results of urinary organic
acid analyses and blood acylcarnitine profiles in the acute phases
of all four cases were regarded as non-specific. Patients 1, 2 and
3 were suspected of having HMGCS2 deficiency, because they
presented with hypoketotic hypoglycemia and a high ratio of
FFAs/TKBs, which prompted sequencing of their HMGCS2 alleles
immediately. In the case of patient 4, because the value of TKBs
during hypoglycemia was unknown, a gene panel analysis was instead
performed. After their critical episode, each patient was advised
to avoid prolonged fasting and to receive glucose infusion
prophylactically during anorexia, to prevent another hypoglycemic
episode. Patients 1, 2 and 4 have been followed for 6, 5 and 3
years, respectively. They have grown and developed normally without
neurological sequelae and fatty liver disappeared in all three. No
data were obtained regarding the prognosis of patient 3 due to the
loss of follow-up.
Mutational analysis
To obtain the white blood cells, ~4 ml whole blood
sample was added to an equivalent volume of solution consisting of
3% (W/V) dextran and 0.9% (W/V) NaCl, which was incubated for 1 h
at room temperature to precipitate the red blood cells and to
obtain the supernatant. The supernatant was then centrifuged at
1,730 x g for 10 min at room temperature to precipitate the white
blood cells. Genomic DNA was purified from the white blood cells
using a Sepa Gene kit (EIDIA Co., Ltd.) following the
manufacturer's protocol. The genomic HMGCS2 sequence was
obtained from the National Center for Biotechnology (NCBI)
Reference Sequence Database (accession no.
NG_013348.1/NM_005518.3). The 10 exons of HMGCS2 from
patients 1, 2 and 3 were amplified by PCR using a TaKaRa Taq™
(Takara Bio Inc.) and flanking intronic primers (listed in Table SII) under the following
thermocycling conditions: Initial denaturation at 94˚C for 1 min,
followed by 35 cycles of 94˚C for 1 min, 54˚C for 1 min and 72˚C
for 1 min. DNA electrophoresis in 2% agarose gel was used to verify
successful amplification. The amplified DNA in the gel was
visualized with EtBr solution (cat. no. 315-90051; Nippon Gene Co.,
Ltd.). Each amplified DNA was sequenced by bidirectional Sanger
sequencing using BigDye™ Terminator v1.1 Cycle Sequencing kit (cat.
no. 4337450; Applied Biosystems; Thermo Fisher Scientific, Inc.)
and a 3130xl Genetic Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The same primers as those listed in
Table SII were used to sequence
the amplified DNA.
In the case of Patient 4, the NextSeq Sequencing
System (Illumina, Inc.) at the Kazusa DNA Research Institute
(Kisarazu, Japan) was used to perform a mutation analysis based on
a DNA panel consisting of 193 genes (Table SIII). In the present study,
additional 25 genes were added to a previous DNA panel containing
168 genes to detect various inherited metabolic diseases, including
fatty acid oxidation, ketone body metabolism and transport and
glycogen storage diseases (15).
The customized probe was designed for target enrichment of the
panel genes by SureSelect DNA Advanced Design Wizard (v5.5;
https://earray.chem.agilent.com/suredesign/) and
obtained from Agilent technologies, Inc. (cat. no. 5190-4859;
Agilent technologies, Inc.). Because genomic DNA was subjected to
enzymatic fragmentation using a KAPA hyperplus library construction
kit (cat. no. KK8510; Roche Diagnostics) for Illumina short-read
next-generation sequencing, the insert DNA size of the resultant
library, but not the integrity of genomic DNA, was monitored on a
MultiNA microchip electrophoresis system (cat. no.
MCE®-202; Shimadzu Corporation). DNA sequencing was done
using a NextSeq 500/550 Mid Output kit v2.5 (75 bp paired end; 150
cycles; cat. no. 20024904; Illumina, Inc.) using a NextSeq 500
system (Illumina, Inc.). The loading concentration of the library
was 1.8 pM, which was quantified with a KAPA library quantification
kit (cat. no. KK4824; KAPA Biosystems; Roche Diagnostics). Variants
in protein-coding exonic regions and their 10-base flanking regions
were detected with the pipeline that was previously described
(15). Briefly, sequence reads were
aligned with the reference human genome (hg38; GCA_000001405.2)
using the Burrows-Wheeler Aligner ‘MEM’ v0.7.5a35 algorithm
(16). The duplicate reads were
removed using Picard-1.84's ‘MarkDuplicates’ command (http://broadinstitute.github.io/picard/). SNPs and
indels were called using VarScan2's v.2.3.3 (http://varscan.sourceforge.net) (17) and Genome Analysis Toolkit-3.6.0
(GATK) HaplotypeCaller and UnifiedGenotyper (18,19).
In silico analysis of identified novel rare
variants in the HMGCS2 gene was performed for all 4 cases
using MutationTaster (https://www.mutationtaster.org/) (20), Polymorphism Phenotyping version 2
(http://genetics.bwh.harvard.edu/pph2/), Protein
Variation Effect Analyzer (PROVEAN) and Sorting Intolerant from
Tolerant (SIFT) (11 Dec, 2019; https://provean.jcvi.org/index.php).
Construction of two HMGCS2 expression
vectors: One eukaryotic and one prokaryotic
The first step was to clone wild-type full-length
HMGCS2 cDNA (NCBI sequence no.: NM_005518.3), which contains
a mitochondrial targeting sequence, into the EcoRI site of
the eukaryotic expression vector pCAGGS (21), which was performed by DNA Synthesis
Services (GenScript Japan, Inc.). Next, the cDNA fragment encoding
HMGCS2 without the mitochondrial targeting sequence was
amplified by PCR from the eukaryotic expression vector using KOD FX
Neo (Toyobo Life Science) and primers containing BamHI and
EcoRI restriction sequences (italics), respectively:
Forward,
5'-CGCGGATCCCTGGAAGTTCTGTTCCAGGGTCCTACAGCCTCTGCTGTCC-3'; and
reverse, 5'-GAGGAGTGAATTCTTAAACGG-3' under the following
cycling conditions: Initial denaturation at 95˚C for 2 min,
followed by 45 cycles of 98˚C for 10 sec, 58˚C for 30 sec and 68˚C
for 45 sec. The underlined segment of the forward primer is the
recognition sequence of PreScission protease (GE Healthcare), which
was used in subsequent experiments. The amplified fragment was
digested by BamHI and EcoRI, then subcloned into the
bacterial expression vector pGEX-6P-1 (GE Healthcare), which
included the coding sequence of the fusion protein glutathione
s-transferase (GST). The synthesized constructs were transformed
into the E. coli strain JM109 (Toyobo Life Science).
