Introduction
Coronary heart disease (CHD) is a significant
challenge to human health, and the morbidity and mortality rates
are rising rapidly in the United States (1). Currently, treatment for CHD is early
reperfusion therapy to open occluded vessels and to prevent
ischemia and myocardial infarction (2). However, myocardial
ischemia-reperfusion (I/R) injury (MIRI) may result in additional
cardiomyocyte dysfunction, which further aggravates myocardial cell
damage (3), and constitutes a
significant part of the pathology, contributing to heart failure in
patients with CHD (4). The
mechanisms underlying MIRI are complex and are associated with a
variety of pathologies, such as oxidative stress, calcium overload,
inflammatory response and energy metabolism disorders (4-6). Among these
contributors, a number of studies have demonstrated that oxidative
stress induced by a rapid increase in the levels of reactive oxygen
species (ROS), which can be observed during reperfusion, is a key
initiator of MIRI (6,7).
ROS are metabolites that are produced primarily by
mitochondria (8). Under
physiological conditions, ROS serve as second messenger signaling
molecules, which modulate the intracellular signaling cascade and
maintain the normal physiological functions of the cells (9). However, excessive ROS generation
during MIRI results in the activation of nuclear factor erythroid
2-related factor 2-associated factors, such as forkhead box O3a and
NF-κB, thereby resulting in an imbalance between oxidation and
anti-oxidation in the cells, leading to oxidative stress and
ultimately, apoptosis, necrosis or autophagy (10).
Autophagy is an adaptive response of cells to
metabolic stresses and environmental alterations. Via lysosome
degradation, abnormal proteins, such as misfolded proteins and
damaged organelles, are degraded and recycled to maintain the
homeostasis of the intracellular environment (11). Autophagy involves four steps:
Induction, formation of autophagosomes, autophagosome and lysosome
fusion and degradation of the enclosed substances (11,12).
Under normal conditions, the levels of autophagy in cardiomyocytes
is low (11). Scherz-Shouval et
al (13) revealed that
increased ROS levels during cellular I/R injury resulted in the
upregulation of autophagy, which promoted the degradation of
damaged organelles and proteins, thereby reducing the damage caused
by oxidative stress to the cells. However, the overactivation of
autophagy during reperfusion may destroy important cellular
components, resulting in abnormal alterations to the cell structure
and the promotion of cell death (11,14). A
number of studies have indicated that autophagy serves an important
role in MIRI (15,16).
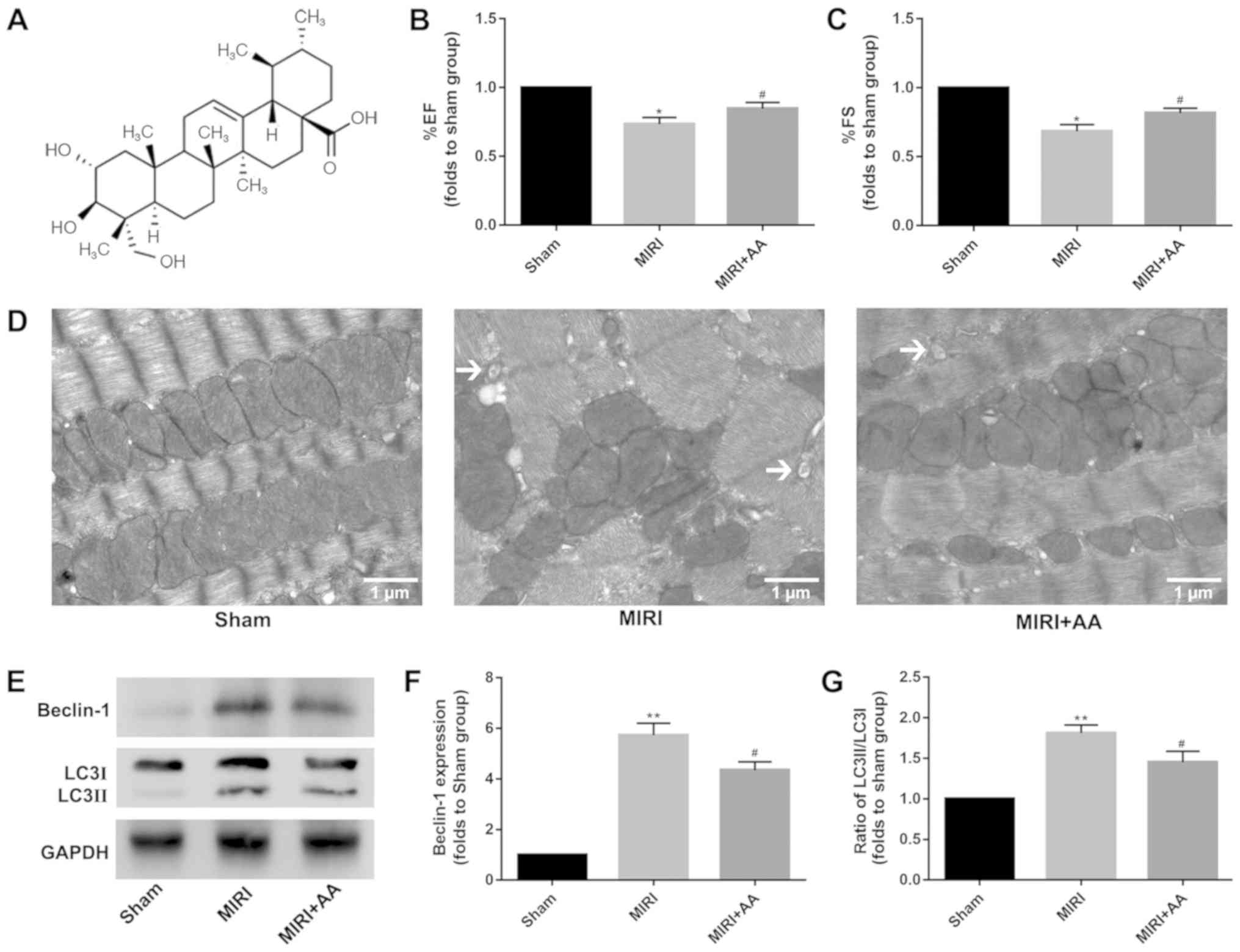
Asiatic acid (AA;
C30H48O5; Fig. 1A), which is chemically known as
2,3,23-trihydroxyurs-12-ene-28-oic-acid, is a pentacyclic
triterpenoid compound extracted from the traditional Chinese
medicinal herb Centella Asiatica (17). Previously, numerous studies have
demonstrated that AA possesses various pharmacological properties,
including hepatoprotective, neuroprotective, antioxidant,
anti-inflammatory, anti-hyperglycemic and anticancer properties
(18-22). In
previous studies, AA was indicated to serve an important role in
myocardial protection (23,24). However, to the best of our
knowledge, the association of AA with autophagy in MIRI has not
been fully investigated. In the present study, in vivo and
in vitro experiments were used to examine the effects of AA
on autophagy during MIRI, with the aim of demonstrating the
potential pharmacological properties of AA to support its use in
the treatment of MIRI.
