Introduction
Gilbert syndrome (GS) is a common autosomal dominant
disorder that results in intermittent hyperbilirubinemia in the
absence of any signs or symptoms of liver disease (1). GS usually manifests in decreased
activity of the uridine diphosphate-glucuronosyltransferase
(UGT1A1) gene with an incidence of ~5-10% in the global population
from 2018 (1,2). UGT1A1 gene encodes a
UDP-glucuronosyltransferase, which transforms small lipophilic
molecules, including bilirubin into water-soluble, excretable
metabolites. Several variations in the UGT1A1 gene have been
described, including UGT1A1*28, UGT1A1*60 and UGT1A1*93 (3,4). GS in
combination with diseases, such as thalassemia, glucose-6-phosphate
dehydrogenase (G6PD) deficiency, spherocytosis and acute
lymphoblastic leukemia may potentiate severe hyperbilirubinemia
(5-9).
In addition, GS may decrease plasma oxidation and affect drug
metabolism, such as irinotecan hydrochloride by decreasing the
ability to conjugate drugs (10).
However, to the best of our knowledge, there are currently no
reports about patients with systemic lupus erythematosus (SLE)
coexisting with GS.
SLE is a chronic multisystem inflammatory disease
characterized by the production of various autoantibodies, such as
anti-double-stranded DNA antibodies (anti-dsDNA antibodies),
anti-Sm antibodies and anti-SSA/SSB antibodies. Fang et al
(11) indicated that hepatic
manifestation triggered by SLE itself is controversial and usually
asymptomatic. This is due to the fact a variety of causes need to
be differentiated, such as i) an overlap of SLE with autoimmune
hepatitis (AIH) or primary biliary cirrhosis; ii) an overlap of SLE
with non-autoimmune hepatopathy; iii) the existence of liver injury
that only relates to SLE. And elevated liver parameters seem to be
common, accounting for 25-50% patients with SLE (11); however, the etiology of hepatic
damage remains unclear (12). A
study by Vitek et al (13)
that involved 259 patients with SLE revealed that SLE disease
activity was accompanied by very low serum bilirubin levels, which
were caused by severe oxidative stress. Patients with GS may be
protected from the development of SLE. The present study aimed to
summarize the clinical characteristics, genetic type and treatment
of a patient with SLE coexisting with GS.
Materials and methods
Patient characteristics, examination
and treatment
A 27-year-old Chinese female patient was referred to
Ruijin Hospital (Shanghai, China) displaying jaundice in March
2016. The jaundice began 6 years prior to admission. The patients
parents did not have jaundice, and she denied the use of potential
cholestasis-inducing medication. The patient presented with a malar
rash, arthritis, thrombocytopenia and decreased hemoglobin, but
without kidney and nervous system involvement. The patient had no
symptoms of photosensitivity, alopecia or oral ulcers. Autoantibody
testing revealed that the patient had a titer of 1:100 for
antinuclear antibody and that anti-Sjogren's syndrome A and B
antibodies were positive as well. On this basis, the patient was
diagnosed with SLE according to the 2011 classification criteria of
the Systemic Lupus International Collaborating Clinics (14). The patient's laboratory findings
were as follows: Hemoglobin, 80 g/l (reference value: 110-150 g/l);
white blood cell count, 9.0x109/l (reference value:
4.0-10.0x109/l); and platelet (PLT) count,
50.0x109/l (reference value: 100-300x109/l).
The liver function test revealed that serum levels of aspartate
aminotransferase, alanine aminotransferase, alkaline phosphatase
and γ-glutamyl transpeptidase were normal and total bilirubin (TB)
was 91.1 µmol/l (3.4-20.5 µmol/l), and direct bilirubin (DB) was
12.5 µmol/l (0.0-6.8 µmol/l). Therefore, the patient was
characterized with persistent elevated indirect (unconjugated)
bilirubin (IB). For the immunosuppressive therapy,
hydroxychloroquine at a dose of 200 mg twice/day, prednisone 40
mg/day and cyclosporine 50 mg twice/day orally were used. The
patient was in a stable condition with regard to the SLE disease
activity. However, jaundice still existed. TB, DB, erythrocyte
sedimentation rate (ESR) and PLT were detected in Ruijin Hospital
(Shanghai, China), consecutively. The Coombs test was negative,
which excluded hemolytic anemia. Transaminases and serological
tests for hepatitis B and C were negative, which excluded virus
hepatitis. There was no obvious abnormality on the abdominal
ultrasound or bone marrow biopsy. Informed written consent and
consent for publication were obtained from the patient. This study
was approved by the Ethics Committee of Ruijin Hospital (ID:
2016-62).