Positive clones were confirmed by the Sanger method and agarose gel
electrophoresis to analyze the size of DNA fragments after
enzymatic digestion of plasmid DNA (data not shown). These plasmid
constructs were purified by an Automatic DNA Isolation system PI-50
(Kurabo Industries, Ltd.) and stored for subsequent experiments.
The simplified map of the bacterial expression vector is
illustrated in Fig. S1. A
pGEX-6P-1 empty vector was used for the expression of an isolated
GST protein. Each variant was introduced into the two HMGCS2
expression vectors (one eukaryotic and one prokaryotic) using a
KOD-Plus mutagenesis kit and KOD-Plus-Neo by following the
manufacturer's instructions (Toyobo Life Sciences, Ltd.).
Transient expression of HMGCS2 in
HEK293T cells and western blotting
293T cells (RIKEN Bioresource Center) were used for
expression experiments. Mycoplasma testing was done using an EZ-PCR
Mycoplasma test kit (Biological Industries) according to the
manufacturer's instructions. Each eukaryotic expression construct
(2 µg) containing wild-type or variant HMGCS2, or empty
vector was transfected into 3x105 293T cells using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.).
After 1 day, the transfected cells were lysed with 200 µl hypotonic
lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 0.5% Triton X-100, 10 mM
EDTA, pH 7.5). Protein in the soluble fraction was quantified by
the Bradford protein assay using Bio-Rad Protein Assay Dye Reagent
Concentrate (cat. no. 500-0006; Bio-Rad Laboratories, Inc.)
(22). Each protein sample (HMGC2,
20 µg; β-actin, 5 µg) was subjected to electrophoresis in a 10%
polyacrylamide gel containing sodium dodecyl sulfate (SDS-PAGE),
then transferred to 0.2-µm PVDF membrane using iBlot 2 Dry Blotting
System (Thermo Fisher Scientific, Inc.). The membranes were then
blocked in TBS supplemented with 0.1% Tween 20 and 5% BSA (cat. no.
015-21274; FUJIFILM Wako Pure Chemical Corporation) for 70 min at
room temperature. The membranes were probed with a rabbit
polyclonal antibody (1:795; cat. no. ab104807; Abcam) against a
synthetic peptide corresponding to a region within C-terminal amino
acids 444-493 of human HMGCS2 (NCBI reference sequence: NP_005509)
or a monoclonal anti-β-actin antibody produced in mouse (1:10,000;
cat. no. A5441; Sigma-Aldrich; Merck KGaA) for 1 h at room
temperature. This is followed by incubation with the secondary
antibodies for 1 h at room temperature and Amersham™ ECL™ Prime
Western Blotting Detection Reagent (GE Healthcare) for detection.
The secondary antibody used to detect HMGCS2 was horseradish
peroxidase (HRP)-conjugated anti-rabbit IgG H + L (cat. no. W4011;
Promega Corporation), whilst that used to detect β-actin was
Amersham ECL sheep anti-mouse IgG, HRP-linked whole Ab (cat. no.
NA931-1ML; Cytiva). Both antibodies had been diluted to 1:10,000
before use.
Expression in E. coli, purification
and western blotting of HMGCS2
Wild-type and mutant HMGCS2 expression vectors were
transformed into the E. coli strain BL21 (DE3; BioDynamics
Laboratory Inc.) grown in lysogeny broth (LB) medium (0.5% NaCl, 1%
tryptone and 0.5% yeast extract; Thermo Fisher Scientific, Inc.)
containing 0.1 mg/ml ampicillin to an A600 of 0.9-1.1 at 37˚C.
Optimal protein expression was induced with 0.3 mM
isopropyl-β-D-thiogalactopyranoside (FUJIFILM Wako Pure Chemical
Corporation) at 18˚C for 16 h. Cells were recovered by
centrifugation at 1,800 x g at 4˚C for 10 min, resuspended in lysis
buffer A [50 mM HEPES, 200 mM NaCl, pH 7.5, 10% glycerol, 1 mM
dithiothreitol (DTT), 1.1 mM 4-(2-Aminoethyl)-benzenesulfonyl
fluoride] and disrupted by sonication at 0˚C, with 10-sec
sonications repeated 25 times. The interval between each sonication
was 50 sec. After centrifugation at 15,000 x g at 4˚C for 30 min,
the soluble fraction was loaded into a column containing
Glutathione Sepharose 4 Fast Flow (GE Healthcare) before the column
was washed with lysis buffer B (50 mM HEPES, 200 mM NaCl, pH 7.5,
10% glycerol, 1 mM DTT) followed by wash buffer (50 mM HEPES, 200
mM NaCl, pH 7.5, 10% glycerol, 1 mM DTT, 0.5% [v/v] Triton X-100).
Triton X-100 was removed by washing the column again with lysis
buffer B. The GST fusion protein was eluted from the column using
elution buffer (50 mM Tris-HCl, 9.8 mM reduced glutathione, pH
8.0). After exchanging the buffer with cleavage buffer (50 mM
Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7.5, 1 mM DTT) by filtration
and dialysis, GST was separated from HMGCS2 by treatment with
PreScission protease (GE Healthcare) at 4˚C for 12 h. PreScission
protease and separated GST were removed from the mixture by running
it through the Glutathione Sepharose 4 Fast Flow column again.
Purified HMGCS2 was quantified by absorbance at 280 nm using a
NanoDrop™ 1,000 Spectrophotometer (Thermo Fisher Scientific, Inc.).