Materials and methods
Materials
Purified natural extracts of AA (97%) and DMSO were
purchased from Merck KGaA. Gibco DMEM and FBS were purchased from
Thermo Fisher Scientific, Inc. Cell Counting Kit-8 (CCK-8) was
obtained from Dojindo Molecular Technologies, Inc. Total superoxide
dismutase (SOD) and lipid peroxidation malondialdehyde (MDA) assay
kits were purchased from Beyotime Institute of Biotechnology. All
antibodies were purchased from Cell Signaling Technology, Inc. An
ECL detection substrate was purchased from Thermo Fisher
Scientific, Inc. Unless otherwise indicated, all other chemicals
and materials were purchased from Merck KGaA.
Animals and establishment of the mouse
MIRI model
A total of 30 Male C57BL/6 mice [purchased from the
Experimental Animal Center of Jiangsu Province (Nanjing, China)],
weighing 20-25 g and aged 4-6 weeks, were given free access to food
and water and were kept under 12:12 h light-dark cycles at room
temperature with 40-60% humidity for 1 week for adaptation, after
which time all mice were randomized and divided into three groups
(n=10 per group) as follows (all treatments were performed once per
day for 7 days): Sham operation group, pretreated with PBS for 7
days before the operation; MIRI group, pretreated with PBS for 7
days before the operation; and MIRI + AA group, pretreated with AA
(100 mg/kg/day via oral gavage) for 7 days before the
operation.
C57BL/6 mice were anesthetized with an
intraperitoneal injection of 1% pentobarbital sodium (30 mg/kg) and
fixed in the supine position (25).
An oral tracheal tube was inserted and connected to an animal
ventilator, and the limb-lead electrocardiogram was recorded. The
ventilator was assessed to be functioning properly to ensure
accurate thoracic surgery could be performed. A lateral incision
was performed along the left sternal margin between the third and
fourth intercostal muscles to open the skin, intercostal muscles,
pleura and to expose the heart. The left anterior descending (LAD)
artery was ligated below the junction of the left atrial appendage
with a 7-0 prolene thread. ST-segment elevation in the
electrocardiogram indicated successful establishment of the model.
In the MIRI and MIRI + AA groups, the ligature was removed 30 min
after infarction, followed by reperfusion for 24 h. In the sham
operation group, the LAD artery was threaded but not ligated, and
the remaining operations followed the same procedures as the
aforementioned groups.
All aspects of animal care and the experimental
protocols to which the animals were subjected were approved by the
Animal Care and Use Committee of Nanjing Medical University and
performed in accordance with the Guide for the Care and Use of
Laboratory Animals, which was published by the United States
National Institutes of Health (26). All efforts were made to minimize
animal suffering.
Echocardiography
Cardiac function was determined by performing
echocardiography on days 0 and 1 after MIRI, using a
Vevo® 2100 echocardiograph equipped with a 30-MHz
high-resolution phase array transducer (VisualSonics, Inc.). M-mode
images were used to obtain the left ventricular parameters [left
ventricular end-diastolic diameter (LVEDD), left ventricular
end-systolic diameter (LVESD)] that were recorded from
two-dimensional images using M-mode interrogation in the short-axis
view.
Autophagy measurement
Following cardiac function determination, the
animals were sacrificed with intraperitoneal administration of an
overdose of pentobarbital sodium (150 mg/kg), death was confirmed
by absence of vital signs. The left ventricular cardiac tissue was
obtained immediately and fixed with 2.5% glutaraldehyde and 1%
citric acid at 4˚C overnight. After dehydration with ethanol (50,
70, 80, 90, 95 and 100%) and acetone (100%) in gradient, the
tissues were embedded in epoxy resin. Polymerization was performed
at 80˚C for 24 h. Ultrathin sections (50 µm) were prepared using a
ultramicrotome and double-stained with 2% uranium acetate and 10%
lead citrate for 1 h at room temperature. Ultrastructural
alterations and autophagy in cardiomyocytes were observed under a
transmission electron microscope (TEM; cat. no. H-7650; Hitachi,
Ltd., magnification, x3,000) at an acceleration voltage of 80 kV.
Electron microscopy images were analyzed with ImageJ software v1.46
(National Institutes of Health).
Cell culture and treatment
H9c2 cells were purchased from The Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences and cultured
as previously described (27).
Briefly, H9c2 cells were cultured in high-glucose DMEM supplemented
with 10% FBS and 1% penicillin/streptomycin (v/v), in an incubator
with 5% CO2 at 37˚C for 48 h.
H9c2 cells in logarithmic growth period were plated
in a 60 mm culture dish (1x106 cells/dish), cultured in
an incubator with 5% CO2 at 37˚C for 24 h, and the cell
cycle was synchronized using serum-free DMEM for 24 h prior to cell
modeling. An oxygen glucose deprivation/reperfusion (OGD) model was
established as described previously (28). Cells were placed in an anoxic
chamber (Mitsubishi Gas Chemical Company, Inc.) containing
AnaeroPack® system to create a hypoxic atmosphere. Cells
were maintained in hypoxic conditions at 37˚C for 6 h, following
which pre-equilibrated DMEM containing 10% FBS was added to the
cells, and the cells were cultured in a humidified 95% air-5%
CO2 atmosphere at 37˚C for an additional 24 h. A total
of three groups were used for the in vitro experiments: In
the control and OGD groups the cells were pretreated with PBS for
24 h prior to OGD modeling at room temperature, and in the OGD + AA
group the cells were pretreated with AA (20 µM) for 24 h prior to
OGD modeling at room temperature.