PCR amplification and sequencing
A total of 2 ml patient peripheral blood was
collected in a tube containing ethylenediaminetetraacetic acid
(EDTA). Genomic DNA was extracted from the peripheral blood sample
using the membrane-based QIAamp DNA extraction kit (Qiagen GmbH)
according to the manufacturer's instructions. DNA concentration and
purity were measured with a spectrophotometer (NanoDrop 2000;
Thermo Fisher Scientific, Inc.). Primer pairs were the same as used
in a previous study (15). The
promoter, exons 1-5, adjacent intronic regions and the
phenobarbital response enhancer module of the UGT1A1 gene were
analyzed by polymerase chain reaction (PCR). PCR mixtures were
initially denatured at 95˚C for 5 min, followed by 35
cycles of 30 sec 95˚C denaturation, 30 sec
54-64˚C annealing according to the primer pair being
used and 45 sec extension at 72˚C, with a final
extension at 72 for 10 min. PCR products (5 µl) were sequenced with
the Big-Dye Terminator Sequencing kit and an ABI 377 automated DNA
sequencer (Applied Biosystems, Thermo Fisher Scientific Inc.).
Follow-ups
The follow-up of this patient including clinical
symptoms, signs and laboratory examinations, such as ESR, PLT,
liver function tests was conducted every 1 to 3 months. In
addition, the SLEDAI score was evaluated each time (16,17).
The total follow-up period was 46 months.
Results
Laboratory data and treatment
Laboratory data were recorded consecutively and were
presented in Table I. TB, IB and
ESR levels were elevated at admission. Then, TB and IB levels
remained stable from June 2016 to October 2016. Treatment using
phenobarbital at a dose of 30 mg/day was started on November 4,