An isolated GST protein was created using the same procedure for
HMGCS2 proteins. To compare the purity of each enzyme, 600 ng each
partially purified wild-type or variant HMGCS2 was separated by 10%
SDS-PAGE transferred onto a nitrocellulose membrane. The membrane
was used for Ponceau staining and subsequent Western blotting. TBS
supplemented with 5% BSA was used as the blocking agent. The
nitrocellulose membrane was then blocked for 3 h at room
temperature. The membrane was probed with a rabbit polyclonal
antibody (cat. no. ab104807; Abcam) against a synthetic peptide
corresponding to a region within C-terminal amino acids 444-493 of
human HMGCS2 (NCBI reference sequence: NP_005509) for 17 h at room
temperature before being probed with the alkaline
phosphatase-conjugated anti-rabbit IgG (Fc) secondary antibody,
(1:7,500; cat. no. S3731; Promega Corporation) for 70 min at room
temperature.
In another experiment, 450 ng each sample was
separated by 10% SDS-PAGE before the proteins in the gel were
stained with Coomassie brilliant blue R-250 (cat. no. 031-17922;
FUJIFILM Wako Pure Chemical Corporation) for 20 min at room
temperature. These samples were not transferred onto a
membrane.
Enzymatic activity assay for purified
HMGCS2
The spectrophotometric method described by
Clinkenbeard et al (23) was
used with modifications. Each protein sample (16.4-43.2 µg) was
incubated in 900 µl enzyme assay buffer (100 mM Tris-HCl, 100 µM
EDTA, 0.2% v/v Triton X-100, pH 8.2) containing 300 µM acetyl-CoA
(Sigma-Aldrich; Merck KGaA) for 17 min at 30˚C to prevent
inactivation of HMGCS2 by succinylation (24,25).
Next, 35 nmol acetoacetyl-CoA (Sigma-Aldrich; Merck KGaA) were
added, where HMGCS2 activity was calculated from the rate of
decrease of acetoacetyl-CoA measured by spectrophotometry at 300
nm, at which the absorbance of the enolate form of acetoacetyl-CoA
was maximum (26). The molar
extinction coefficient of acetoacetyl-CoA is 3.6x103 in
this enzyme assay buffer (23).
Activity measurements were performed in three independent
experiments. Data are expressed as the mean ± SD.
Statistical analysis
One-way analysis of variance (ANOVA) with Dunnett's
multiple comparison test was performed using Prism 8 software
(GraphPad Software, Inc.) to compare enzyme activities. P<0.05
was considered to indicate a statistically significant
difference.
Results
Mutational analysis
Six rare, non-synonymous variants of HMGCS2
were detected in the four study cases, comprising compound
heterozygotes of two variants (Fig.
S2; Table I). Two of the
variants, c.704T>C (p.M235T) and c.1175C>T (p.S392L), were
identified in two unrelated patients. Five of the variants are
novel mutations: c.656G>A (p.G219E), c.704T>C (p.M235T),
c.758T>C (p.V253A), c.1175C>T (p.S392L), and c.1498C>T
(p.R500C). Only the c.1499G>A (p.R500H) variant had been
reported previously as a pathogenic mutation (27,28).
In the mutation analysis of patient 4, the presence of any rare
variants in other genes known to be related to abnormal blood
glucose levels was not observed (Table
SIII). All five of the novel HMGCS2 variants were
evaluated in silico by MutationTaster, PolyPhen-2, PROVEAN
and SIFT to be disease-causing, probably damaging, deleterious and
damaging, respectively.
Transient expression and
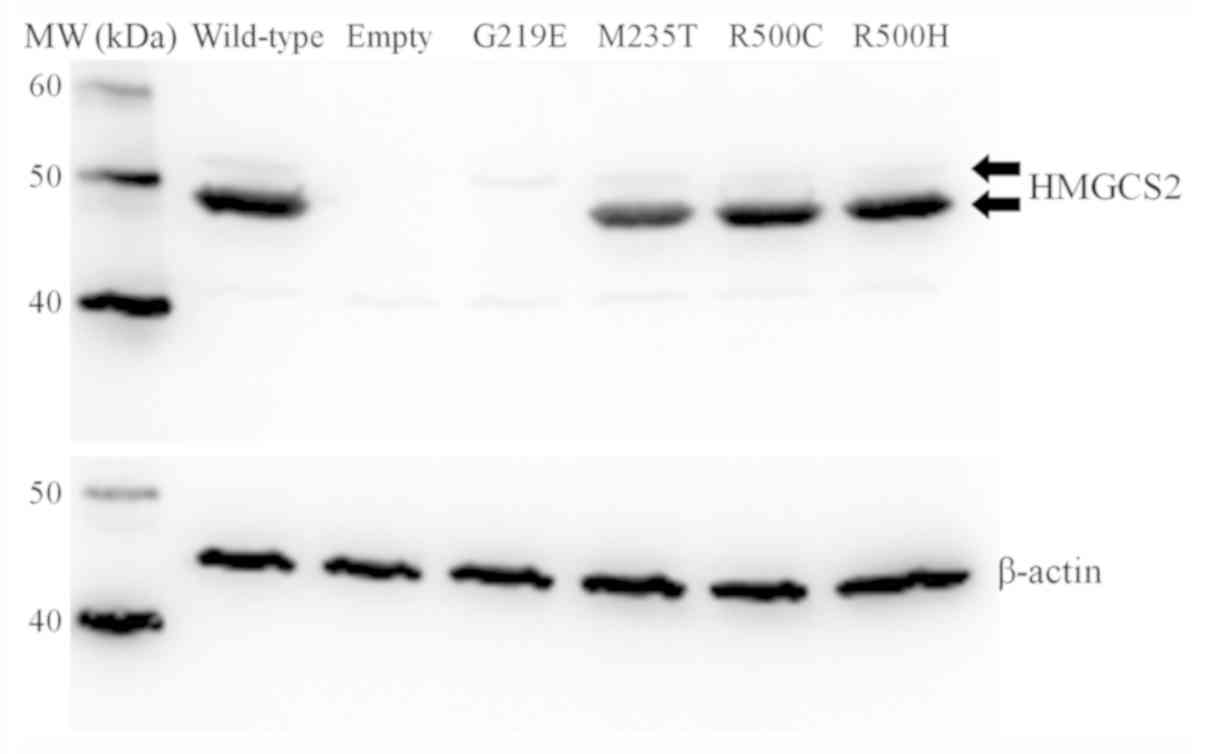
immunoblotting of HMGCS2 in 293T cells
Full-length HMGCS2 cDNA contains a
mitochondrial targeting sequence. HMGCS2 is expressed in cytosol as
an immature protein with mitochondrial targeting peptide, which is
cleaved in the mitochondrial matrix (5,29).