Cell viability assay
Cell viability were detected using CCK-8 kit
according to the manufacturer's instructions. In brief, the H9c2
cells were initially cultured at a density of 1x104
cells/well in 96-well plates at 37˚C. The cells were then
pretreated with various concentrations of AA (2.5-100 µM) for 24 h
at 37˚C. CCK-8 solution (10 µl) was then added to each well and
incubated for an additional 2 h at 37˚C. The absorbance at 450 nm
was measured using a microplate reader (Bio-Rad Laboratories,
Inc.). Also, CCK-8 assay was performed after OGD or control
treatment. All experiments were performed in triplicate.
Measurement of intracellular ROS
levels
ROS generation was determined using the fluorescent
probe 2',7'-dichlorofluorescin diacetate (DCFH-DA; Beyotime
Institute of Biotechnology). Cell-permeable non-fluorescent DCFH-DA
is oxidized to the fluorescent 2',7'-dichlorofluorescein (DCF) in
the presence of ROS. H9c2 cells were harvested after the various
aforementioned treatments using trypsin. After washing with PBS, 10
µM DCFH-DA was added to the cells for 20 min in the dark at 37˚C.
The fluorescence intensity was observed under a fluorescence
microscope (magnification, x100; Nikon Corporation) and analyzed
using ImageJ software. All experiments were performed in
triplicate.
Assessment of oxidative damage
SOD activity and MDA content were measured as the
decomposition products of lipid hydroperoxides. These are often
used as indicators of oxidative damage to cells and tissues
(25,29,30),
and were used in the current in vitro study by SOD and MDA
assay kits to detect the antioxidant performance of AA according to
the manufacturer's protocol. In brief, the H9c2 cells were
initially cultured at a density of 1x104 cells/well in
96-well plates at 37˚C. The cells were sufficiently lysed and
centrifuged at 12,000 x g for 5 min at 4˚C after OGD or control
treatment. Supernatants were collected to assess SOD activity and
MDA content using a microplate reader (Bio-Rad Laboratories, Inc.).
All experiments were performed in triplicate.
Western blot analysis
Proteins were extracted from myocardial tissues and
H9c2 cells following treatment by ice-cold RIPA lysis buffer
(Beyotime Institute of Biotechnology) and the protein concentration
was measured using BCA assay (Beyotime Institute of Biotechnology).
Protein samples (30 µg) were loaded onto an 10% SDS gel resolved
using SDS-PAGE, transferred to a PVDF membrane, and blocked with 5%
skimmed milk for 90 min at room temperature. Subsequently, the
membranes were incubated with one of the following antibodies:
Anti-microtubule-associated proteins 1A/1B light chain 3B (LC3;
cat. no. 3868; Cell Signaling Technology, Inc), anti-B-cell
lymphoma-2 (Bcl-2; cat. no. 15071; Cell Signaling Technology, Inc),
anti-beclin-1 (cat. no. 3495; Cell Signaling Technology, Inc),
anti-phosphorylated-p38 (cat. no. 4511; Cell Signaling Technology,
Inc) and anti-p38 (cat. no. 8690; Cell Signaling Technology, Inc)
or GAPDH (cat. no. 5174; Cell Signaling Technology, Inc) rabbit
polyclonal antibodies, all at 1:1,000 dilution at 4˚C overnight.
Following incubation with the primary antibodies, the membranes
were incubated with the horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody (1:2,000; cat. no. 7074; Cell
Signaling Technology, Inc) at room temperature for 90 min. Signals
were visualized using a chemiluminescence reagent and densitometry
analysis was performed using ImageJ software v1.46 (National
Institutes of Health). GAPDH was used as the loading control. All
experiments were performed in triplicate.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Differences between groups were compared using one-way ANOVA.
Statistical analysis was performed using GraphPad Prism v7
(GraphPad Software, Inc.) and PASW Statistics v18.0 (SPSS, Inc.).
Comparisons between two groups were performed using unpaired
two-tailed Student's t-test. Comparisons between more than two
groups were performed using one-way ANOVA and Tukey's post hoc
tests. P<0.05 was considered to indicate a statistically
significant difference. All experiments were performed in
triplicate.
Results
AA improves cardiac function in mice
with MIRI
Cardiac function was similar among mice in all
groups prior to MIRI treatment (data not shown). However, the
echocardiography evaluation revealed significant decreases in
percent ejection fraction and percent fractional shortening in MIRI
mice at 24 h post-reperfusion compared with sham mice, and
pretreatment with AA partially rescued MIRI-induced cardiac
impairment (Fig. 1B and C).
MIRI initiates an autophagic response
in murine hearts
TEM is considered as the gold standard for
identifying double-membrane vacuole structures (31-33). Autophagy
activation during MIRI was first measured via ultrastructural
analysis using TEM. As demonstrated in Fig. 1D, the degree of mitochondrial
destruction and the number of autophagosomes in myocardial cells
that underwent MIRI were higher compared with the sham group. AA
pretreatment was indicated to attenuate the mitochondrial damage
and autophagosome formation in injured cardiomyocytes. The
autophagy-associated proteins beclin-1 and LC3 were used to
evaluate the levels of intracellular autophagy (34,35),
and their expression was examined via western blotting. The
expression levels of beclin-1 and the ratio of LC3 II/I were
increased following MIRI compared with the sham group, whereas AA
pretreatment lowered these levels (Fig.
1E-G). Taken together, these results suggest that autophagy is
associated with MIRI, and that AA pretreatment partially reduces
autophagy following MIRI.
MIRI decreases and AA restores cell
viability
The chemical structure of AA is presented in
Fig. 1A. To determine the optimal
AA concentration for treatment of cardiomyocytes in vitro,
an AA dose-response curve was plotted, and it was demonstrated that
the viability of H9c2 cells was impaired when treated with AA
concentrations >20 µM. Therefore, 20 µM AA was used for all
subsequent in vitro experiments (Fig. 2A). Based on the CCK-8 assay
(Fig. 2B), H9c2 viability was
reduced by OGD treatment compared with the control cells, whereas
AA pre-treatment protected myocardial cells from OGD treatment.