2016 until November 10, 2016.
 | Table IVariation of laboratory data during
follow-ups for the patient with systematic lupus erythematosus and
coexisting Gilbert syndrome. |
Table I
Variation of laboratory data during
follow-ups for the patient with systematic lupus erythematosus and
coexisting Gilbert syndrome.
| Time,
day/month/year | ESR, mm/h | PLT,
x109/l | TB, µmol/l | DB, µmol/l | IB, µmol/l |
|---|
| 28/03/2016 | 23 | 106 | 96.2 | 9.4 | 86.8 |
| 29/06/2016 | 9 | 108 | 87.2 | 11.4 | 75.8 |
| 19/07/2016 | 13 | 90 | 90.7 | 10.5 | 80.2 |
| 10/08/2016 | 5 | 88 | 89.5 | 12.4 | 77.1 |
| 11/10/2016 | 6 | 75 | 90.7 | 10.5 | 80.2 |
| 04/11/2016 | 8 | 54 | 77.2 | 8.7 | 68.5 |
| 10/11/2016 | 7 | 50 | 54.2 | 8.9 | 45.3 |
| 29/11/2017 | 22 | 15 | 80.2 | 9.0 | 71.2 |
| 30/05/2018 | 7 | 43 | 66.5 | 8.8 | 57.7 |
| 01/03/2019 | 13 | 27 | 67.6 | 9.1 | 58.5 |
| 06/01/2020 | 12 | 32 | 81.5 | 9.8 | 71.7 |
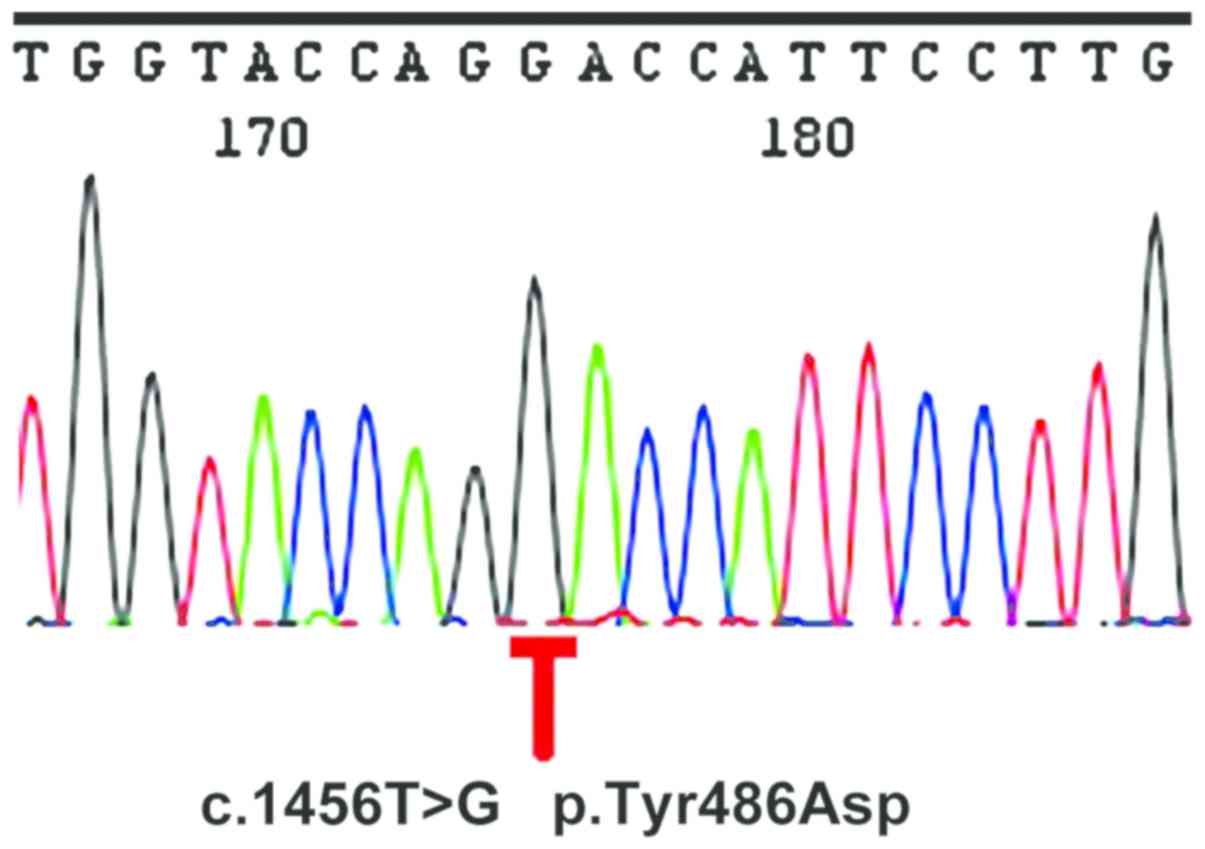
Mutation in the UGT1A1 gene
A direct sequencing analysis was conducted to
identify the mutation in the UGT1A1 gene of the patient. The
analysis revealed a homozygous mutation from a T to G at nucleotide
position 1456 in UGT1A1 exon 5 (c.1456T>G), resulting in the
substitution of aspartate to tyrosine at position 486 of the UGT1A1
protein (p.Y486D) (Fig. 1).
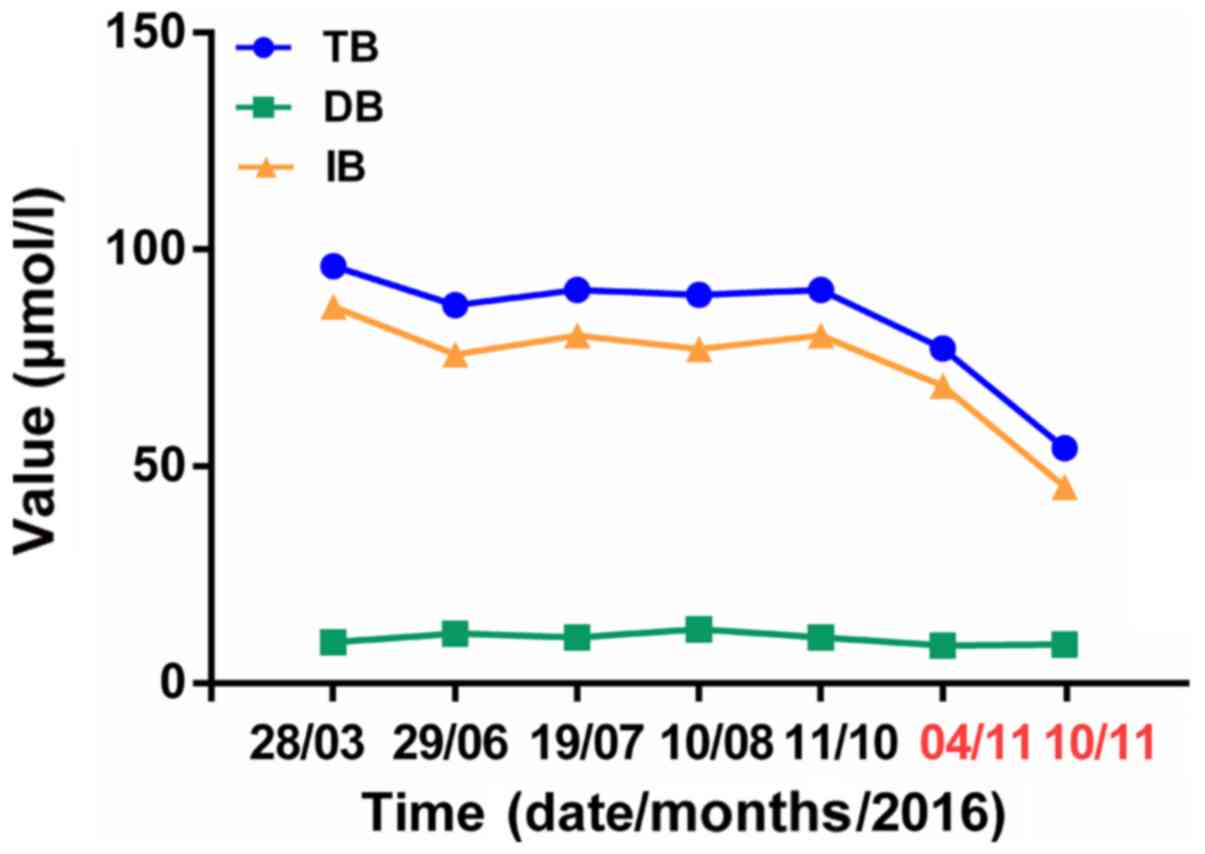
Outcome and follow-up period
SLE activity indicators, such as ESR,
high-sensitivity C-reactive protein and anti-dsDNA antibody
remained normal during the follow-up period. SLEDAI score varied
from 0 to 1. Following phenobarbital treatment for 1 week, there