Western blot analysis confirmed that immature HMGCS2 protein with
mitochondrial targeting peptide was expressed in all transfected
cells except for the empty vector-transfected cells, and each
expression level was similar (upper-band of the suitable molecular
weight of immature HMGCS2 protein with mitochondrial targeting
peptide). This result indicated by the western blot analysis
suggested that each transfection was successful (Fig. 1). An antibody against β-actin was
used as a protein loading control, confirming that the amount of
proteins in applied samples were similar (Fig. 1). The levels of mutant M235T, R500C
and R500H proteins without mitochondrial targeting peptide were
clearly detectable (lower-band). However, mutant G219E protein
without mitochondrial targeting peptide was not detectable, even
though immature protein with mitochondrial targeting peptide was
detected, as were wild-type and other mutants.
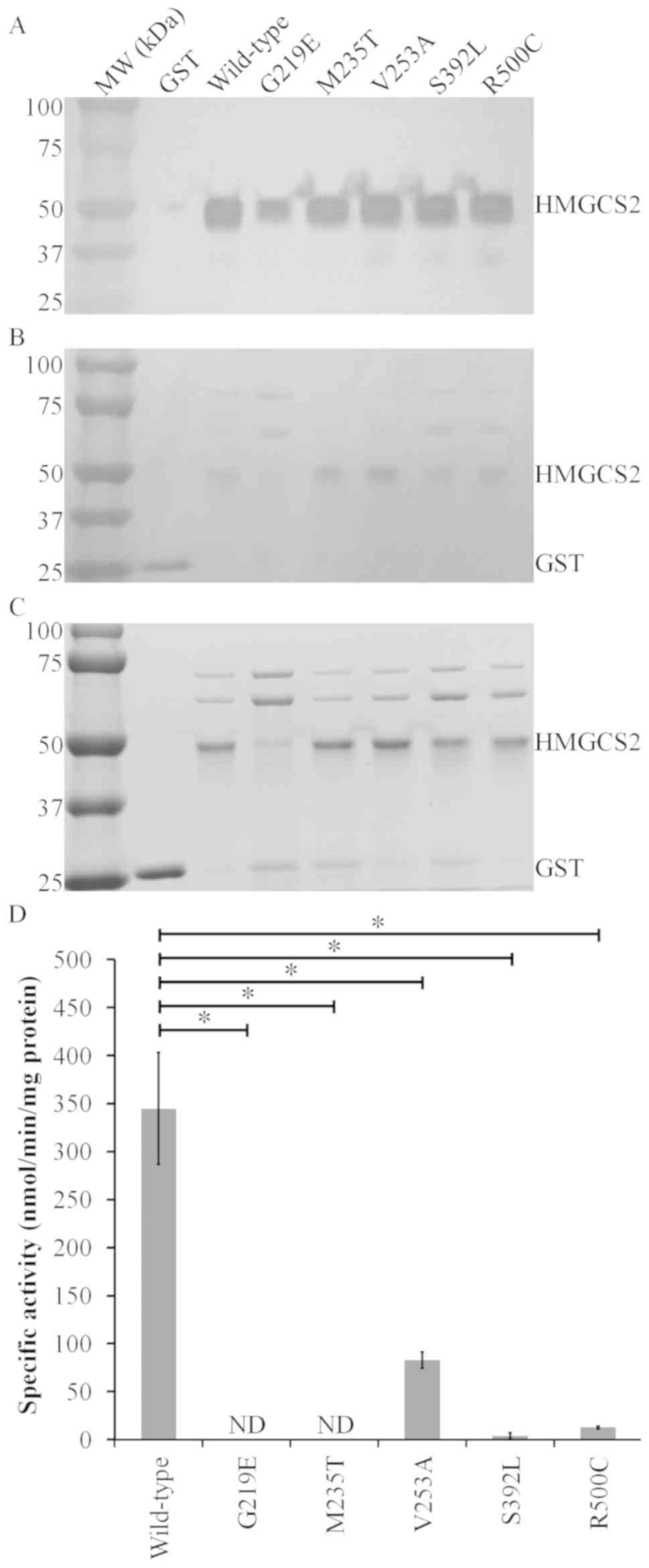
Expression and purification of HMGCS2
from E. coli
A wild-type HMGCS2-GST fusion protein that was
expressed in E. coli was successfully constructed. Wild-type
HMGCS2 protein was then purified using Glutathione Sepharose column
chromatography, PreScission protease cleavage and a second
Glutathione Sepharose column (Fig.
S3). Although extra protein bands remained in the purified
wild-type HMGCS2 preparation, a further purification step decreased
its enzyme activity, this partially purified preparation was used
in the enzymatic activity assay. From 1 g of cultured E.
coli 147 µg of wild-type HMGCS2 was purified.
The variant HMGCS2 proteins were expressed and
purified using the same procedure. The amounts of purified proteins
(µg) obtained from 1 g of E. coli expressing the HMGCS2
variants G219E, M235T, V253A, S392L and R500C were 42.2, 258,
147.8, 66.5 and 99.6, respectively.
Fig. 2A-C shows
western blotting results, ponceau staining and coomassie brilliant
blue R-250 staining of each partially purified HMGCS2. The G219E
variant had a weaker signal compared with the wild-type and other
HMGCS2 variants, indicating that the purity of the G219E
preparation was lower than the others (Fig. 2A). The enzymatic activity of the
wild-type, V253A, R500C and S392L HMGCS2 were 345±58.2, 83±8.45,
12.7±1.30, and 3.6±3.43 nmol/min/mg protein, respectively, and
these activities of the variants were significantly lower than that
of wild-type. Only the V253A variant had some residual activity.
The other four variants had either no detectable activity or
negligible enzymatic activity. (Fig.