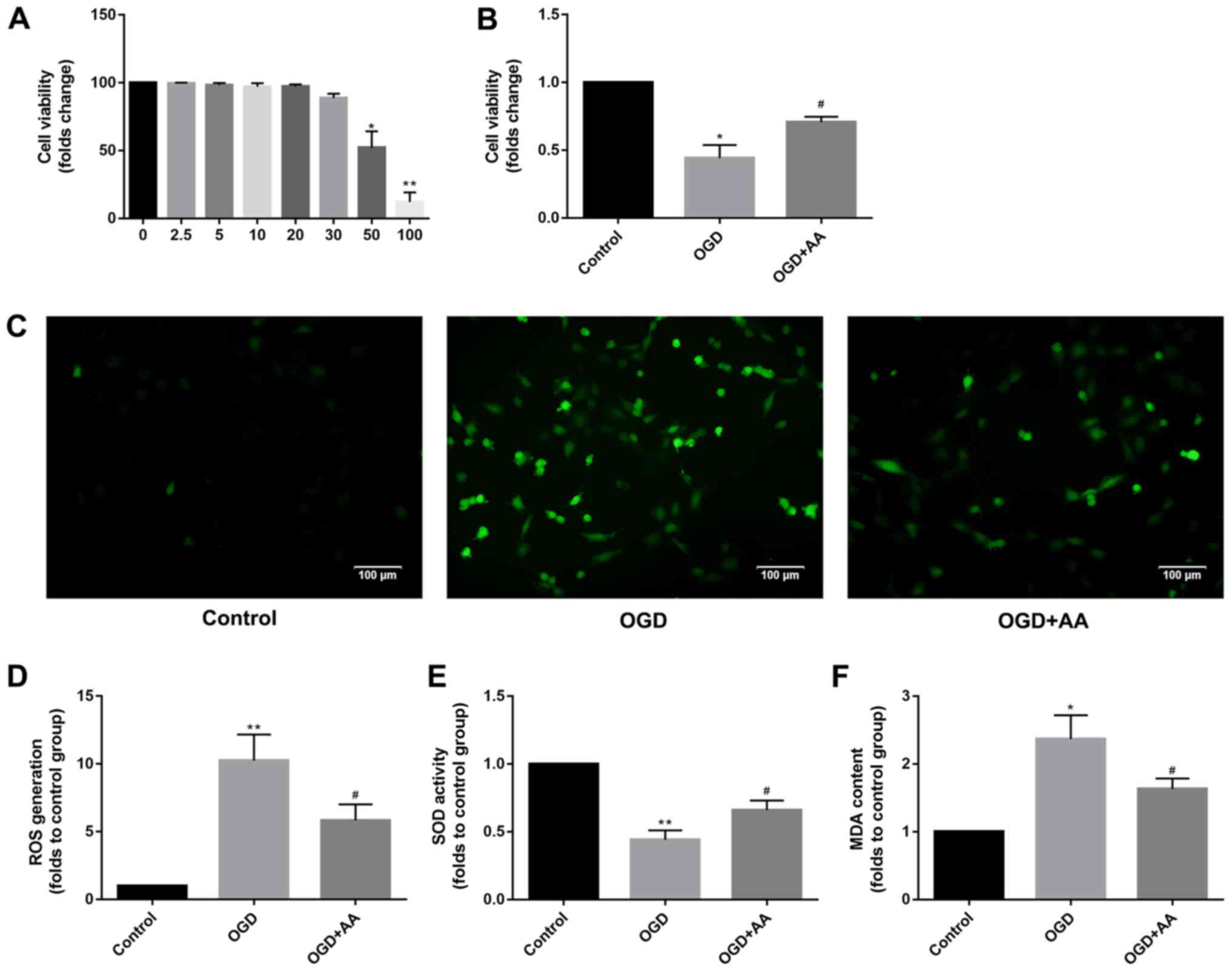 | Figure 2Protective effects of AA on
OGD-induced oxidative stress in H9c2 cells. (A) The cytotoxicity of
AA on H9c2 cells was assessed via the CCK-8 assay. 20 µM was chosen
as the dose of AA for the following study. The data are presented
as the mean ± SD. *P<0.05 and **P<0.01
vs. 0 mM. (B) The viability of H9c2 cells after OGD or control
treatment was measured via the CCK-8 assay. The data are presented
as the mean ± SD. *P<0.05 vs. Control;
#P<0.05 vs. OGD. (C) ROS mediate the OGD-induced
autophagy in H9c2 cells. H9c2 cells were subjected to OGD in the
absence or presence of the AA. The cells were incubated with
2',7'-dichlorofluorescin diacetate and fluorescence was measured
using a fluorescence microscope. Representative images
(magnification, x100) are presented to indicate ROS levels. Scale
bars, 100 µm. The data are presented as the mean ± SD. (D) ImageJ
software was used to perform a quantitative analysis of
intracellular ROS production. **P<0.01 vs. Control;
#P<0.05 vs. OGD. The (E) SOD activity and (F)
intracellular MDA production was measured in H9c2 cells after OGD
or control treatment. The data are presented as the mean ± SD.
*P<0.05 and **P<0.01 vs. Control;
#P<0.05 vs. OGD. AA, asiatic acid; CCK-8, cell
counting kit-8; OGD, glucose deprivation/reperfusion; ROS, reactive
oxygen species; SD, standard deviation; SOD, superoxide dismutase;
MDA, malondialdehyde. |
AA attenuates the increased production
of intracellular ROS induced by MIRI
DCF intensity was used to measure the intracellular
ROS production. The effects of OGD treatment on ROS production in
cardiomyocytes are presented in Fig.
2C and D. H9c2 cells treated
with OGD exhibited a prominent increase in fluorescence intensity
compared with the control group, and AA pretreatment reduced the
production of ROS, which was mediated by OGD. The intracellular MDA
levels and SOD activity were additionally measured to support the
antioxidant function of AA. As demonstrated in Fig. 2E and F, increased levels of MDA and reduced SOD
activity were observed in OGD-treated cells compared with the
control group, suggesting the presence of an increased oxidative
stress and the failure of the antioxidant responses to alleviate
stress resulting from OGD. However, AA pretreatment decreased the
MDA levels and increased SOD activity, compared with the OGD group.
Taken together, these results suggest that the protective effects
of AA against the OGD-induced cardiomyocyte injury were associated
with the alleviation of oxidative stress, which was mediated by
ROS.