was a rapid decrease in bilirubin levels (Fig. 2). TB and IB were decreased and
therapy was well tolerated without any side effects. After 46
months of follow up, the patient remained stable on low-dose oral
prednisone (10 mg/day) and cyclosporin (50 mg twice/day). The
latest TB and IB values were 81.5 and 71.7 µmol/l, respectively
(Table I).
Discussion
GS is caused by a mutation in the UGT1A1 gene
resulting in impairment of glucuronidation of unconjugated
bilirubin within hepatocytes. Several studies have reported the
coexistence of GS and hereditary spherocytosis, G6PD deficiency,
gallstone disease and other diseases (8,18,19).
The reports were summarized in Table
II for UGT1A1 genetic mutations and related diseases during the
past 4 years. Butorac et al (7) reported the coexistence of hereditary
spherocytosis and GS in a 21-month-old girl with unconjugated
hyperbilirubinemia. Li et al (20) reported the combination of
myeloproliferative neoplasms and the presence of the insertion
mutation with the (TA)6TAA box and the missense mutation (G→A) at
211 bp of exon 1 in the UGT1A1 gene. Recently, over 130 genetic
variants in the UGT1A1 gene were associated with GS after assessing
the presence of genetic polymorphisms among different ethnicities
(21). East Asian individuals had a
prevalence of ~2% for the genetic variants in the UGT1A1 gene,
while Caucasian individuals had a prevalence of 2-10%, and Southern
Asian and Middle Eastern individuals demonstrated a significantly
increased prevalence of 20% (22-24).
A TA insertion mutation in the TATA box [A (TA) 7TAA] (UGT1A1*28)
and c0.211 G>A (p.G71R) in exon 1 (UGT1A1*6) were
common (25-27).