2D). Based on these expression analyses, it was concluded that
all five variants were pathogenic.
Discussion
In the present report, four cases of HMGCS2
deficiency have been described in Japanese patients. All four
patients presented with severe hypoketotic hypoglycemia with severe
metabolic acidosis, hepatomegaly or fatty liver with elevated liver
enzymes, elevated C2 and C2/C0 ratio in acylcarnitine analysis and
an elevated FFA level during an acute episode of metabolic
decompensation (Table I). Although
the TKBs measurement during the acute episode in Patient 4 was not
available, the other three patients had high FFAs/TKBs ratios.
Genetic analyses revealed that these patients were compound
heterozygotes of two variants. Five out of the six identified
variants were novel variants (Table
I). Initially a transient expression analysis of wild-type and
variant HMGCS2 was conducted in human fibroblasts, which was
established and SV40-transformed in the laboratory from a patient
with beta-ketothiolase deficiency and used in previous report
(30). In this earlier study it was
difficult to detect wild-type HMGCS enzyme activity. Using a
bacterial expression system that was previously used for
characterization of HMGCS2 mutants by other groups (28,31) it
was possible to confirm that these novel variants are
disease-causing missense mutations. The p.G219E mutant enzyme
appeared to be less stable than the wild-type enzyme, which was
also confirmed by the transient expression experiment in HEK293T
cells.
Clinically it is still challenging to recognize
HMGCS2 deficiency (10). A
non-ketotic hypoglycemic episode with a high FFAs/TKBs ratio is the
most important feature of HMGCS2 deficiency as well as
defects in fatty acid oxidation (6). Each fatty acid oxidation defect has a
characteristic profile in blood acylcarnitine analysis (32). In cases of HMGCS2 deficiency,
some reports have described that elevated C2 and C2/C0 ratios were
observed during acute episodes (11,12,33).
In the present study high C2 and relatively low C0 was also
observed in the patients. In HMGCS2 deficiency, the
β-oxidation pathway is intact and produces plenty of acetyl-CoA in
times of ketogenic stress, but acetyl-CoA cannot be used for
ketogenesis. Hence, it is reasonable that acetyl-CoA is accumulated
and produces acetylcarnitine (C2) in hepatocytes. However, in
patients with fatty acid oxidation defects, acetyl-CoA production
via β-oxidation is impaired. It may be hypothesized that
hypoketotic hypoglycemia with elevated C2 and C2/C0 ratios during
an acute crisis may be a promising indicator to identify
HMGCS2 deficiency. Urinary organic acid analysis shows
non-ketotic dicarboxylic aciduria in both HMGCS2 deficiency
and fatty acid oxidation defects. Recently, Pitt et al
(10) reported the presence of
characteristic urinary organic acids such as 4HMP during acute
episodes in patients with HMGCS2 deficiency.
Retrospectively, 4HMP was also detected in the urine samples of the
patients in the present study. It should be stressed that such
findings are only detected during acute episodes in both
acylcarnitine and urinary organic acid analyses (10).
Enzymatic assay of HMGCS2 activity is reported to be
challenging (5) and is currently
measured by the reduction of acetoacetyl-CoA spectrophotometrically
(13,28,31,34).
However, another cytosolic form of HMGCS and several other enzymes
may influence the assay because these enzymes utilize
acetoacetyl-CoA (34). The latter
might include mitochondrial acetoacetyl-CoA thiolase, mitochondrial
3-ketoacyl-CoA thiolase, cytosolic acetoacetyl-CoA thiolase, and
acyl-CoA hydrolase. Lascelles and Quant (34) measured HMGCS2 activity in human
liver samples. Whole lysates of transfected SV40-transformed
fibroblasts, which were derived from a patient with mitochondrial
acetoacetyl-CoA thiolase (T2) deficiency (30) and chosen because T2 is one of the
major enzymes that catalyzes acetoacetyl-CoA and thus could affect
the HMGCS2 enzyme assay, failed to show HMGCS2 enzymatic activity.
The expression level of HMGCS2 protein might have been too low to
overcome the other intrinsic enzymes that use acetoacetyl-CoA as a
substrate.
When the research strategy was changed to assay the
activity of HMGCS2 purified from a bacterial expression system
influences from other enzymes no longer needed to be considered.
Previously two reports described the successful characterization of
HMGCS mutants using bacterial expression systems (28,31).
In the bacterial expression system in the present study, enzyme
assays were performed without adding Mg2+.
Mg2+ can increase the absorbance of acetoacetyl-CoA
(26), but it inhibits HMGCS2 in a
concentration-dependent manner; for example, 10 mM Mg2+
in the assay buffer inhibits the reaction by 50% (35). The concentration of free
Mg2+ in the matrix of liver mitochondria is estimated to
range from 0.8 to 1.2 mM (36), so
the in vivo inhibition effect of Mg2+ would seem
to be quite small. While adopting this physiological concentration
of Mg2+ would have been ideal, a reliable molar
extinction coefficient of acetoacetyl-CoA at this physiological
condition was not found in the literature. Thus, in the present
study no Mg2+ was added to the enzyme assay buffer, even
though a previous report adopted 5-10 mM Mg2+ (34).
Enzymatic activity of the V253A variant was
approximately one quarter that of the wild-type enzyme, which was
much higher than the other four variants. The V253A variant was
found in patient 4. Patient 4 C2/C0 and C2 (acetylcarnitine) levels
were the lowest of all four patients, which is compatible with the
fact that this variant showed some residual activity in the
experiments. It may be hypothesized that a small amount of
acetyl-CoA in hepatocytes was transformed into ketone bodies in
Patient 4 by the V253A variant. The data suggest that the V253A
variant may be a disease-causing mutation. It shows lower enzymatic
activity than the R505Q variant, which was reported to be a
disease-causing mutation (31).
The enzyme kinetics of HMGCS2 were also challenging.
The V253A variant showed some residual activity, which led to
further analysis. Kinetic analysis of the V253A variant and
wild-type protein was performed. However, Km values for
acetoacetyl-CoA were too low to be evaluated accurately (data not
shown). An attempt to identify the Km value for
acetyl-CoA was also made. The Km value for acetyl-CoA in
this experiment was higher compared with that described previously
(data not shown) (28). This could
be because large amounts of acetyl-CoA were consumed during
preincubation. Enzyme kinetics of the variant should be further
researched to elucidate its pathophysiology in more detail.