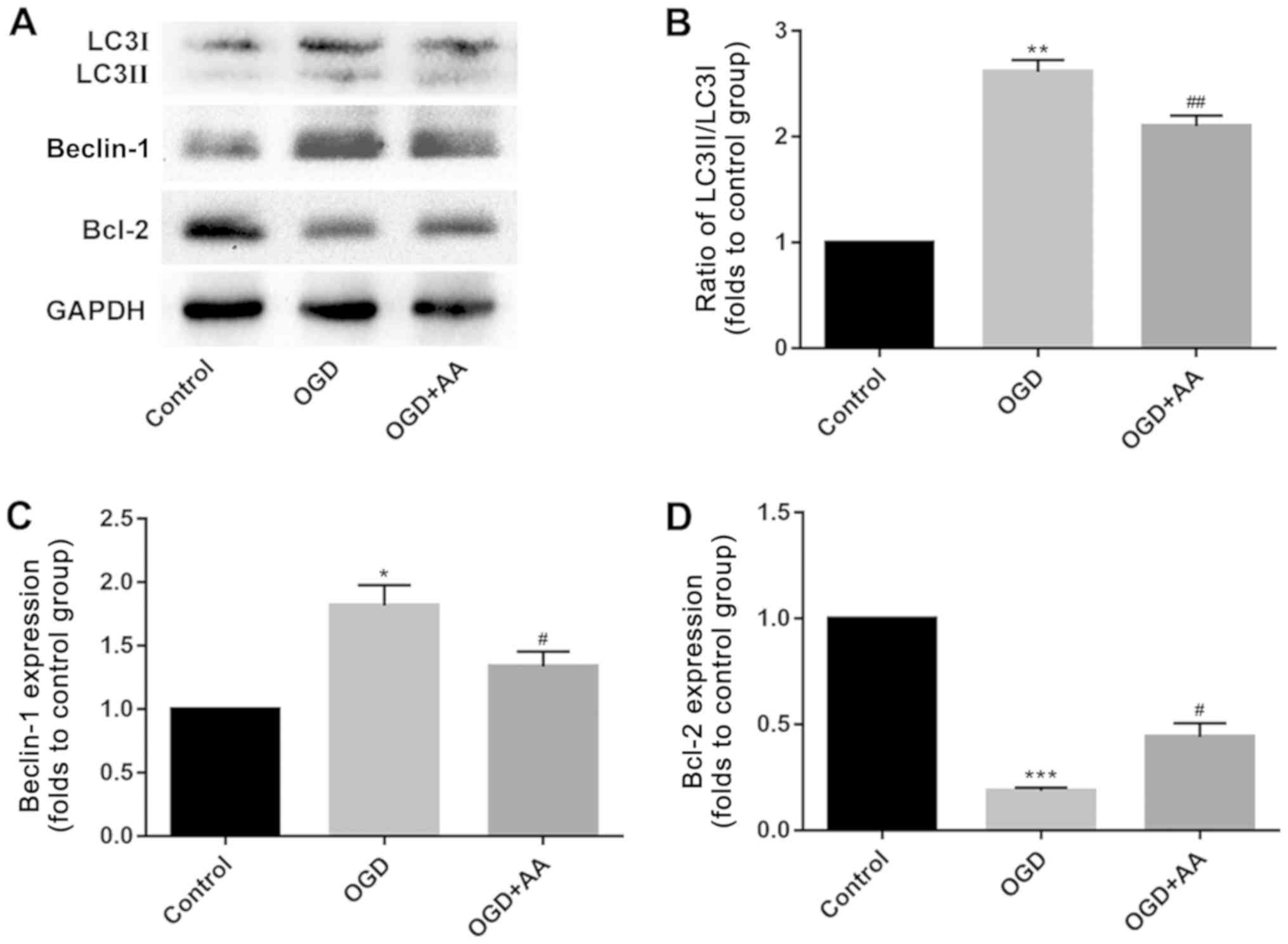
OGD induces autophagy in vitro
The expression of autophagy-associated proteins was
measured in OGD-treated cardiomyocytes. Compared with
cardiomyocytes in the control group, OGD induced cardiomyocyte
autophagy, which was reflected by the increased beclin-1 expression
and LC3 II/I ratio, as indicated via western blotting. AA
pretreatment partially suppressed the effects of OGD on autophagy
in cardiomyocytes in vitro (Fig.
3A-C).
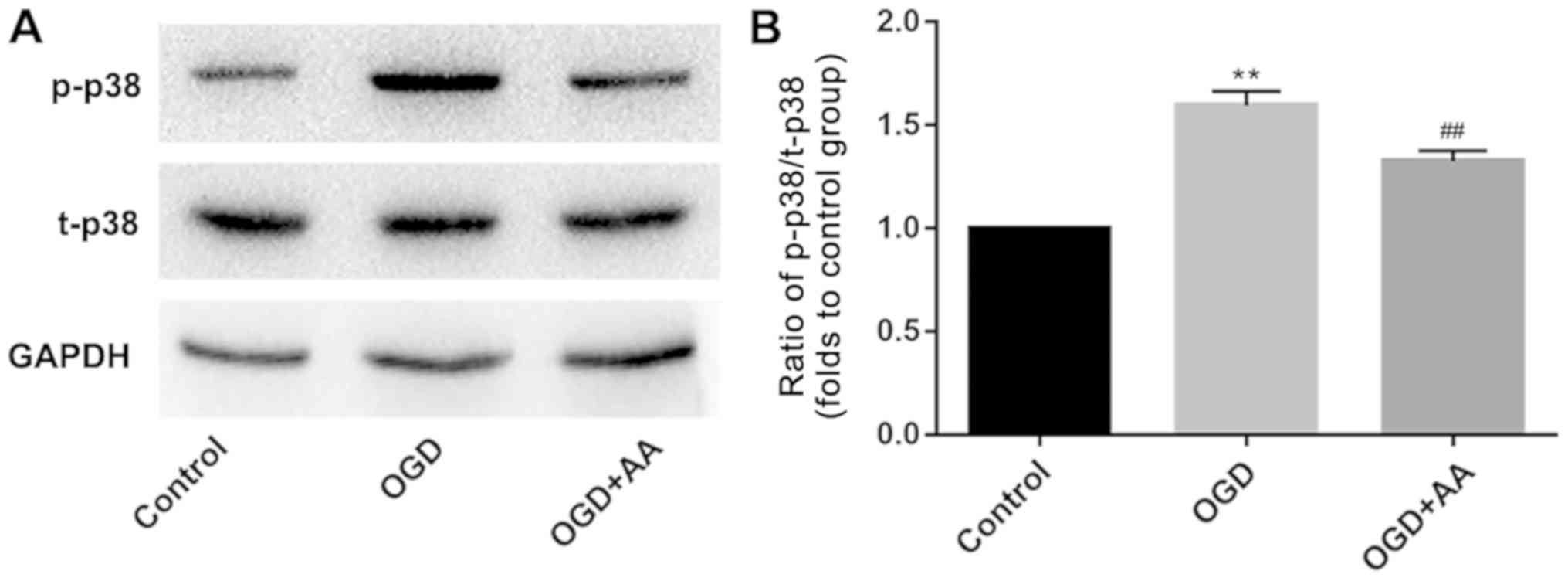
OGD regulates p38/Bcl-2 signaling,
which is attenuated by AA
The Bcl-2/beclin-1 complex is a key regulator of
autophagy (36). As indicated via
western blot analysis, MIRI reduced the protein expression levels
of Bcl-2 (Fig. 3A and D). This may underlie the increased
expression of beclin-1 in MIRI. A variety of protein kinases,
including mitogen-activated protein kinase (MAPK) family members,
have been reported to induce Bcl-2 phosphorylation in other cell
systems (37,38). In the present study, p38-MAPK was
selected for subsequent analysis, and phosphorylation of p38 was
increased following OGD stimulation. AA pretreatment reduced the
degree of p38 phosphorylation (Fig.