A(TA)7TAA was the most common mutation in the UGT1A1 gene seen in
Caucasian individuals, accounting for ~35-40% (28). A study from a Romanian cohort
demonstrated that the polymorphism with the highest frequency was
the UGT1A1 7TA (UGT1A1*28) (29). However, a Chinese study revealed
that 36.3% of patients with GS had the c.3279T>G mutation
(30). And the frequency of A (TA)
7TAA was 30.6%, which was lower compared with Caucasians (21). To the best of our knowledge, the
present study demonstrated the first patient diagnosed as SLE with
GS who had the homozygous mutation c.1456 T>G (p.Y486D) in the
UGT1A1 gene. The patient in the present study presented with
persistent unconjugated hyperbilirubinemia and had a good response
to phenobarbital with a decrease in bilirubin, which confirmed the
diagnosis of GS. Low-dose phenobarbital can be used continuously in
patients with GS who tolerate it well. During the 46 month
follow-up period, the patient exhibited stable SLE activity and
serum bilirubin level. A limitation of the present study was lack
of family gene verification, as the parents of the patient did not
have jaundice and refused gene testing.
| Table IIUGT1A1 genetic mutations and related
diseases during the 4 year follow up period. |
Table II
UGT1A1 genetic mutations and related
diseases during the 4 year follow up period.
| First author,
year | Number of
patients | Country | Disease | Mutations | (Refs.) |
|---|
| Maruo, et al
2016 | 121 | Japan | GS | p.G71R,p.P229Q,
c.-3279T>G:A(TA)7TAA | (3) |
| Jamwal, et
al 2016 | 3 | India | GS,
G6PD-deficiency, HS | (TA)7/7
repeats | (46) |
| Singer, et
al 2016 | 43 | Israel | GS, type I
diabetes | Not mentioned | (47) |
| Radoi, et al
2017 | 292 | Romania | GS | UGT1A1 (7TA),
UGT1A1 (8TA) | (29) |
| Moyer, et al
2017 | 54 | England | GS, neonatal
jaundice | Novel variants:
c.337T>G (p.Y113D), c.1469A>C(p.D490A) | (48) |
| Li, et al
2017 | 1 | China | GS, MPN | (TA)6TAA,
c.-211G>A | (20) |
| Sun, et al
2017 | 59 | China | GS | c.-3279T>G,
A(TA)7TAA, p.G71R, p.P229Q, p.P364L, p.Y486D | (30) |
| Aiso, et al
2017 | 1 | Japan | GS, HS |
A(TA)7TAA,
c.211G>A:p.G71R | (49) |
| Pasha, et al
2017 | 51 | Iran | GS | UGT1A1 (7TA) | (50) |
| Haddad, et
al 2017 | 1 | Tunisia | GS,
β-thalassemia |
(TA)6/(TA)7 | (51) |
| Butorac, et
al 2018 | 1 | Croatia | GS, HS | UGT1A1 (7TA) | (7) |
| Qian, et al
2018 | 1 | China | GS, gallstone
disease |
A(TA)7TAA, c.-364C>T,
c.-1352A>C | (52) |
| Bale, et al
2018 | 1,191 | India | GS, gallstone
disease | UGT1A1(TA)n | (19) |
| Kamal, et al
2019 | 110 | Egypt | GS |
A(TA)7TAA | (53) |
Nakagawa et al (31) described a single homozygote for the
p.Y486D mutation in UGT1A1 exon 5, one of the shared exons, and
predicted that the p.Y486D mutation may disturb the metabolization
of the antipyretic and acetaminophen. This may affect the activity
of the UGT1A1, UGT1A6 and UGT1A9 genes, which catalyze
acetaminophen glucuronidation (32). Acetaminophen (~85%) is metabolized
by conjugation, mainly glucuronidation through
UDP-glucuronosyltransferase (33).
Ha et al (10) revealed that
UGT1A1 genetic polymorphisms, particularly the UGT1A1*28
allele of GS may alter the metabolism of drugs, such as irinotecan
hydrochloride by decreasing the ability to conjugate drugs.
Attention should be paid to the use of these drugs in patients with
GS.
The coexistence of SLE and GS requires further
investigation. The association between serum bilirubin and SLE
activity remains unclear. Serum bilirubin is the final product of
hemoglobin metabolism (34). Severe
hyperbilirubinemia could lead to cholestasis and neurological
impairments (neurotoxicity or kernicterus), which have considerable
morbidity and mortality risks (35). It was revealed that bilirubin has
potent cytoprotective action due to its anti-inflammatory,
anti-oxidant and immunosuppressive roles at low concentrations, and
mild hyperbilirubinemia prevents the development of ischemic heart
disease by increasing the serum antioxidant capacity (36-39).