The three-dimensional structure of human HMGCS2
(Protein Databank ID: 2WYA) is shown in Fig. 3A. In this structure, two subunits
form a homodimer, which is crucial for its enzymatic activity
(31). The residues Glu132, Cys166
and His301 catalyze the reaction directly (37). The residues Ser392 and Leu374, each
a part of an α-helix, are connected by the only hydrogen bond
between these two alpha-helices (Fig.
3B). Replacing Ser392 with a Leu residue (S392L) would remove
this hydrogen bond and may alter the tertiary structure of HMGCS2.
Arg500 is located on the neighbor region of a beta-turn, and is
also located on the dimerization surface. Because the side chain of
Arg500 forms two hydrogen bonds with Asp496 (Fig. 3C), this beta-turn may become
unstable when these hydrogen bonds are lost due to the replacement
of Arg500 with Cys (variant R500C), which also could affect
dimerization. Met235 is located on the dimerization surface
(Fig. 3D). Because replacement of
this amino acid with Thr (M235T) would change the hydrophilicity of
this surface, it may affect dimerization. Val253 is one of the
residues that compose the active site of this enzyme (Fig. 3E), but is not directly involved in
the chemical reaction. Replacing this amino acid with Ala (V253A)
could alter the interaction of residues that compose the active
site, and would make it difficult to stabilize the substrates in an
appropriate position. In summary, the secondary structure may be
affected by R500C, the tertiary structure may be affected by S392L,
the quaternary structure may be affected by M235T and R500C and the
V253A variant would be expected to affect the binding to
substrates.
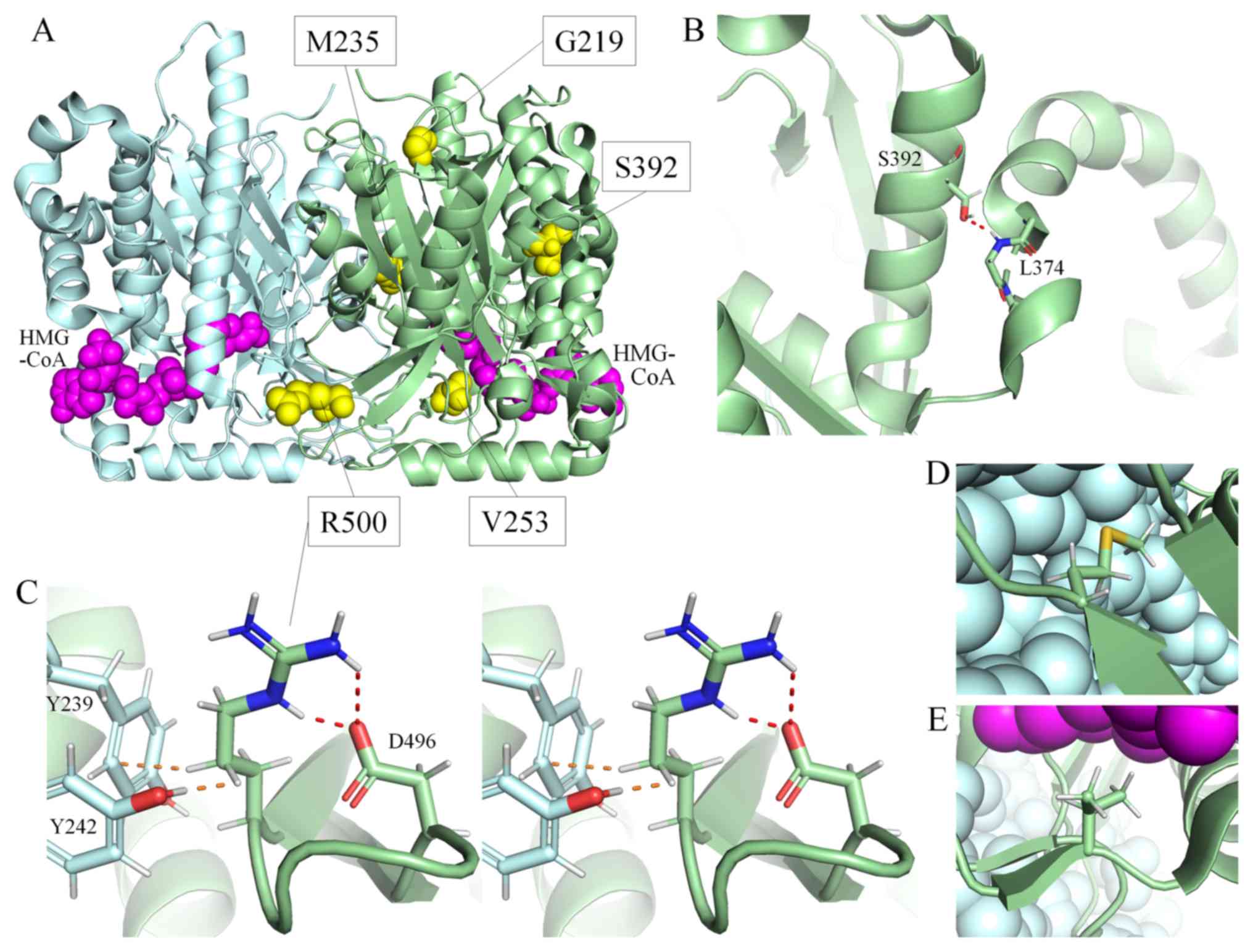 | Figure 3Three-dimensional structure of
HMGCS2, obtained using PyMOL Molecular Graphics System version
2.2.0. (A) The whole structure of wild-type HMGCS2, shown as a
homodimer. Each subunit is indicated by a green or cyan ribbon
diagram. The residues substituted in patients are shown as yellow
spheres. HMG-CoA is indicated by magenta spheres. (B) A partial
structure of wild-type HMGCS2. Ser392 and the main chain of Leu374
are shown as a stick model colored by atom type (cyan or green, C;
red, O; blue, N; white, H; and gold, S). The hydrogen bond between
Ser392 and Leu374 is shown as a dotted red line. (C) A partial
structure of wild-type HMGCS2 in cross-eye stereo view. The side
chains of Asp496 and Arg500, and those of Tyr239 and Tyr242 in
another subunit, are shown as a stick model. Hydrogen bonds are
shown as dotted red lines. The side chain of Arg500 forms two
hydrogen bonds with Asp496. Arg500 is also exposed to Tyr239 and
Tyr242 in another subunit. The shortest interatomic distances to
Arg500 from Tyr242 and Tyr239 (shown as dotted orange lines) are
170 and 280 picometers, respectively. (D) The side chain of Met235
shown as a stick model. Cyan spheres indicate another subunit. (E)
The side chain of Val253 shown as a stick model. HMG-CoA is
indicated by magenta spheres. Cyan spheres indicate another
subunit. HMGCS2, 3-hydroxy-3-methylglutaryl-CoA. |
In conclusion, in vitro analysis has shown
that the p.G219E, p.M235T, p.V253A, p.S392L and p.R500C variants of
HMGCS2 are disease-causing mutations. In acylcarnitine
analysis, C2 and C2/C0 in the acute phase may be promising
indicators to differentiate HMGCS2 deficiency from fatty
acid oxidation defects.