4) and increased Bcl-2 expression (Fig. 3A and D), compared with the OGD group.
Discussion
Thanks to the continuous advances in cell biology,
it has been demonstrated that in addition to necrosis and
apoptosis, MIRI also results in autophagy in cardiomyocytes
(15,39,40).
In 1976, Sybers et al (41)
demonstrated that autophagosomes containing damaged organelles were
detected following reoxygenation and glucose administration in
hypoglycemic cells in vitro. This was the first report of
autophagy in MIRI. Subsequently, Decker et al (42) reported an important increase in
autophagy in rabbit hearts after 40 min of I/R. Matsui et al
(14) demonstrated that the
expression of LC3 was increased in a mouse model of MIRI,
reflecting the enhancement of myocardial autophagy. Several factors
have been reported to be associated with MIRI, including oxidative
stress, and ROS has been indicated to induce autophagy (43); however, how ROS-mediated oxidative
stress initiates the autophagic response remains unclear.
Beclin-1 is an autophagy-associated protein with
homology to autophagy-related protein 6, which is a key factor
regulating autophagy (44).
Beclin-1 initiates autophagy and promotes the formation of
autophagic lysosomes (45).
Structurally, beclin-1 has been indicated to contain three
identifiable domains: A short Bcl-2-homology (BH) 3 motif, a
central coiled-coil domain and a C-terminal half, which encompasses
the evolutionarily conserved domain (46). Beclin-1 has been revealed to form
protein complexes with autophagy regulatory proteins, thereby
regulating autophagy levels (44,45,47).
Matsui et al (14)
demonstrated that autophagy during MIRI was beclin-1-dependent, and
primarily manifested following increased beclin-1 expression during
myocardial reperfusion, whereas knockout of the beclin-1 gene
inhibited the formation of autophagosomes during reperfusion. This
result was also confirmed by Valentim et al (39) who demonstrated that when the
expression of beclin-1 in cardiomyocytes was decreased, autophagy
was also reduced. Bcl-2 is an endogenous inhibitor of
beclin-1(48). Previous studies
have indicated the existence of a direct association between the
balance of Bcl-2 and beclin-1 protein expression, which has been
demonstrated to affect autophagy levels (37,49).
Physiologically, beclin-1 binds to the BH3 domain of Bcl-2 to form
a complex, and this Bcl-2/beclin-1 complex ensures that autophagy
levels remain within a homeostatic range (49). However, when beclin-1 function is
not regulated by Bcl-2 or potentially other Bcl-2 family members,
including Bcl-XL, this may result in excessive levels of autophagy
(49). During MIRI, the excessive
accumulation of ROS results in increased phosphorylation of Bcl-2,
disrupting the balance between Bcl-2 and beclin-1. The subsequent
dissociation of the Bcl-2/beclin-1 complex causes the release of
large quantities of beclin-1, which results in excessive autophagy
and, possibly, autophagy-dependent cell death (25). Therefore, the balance between Bcl-2
and beclin-1 appears to underlie the activation of autophagy.
Multiple signaling pathways are associated with
ROS-mediated autophagy, including the MAPK signaling pathways
(50). MAPK is an important
transmembrane signaling pathway that is composed of three major
MAPK cascades, p38, ERK1/2 and JNK, which serve a critical role
during cell proliferation, differentiation and apoptosis (51-53). Oxidative
stress has been indicated to activate MAPK subfamilies in
cardiomyocytes (54). The dynamic
balance between the effects of MAPKs are important for determining
cell fate (27,43). p38-MAPK appears to be more sensitive
to oxidative stress compared with other MAPKs, such as ERK1/2 and
JNK (37). Additionally, several
studies have demonstrated that ROS-dependent p38 activation
regulates Bcl-2 via phosphorylation, which results in autophagy or
apoptosis (27,55).
AA exerts beneficial effects against I/R injury. For
example, Xu et al (56)
reported that AA was effective in mitigating hepatic I/R injury and
inducing the attenuation of Kupffer cell activation via a
peroxisome proliferator-activated receptor gamma/NACHT, LRR and PYD
domains-containing protein 3 inflammasome signaling pathway. Lu
et al (57) demonstrated
that AA attenuated the I/R-induced liver damage via reducing the
oxidative stress and restoring the mitochondrial function. In MIRI,
Huang et al (58)
demonstrated that AA induced activation of the Akt/glycogen
synthase kinase-3β/hypoxia-inducible factor 1-alpha pathway, and
may contribute to the suppression of ROS accumulation and
mitochondrial dysfunction in MIRI injury. However, the results of
Huang et al (58) were
limited to in vitro experiments and did not illustrate the
roles of AA in vivo. The results of the present study
demonstrated that pretreatment with AA was protective against
MIRI-induced cell damage thanks to its antioxidant and
autophagy-suppressing properties. In vivo experiments
revealed that AA pretreatment improved cardiac function and
attenuated autophagy in injured cardiomyocytes, which was evidenced
by the reduced mitochondrial damage, the decreased autophagosome
formation and the reduced expression of autophagy-associated
proteins. Additionally, the in vitro experiments
demonstrated that AA reduced ROS production, which was induced by
OGD, a cellular model of MIRI, decreased the phosphorylation of p38
and increased the expression of Bcl-2 to stabilize the
Bcl-2/beclin-1 complex, thereby suppressing the dissociation of
beclin-1 and the occurrence of autophagy.