Supplementary Material
Simplified map of the bacterial
expression vector. IPTG, isopropyl β-D-thiogalactoside.
Purification of wild-type HMGCS2
protein from Escherichia coli lysates. Sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and Coomassie brilliant
blue R250 staining of protein fractions obtained by affinity
chromatography of wild-type HMGCS2 expressed in E. coli.
Lane 1, whole bacterial cell extract before induction with IPTG;
lane 2, whole bacterial cell extract after IPTG induction for 16 h;
lane 3, cell pellet after sonication; lane 4, supernatant after
sonication; lane 5, soluble fraction passed through a glutathione
sepharose bead column; lane 6, fraction eluted from the affinity
column; lane 7, solution after cleavage of the GST-HMGCS2 fusion
protein with PreScission protease; lane 8, solution after removal
of GST and PreScission protease. HMGCS2,
3.hydroxy-3-methylglutaryl-CoA; IPTG, isopropyl
β-D-thiogalactoside; GST, glutathione s-transferase.
DNA sequencing data of the four
patients using the Sanger method.
The details of blood acylcarnitine
analyses.
Sequences of the primers used for PCR
amplification of individual exons of the human HMGCS2
gene.
DNA panel of 193 metabolic disease
genes used for mutation analysis in patient 4. The top row refers
to classifications of genes in the panel.
Acknowledgements
The authors would like to express gratitude to Ms
Naomi Sakaguchi and Ms Sachie Hori, the Department of Pediatrics,
Gifu University (Gifu, Japan) assisted in these experiments as
laboratory assistants. Dr. Yuki Hasegawa of the Department of
Pediatrics, Shimane University Faculty of Medicine (Izumo, Japan)
confirmed the mas spectra of 4-HMP in all urine samples.
Funding
This research was supported in part by a
Grant-in-Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology of Japan (grant
no. 16K09962) and by grants from Japan Agency for Medical Research
and Development (grant no. JP17ek0109276), Health and Labor
Sciences Research Grants [H29-nanchitou(nan)-ippan-051] for
Research on Rare and Intractable diseases and The Morinaga
Foundation for Health & Nutrition.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Figshare repository (https://doi.org/10.6084/m9.figshare.10308536.v1). All
datasets are additionally available from the corresponding author
on reasonable request.
Authors' contributions
KA, KF, YW, YN and TI were involved in clinical
management of the patients. MN, YA, HS, HM, RF and OO were involved
in mutation analysis. YA, HO, EA and HO were involved in expression
analyses. YA wrote the first draft of the manuscript. TF initiated
and supervised the study and reviewed and revised the manuscript.
All authors approved the final manuscript for submission to this
journal and agree to be accountable for all aspects of the
work.
Ethics approval and consent to
participate
This study was approved by the Ethical Committee of
the Graduate School of Medicine, Gifu University, Japan and was
carried out in accordance with the principles contained within the
Declaration of Helsinki. Informed consent was obtained from the
parents of all patients.
Patient consent for publication
All parents of the patients agreed to the
publication of these data without identifying information by
signing informed consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mitchell GA and Fukao T: Inborn errors of
ketone body metabolism. In: The metabolic & molecular basis of
inherited disease. Scriver CR, Beaudet AL, Sly WS and Valle D
(eds.). McGraw-Hill, New York, pp2327-2356, 2001.
|
|
2
|
Sass JO: Inborn errors of ketogenesis and
ketone body utilization. J Interit Metab Dis. 35:23–28.
2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hegardt FG: Mitochondrial
3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in
ketogenesis. Biochem J. 338:569–582. 1999.PubMed/NCBI
|
|
4
|
Williamson DH, Bates MW and Krebs HA:
Activity and intracellular distribution of enzymes of ketone-body
metabolism in rat liver. Biochem J. 108:353–361. 1968.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Boukaftane Y and Mitchell GA: Cloning and
characterization of the human mitochondrial
3-hydroxy-3-methylglutaryl CoA synthase gene. Gene. 195:121–126.
1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fukao T, Mitchell G, Sass JO, Hori T, Orii
K and Aoyama Y: Ketone body metabolism and its defects. J Inherit
Metab Dis. 37:541–551. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wolf NI, Rahman S, Clayton PT and Zschocke
J: Mitochondrial HMG-CoA synthase deficiency: Identification of two
further patients carrying two novel mutations. Eur J Pediatr.
162:279–280. 2003.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Carpenter KH, Bhattacharya K, Ellaway C,
Zschocke J and Pitt JJ: Improved sensitivity for HMG CoA synthase
detection using key markers on organic acid screen. J Inherit Metab
Dis. 33(S62)2010.
|
|
9
|
Sass JO, Kuhlwein E, Klauwer D, Rohrbach M
and Baumgartner MR: Hemodiafiltration in mitochondrial
3-hydroxy-3-methylglutaryl coenzyme A synthase (HMG-CoA synthase)
deficiency. J Inherit Metab Dis. 36 (Suppl 2)(S189)2013.
|
|
10
|
Pitt JJ, Peters H, Boneh A, Yaplito-Lee J,
Wieser S, Hinderhofer K, Johnson D and Zschocke J: Mitochondrial
3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Urinary organic
acid profiles and expanded spectrum of mutations. J Inherit Metab
Dis. 38:459–466. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Conboy E, Vairo F, Schultz M, Agre K,
Ridsdale R, Deyle D, Oglesbee D, Gavrilov D, Klee EW and Lanpher B:
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency:
Unique presenting laboratory values and a review of biochemical and
clinical features. JIMD Rep. 40:63–69. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lee T, Takami Y, Yamada K, Kobayashi H,
Hasegawa Y, Sasai H, Otsuka H, Takeshima Y and Fukao T: A Japanese
case of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase
deficiency who presented with severe metabolic acidosis and fatty
liver without hypoglycemia. JIMD Rep. 48:19–25. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Morris AA, Lascelles CV, Olpin SE, Lake
BD, Leonard JV and Quant PA: Hepatic mitochondrial
3-hydroxy-3-methylglutaryl-coenzyme a synthase deficiency. Pediatr
Res. 44:392–396. 1998.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kimura M, Yamamoto T and Yamaguchi S:
Automated metabolic profiling and interpretation of GC/MS data for
organic acidemia screening: A personal computer-based system.
Tohoku J Exp Med. 188:317–334. 1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fujiki R, Ikeda M, Yoshida A, Akiko M, Yao
Y, Nishimura M, Matsushita K, Ichikawa T, Tanaka T, Morisaki H, et
al: Assessing the accuracy of variant detection in cost-effective
gene panel testing by next-generation sequencing. J Mol Diagn.
20:572–582. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–160. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Song XQ, Fukao T, Yamaguchi S, Miyazawa S,
Hashimoto T and Orii T: Molecular cloning and nucleotide sequence
of complementary DNA for human hepatic cytosolic
acetoacetyl-coenzyme A thiolase. Biochem Biophys Res Commun.
201:478–485. 1994.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Clinkenbeard KD, Reed WD, Mooney RA and
Lane MD: Intracellular localization of the
3-hydroxy-3-methylglutaryl coenzme A cycle enzymes in liver.
Separate cytoplasmic and mitochondrial 3-hydroxy-3-methylglutaryl
coenzyme A generating systems for cholesterogenesis and
ketogenesis. J Biol Chem. 250:3108–3116. 1975.PubMed/NCBI
|
|
24
|
Lowe DM and Tubbs PK:
3-Hydroxy-3-methylglutaryl-coenzyme A synthase from ox liver
Properties of its acetyl derivative. Biochem J. 227:601–607.
1985.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Rardin MJ, He W, Nishida Y, Newman JC,
Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, et al: SIRT5
regulates the mitochondrial lysine succinylome and metabolic
networks. Cell Metab. 18:920–933. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Decker K: Acetoacetyl Coenzyme A. In:
Methods of enzymatic analysis. 3rd edition, Vol. 7, Metabolites 2:
Tri- and dicarboxylic acids, purines, pyrimidines and derivatives,
coenzymes, inorganic compounds. Bergmeyer HU, Bergmeyer J and Graßl
M (eds.). Wiley-Blackwell, Hoboken, pp201-206, 1985.
|
|
27
|
Aledo R, Zschocke J, Pié J, Mir C, Fiesel
S, Mayatepek E, Hoffmann GF, Casals N and Hegardt FG: Genetic basis
of mitochondrial HMG-CoA synthase deficiency. Hum Genet. 109:19–23.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ramos M, Menao S, Arnedo M, Puisac B,
Gil-Rodríguez MC, Teresa-Rodrigo ME, Hernández-Marcos M, Pierre G,
Ramaswami U, Baquero-Montoya C, et al: New case of mitochondrial
HMG-CoA synthase deficiency Functional analysis of eight mutations.
Eur J Med Genet. 56:411–415. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Neupert W: Protein import into
mitochondria. Annu Rev Biochem. 66:863–917. 1997.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Fukao T, Yamaguchi S, Scriver CR, Dunbar
G, Wakazono A, Kano M, Orii T and Hashimoto T: Molecular studies of
mitochondrial acetoacetyl-coenzyme A thiolase deficiency in the two
original families. Hum Mutat. 2:214–220. 1993.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Puisac B, Marcos-Alcalde I,
Hernandez-Marcos M, Tobajas Morlana P, Levtova A, Schwahn BC,
DeLaet C, Lace B, Gómez-Puertas P and Pié J: Human mitochondrial
HMG-CoA synthase deficiency: Role of enzyme dimerization surface
and characterization of three new patients. Int J Mol Sci.
19(1010)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kompare M and Rizzo WB: Mitochondrial
fatty-acid oxidation disorders. Semin Pediatr Neurol. 15:140–149.
2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Aledo R, Mir C, Dalton RN, Turner C, Pié
J, Hegardt FG, Casals N and Champion MP: Refining the diagnosis of
mitochondrial HMG-CoA synthase deficiency. J Inherit Metab Dis.
29:207–211. 2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lascelles CV and Quant PA: Investigation
of human hepatic mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme
A synthase in postmortem or biopsy tissue. Clin Chim Acta.
260:85–96. 1997.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Reed WD, Clinkenbeard D and Lane MD:
Molecular and catalytic properties of mitochondrial (ketogenic)
3-hydroxy-3-methylglutaryl coenzyme A synthase of liver. J Biol
Chem. 250:3117–3123. 1975.PubMed/NCBI
|
|
36
|
Romani AM: Cellular magnesium homeostasis.
Arch Biochem Biophys. 512:1–23. 2011.PubMed/NCBI
|
|
37
|
Shafqat N, Turnbull A, Zschocke J,
Oppermann U and Yue WW: Crystal structures of human HMG-CoA
synthase isoforms provide insights into inherited ketogenesis
disorders and inhibitor design. J Mol Biol. 398:497–506.
2010.PubMed/NCBI View Article : Google Scholar
|