In conclusion, the present study suggested that AA
protected cardiomyocytes from ROS-mediated autophagy via a
p38-MAPK/Bcl-2 signaling pathway in MIRI. This may be a novel
mechanism underlying the protective effects of AA in
cardiomyopathy. However, additional studies are required to
determine whether AA may be used as a potential treatment in the
clinical setting.
Acknowledgements
Not applicable.
Funding
The present study was funded by a research project
of Northern Jiangsu People's Hospital (grant no. yzucms201621), the
National Natural Science Foundation of China (grant nos. 81573234
and 81773445) and the ‘333 Project’ Of Jiangsu Province (grant no.
LGY2016006).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CY, LS, JX and JY performed the experiments,
analyzed the data and prepared the manuscript. QW and XW wrote and
revised the manuscript, and designed the experiments. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All aspects of animal care and the experimental
protocols to which the animals were subjected were approved by the
Animal Care and Use Committee of Nanjing Medical University and
conducted in accordance with the Guide for the Care and Use of
Laboratory Animals, published by the United States National
Institutes of Health (23). All
efforts were made to minimize animal suffering.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rai V, Sharma PK, Agrawal S and Agrawal
DK: Relevance of mouse models of cardiac fibrosis and hypertrophy
in cardiac research. Mol Cell Biochem. 424:123–145. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sharma V, Bell RM and Yellon DM: Targeting
reperfusion injury in acute myocardial infarction: A review of
reperfusion injury pharmacotherapy. Expert Opin Pharmacother.
13:1153–1175. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hearse DJ and Bolli R: Reperfusion induced
injury: Manifestations, mechanisms, and clinical relevance.
Cardiovasc Res. 26:101–108. 1992.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Murphy E and Steenbergen CJ: Mechanisms
Underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhao Q, Liu Z, Huang B, Yuan Y, Liu X,
Zhang H, Qiu F, Zhang Y, Li Y, Miao H, et al: PEDF improves cardiac
function in rats subjected to myocardial ischemia/reperfusion
injury by inhibiting ROS generation via PEDF-R. Int J Mol Med.
41:3243–3252. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ferrari RS and Andrade CF: Oxidative
stress and lung ischemia-reperfusion injury. Oxid Med Cell Longev.
2015(590987)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhou T, Chuang CC and Zuo L: Molecular
characterization of reactive oxygen species in myocardial
ischemia-reperfusion injury. Biomed Res Int.
2015(864946)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Toda C and Diano S: Mitochondrial UCP2 in
the central regulation of metabolism. Best Pract Res Clin
Endocrinol Metab. 28:757–764. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Saeed-Zidane M, Linden L, Salilew-Wondim
D, Held E, Neuhoff C, Tholen E, Hoelker M, Schellander K and
Tesfaye D: Cellular and exosome mediated molecular defense
mechanism in bovine granulosa cells exposed to oxidative stress.
PLoS One. 12(e0187569)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Przyklenk K, Dong Y, Undyala VV and
Whittaker P: Autophagy as a therapeutic target for
ischaemia/reperfusion injury? Concepts, controversies, and
challenges. Cardiovasc Res. 94:197–205. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated Protein Kinase and Beclin 1 in Mediating Autophagy.
Circ Res. 100:914–922. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Dong Y, Undyala VV, Gottlieb RA, Mentzer
RM Jr and Przyklenk K: Autophagy: Definition, molecular machinery,
and potential role in myocardial ischemia-reperfusion injury. J
Cardiovasc Pharmacol Ther. 15:220–230. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Abounit K: The process of autophagy in an
in vitro model of myocardial ischemia-reperfusion injury.
Dissertations & Theses-Gradworks 2011.
|
|
17
|
Lv J, Sharma A, Zhang T, Wu Y and Ding X:
Pharmacological review on asiatic acid and its derivatives: A
potential compound. SLAS Technol. 23:111–127. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Duggina P, Kalla CM, Varikasuvu SR, Bukke
S and Tartte V: Protective effect of centella triterpene saponins
against cyclophosphamide-induced immune and hepatic system
dysfunction in rats: Its possible mechanisms of action. J Physiol
Biochem. 71:435–454. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ternchoocheep K, Surangkul D and
Ysothonsreekul S: The recovery and protective effects of asiatic
acid on differentiated human neuroblastoma SH-SY5Y cells
cytotoxic-induced by cholesterol. Asian Pacific J Tropical
Biomedicine. 7:416–420. 2017.
|
|
20
|
Huang SS, Chiu CS, Chen HJ, Hou WC, Sheu
MJ, Lin YC, Shie PH and Huang GJ: Antinociceptive activities and
the mechanisms of anti-inflammation of asiatic acid in mice. Evid
Based Complement Alternat Med. 2011(895857)2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chun F: Asiatic acid protects hearts and
relative mitochondria of streptozotocin-induced diabetic rats. J
Jiangsu University 2010.
|
|
22
|
Goncalves B, Salvador JAR, Marin S and
Cascante M: Synthesis and biological evaluation of novel asiatic
acid derivatives with anticancer activity. RSC Adv. 6:3967–3985.
2016.
|
|
23
|
Si L, Xu J, Yi C, Xu X, Ma C, Yang J, Wang
F, Zhang Y and Wang X: Asiatic acid attenuates the progression of
left ventricular hypertrophy and heart failure induced by pressure
overload by inhibiting myocardial remodeling in mice. J Cardiovasc
Pharmacol. 66:558–568. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xu X, Si L, Xu J, Yi C, Wang F, Gu W,
Zhang Y and Wang X: Asiatic acid inhibits cardiac hypertrophy by
blocking interleukin-1β-activated nuclear factor-κB signaling in
vitro and in vivo. J Thorac Dis. 7:1787–1797. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tang Z, Yang L and Zhang X: Vitexin
mitigates myocardial ischemia reperfusion-induced damage by
inhibiting excessive autophagy to suppress apoptosis via the
PI3K/Akt/mTOR signaling cascade. RSC Adv. 7:56406–56416. 2017.
|
|
26
|
Clark JD, Gebhart GF, Gonder JC, Keeling
ME and Kohn DF: Special report: The 1996 Guide for the care and use
of laboratory animals. ILAR J. 38:41–48. 1997.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu J, Chang F, Li F, Fu H, Wang J, Zhang
S, Zhao J and Yin D: Palmitate promotes autophagy and apoptosis
through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun.
463:262–267. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kamiya T, Kown AH, Kanemaki T, Matsui Y,
Uetsuji S, Okumura T and Kamiyama Y: A simplified model of hypoxic
injury in primary cultured rat hepatocytes. In Vitro Cell Dev Biol
Anim. 34:131–137. 1998.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Jomova K and Valko M: Advances in
metal-induced oxidative stress and human disease. Toxicology.
283:65–87. 2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ray G, Batra S, Shukla NK, Deo S, Raina V,
Ashok S and Husain SA: Lipid peroxidation, free radical production
and antioxidant status in breast cancer. Breast Cancer Res Treat.
59:163–170. 2000.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Eskelinen E, Reggiori F, Baba M, Kovacs AL
and Seglen PO: Seeing is believing: The impact of electron
microscopy on autophagy research. Autophagy. 7:935–956.
2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Martinet W, Timmermans J and De Meyer GR:
Methods to assess autophagy in situ-transmission electron
microscopy versus immunohistochemistry. Methods Enzymol.
543:89–114. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Nadal M and Gold SE: Assessment of
autophagosome formation by transmission electron microscopy.
Methods Mol Biol. 835:481–489. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Pattingre S, Espert L, Biard-Piechaczyk M
and Codogno P: Regulation of macroautophagy by mTOR and Beclin 1
complexes. Biochimie. 90:313–323. 2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Marquez RT and Xu L: Bcl-2:Beclin 1
complex: Multiple, mechanisms regulating autophagy/apoptosis toggle
switch. Am J Cancer Res. 2:214–221. 2012.PubMed/NCBI
|
|
37
|
Markou T, Dowling AA, Kelly T and Lazou A:
Regulation of Bcl-2 phosphorylation in response to oxidative stress
in cardiac myocytes. Free Radic Res. 43:809–816. 2009.PubMed/NCBI View Article : Google Scholar
|
|
38
|
De Chiara G, Marcocci ME, Torcia M,
Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G,
Armirotti A, Amodei S, et al: Bcl-2 Phosphorylation by p38 MAPK:
Identification of target sites and biologic consequences. J Biol
Chem. 281:21353–21361. 2006.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Valentim L, Laurence KM, Townsend PA,
Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS and
Stephanou A: Urocortin inhibits Beclin1-mediated autophagic cell
death in cardiac myocytes exposed to ischaemia/reperfusion injury.
J Mol Cell Cardiol. 40:846–852. 2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Aghaei M, Motallebnezhad M, Ghorghanlu S,
Jabbari A, Enayati A, Rajaei M, Pourabouk M, Moradi A, Alizadeh AM
and Khori V: Targeting autophagy in cardiac ischemia/reperfusion
injury: A novel therapeutic strategy. J Cell Physiol.
234:16768–16778. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Sybers HD, Ingwall J and Deluca M:
Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct
Metab. 12:453–463. 1976.PubMed/NCBI
|
|
42
|
Decker RS and Wildenthal K: Lysosomal
alterations in hypoxic and reoxygenated hearts. I. Ultrastructural
and cytochemical changes. Am J Pathol. 98:425–444. 1980.PubMed/NCBI
|
|
43
|
Guo C, Yang M, Jing L, Wang J, Yu Y, Li Y,
Duan J, Zhou X, Li Y and Sun Z: Amorphous silica nanoparticles
trigger vascular endothelial cell injury through apoptosis and
autophagy via reactive oxygen species-mediated MAPK/Bcl-2 and
PI3K/Akt/mTOR signaling. Int J Nanomedicine. 11:5257–5276.
2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Cao Y and Klionsky DJ: Physiological
functions of Atg6/Beclin 1: A unique autophagy-related protein.
Cell Res. 17:839–849. 2007.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-Induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via Their BH3 Domains. Mol Cell Biol.
29:2570–2581. 2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fu LL, Cheng Y and Liu B: Beclin-1:
Autophagic regulator and therapeutic target in cancer. Int J
Biochem Cell Biol. 45:921–924. 2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Matsunaga K, Saitoh T, Tabata K, Omori H,
Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe
T, et al: Two Beclin 1-binding proteins, Atg14L and Rubicon,
reciprocally regulate autophagy at different stages. Nat Cell Biol.
11:385–396. 2009.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Kulisz A, Chen N, Chandel NS, Shao Z and
Schumacker PT: Mitochondrial ROS initiate phosphorylation of p38
MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell
Mol Physiol. 282:1324–1329. 2002.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kong D, Zheng T, Zhang M, Wang D, Du S, Li
X, Fang J and Cao X: Static mechanical stress induces apoptosis in
rat endplate chondrocytes through MAPK and mitochondria-dependent
caspase activation signaling pathways. PLoS One.
8(e69403)2013.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Becatti M, Taddei N, Cecchi C, Nassi N,
Nassi PA and Fiorillo C: SIRT1 modulates MAPK pathways in
ischemic-reperfused cardiomyocytes. Cell Mol Life Sci.
69:2245–2260. 2012.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ranawat P and Bansal MP: Apoptosis induced
by modulation in selenium status involves p38 MAPK and ROS:
Implications in spermatogenesis. Mol Cell Biochem. 330:83–95.
2009.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Xu Y, Yao J, Zou C, Zhang H, Zhang S, Liu
J, Ma G, Jiang P and Zhang W: Asiatic acid protects against hepatic
ischemia/reperfusion injury by inactivation of Kupffer cells via
PPARγ/NLRP3 inflammasome signaling pathway. Oncotarget.
8:86339–86355. 2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Lu Y, Kan H, Wang Y, Wang D, Wang X, Gao J
and Zhu L: Asiatic acid ameliorates hepatic ischemia/reperfusion
injury in rats via mitochondria-targeted protective mechanism.
Toxicol Appl Pharmacol. 338:214–223. 2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Huang X, Zuo L, Lv Y, Chen C, Yang Y, Xin
H, Li Y and Qian Y: Asiatic acid attenuates myocardial
ischemia/reperfusion injury via Akt/GSK-3β/HIF-1α signaling in rat
H9c2 cardiomyocytes. Molecules. 21(1248)2016.PubMed/NCBI View Article : Google Scholar
|