Patients with GS had low levels of oxidative stress associated with
hyperbilirubinemia (40). In
addition, GS was associated with a decreased prevalence of
cardiovascular disease, diabetes, endometrial cancers and with a
better prognosis for Hodgkin's lymphoma (1,39,41-43). In the present study, unconjugated
bilirubin levels were elevated, while the SLE activity indicators
remained normal and stable, such as the SLEDAI score. dos Santos
et al (44) reported that
unconjugated bilirubin level in SLE was negatively correlated with
disease activity, which is consistent with the present study.
Although the activity of SLE increases oxidative stress, serum
bilirubin plays an important role in controlling it (45). Patients with GS can be treated with
phenobarbital or no medication. When a patient experiences any
other factors, such as menstruation, infection, surgery or
overexertion, the degree of jaundice could be aggravated.
To conclude, the present study identified a
homozygous mutation, c.1456T>G, in a patient with SLE with
persistent hyperbilirubinemia coexisting with GS. It is pivotal
that elevated unconjugated hyperbilirubinemia in SLE should be
differentiated from other diseases, such as GS.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81801592),
Shanghai Sailing Program (grant no. 18YF1414100), and Excellent
Youth B Project (grant no. GCQN-2017-B05) and Innovative Research
Team of high-level local universities in Shanghai.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
NY and ZZ drafted the manuscript. HG performed the
laboratory tests. YH performed the PCR amplification and
sequencing. JT performed the follow-up task and analysed clinical
data. The experiments were designed by JY and CY. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Ruijin Hospital (ID: 2016-62) and signed informed
consent was obtained from the patient.
Patient consent for publication
The patient referred to in this study provided
consent for the publication of her information.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wagner KH, Shiels RG, Lang CA, Seyed Khoei
N and Bulmer AC: Diagnostic criteria and contributors to Gilbert's
syndrome. Crit Rev Clin Lab Sci. 55:129–139. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sieg A, Arab L, Schlierf G, Stiehl A and
Kommerell B: Prevalence of Gilbert's syndrome in Germany. Dtsch Med
Wochenschr. 112:1206–1208. 1987.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
3
|
Maruo Y, Nakahara S, Yanagi T, Nomura A,
Mimura Y, Matsui K, Sato H and Takeuchi Y: Genotype of UGT1A1 and
phenotype correlation between Crigler-Najjar syndrome type II and
Gilbert syndrome. J Gastroenterol Hepatol. 31:403–408.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
D'Angelo R, Rinaldi C, Donato L, Nicocia G
and Sidoti A: The combination of new missense mutation with
[A(TA)7TAA] dinucleotide repeat in UGT1A1 gene promoter causes
Gilbert's syndrome. Ann Clin Lab Sci. 45:202–205. 2015.PubMed/NCBI
|
|
5
|
Fretzayas A, Moustaki M, Liapi O and
Karpathios T: Gilbert syndrome. Eur J Pediatr. 171:11–15.
2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Berrueco R, Alonso-Saladrigues A,
Martorell-Sampol L, Catala-Temprano A, Ruiz-Llobet A, Toll T,
Torrebadell M, Naudo M, Camos M and Rives S: Outcome and toxicities
associated to chemotherapy in children with acute lymphoblastic
leukemia and Gilbert syndrome. Usefulness of UGT1A1 mutational
screening. Pediatr Blood Cancer. 62:1195–1201. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Butorac Ahel I, Baraba Dekanic K,
Palcevski G and Roganovic J: An infant with unusually high
unconjugated hyperbilirubinemia due to coexistence of hereditary
spherocytosis and gilbert syndrome. J Pediatr Hematol Oncol.
40:e127–e128. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zahedpasha Y, Ahmadpour M, Niaki HA and
Alaee E: Relation between neonatal icter and gilbert syndrome in
gloucose-6-phosphate dehydrogenase deficient subjects. J Clin Diagn
Res. 8:63–65. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tzetis M, Kanavakis E, Tsezou A, Ladis V,
Pateraki E, Georgakopoulou T, Kavazarakis E, Maragoudaki E,
Karpathios T and Kitsiou-Tzeli S: Gilbert syndrome associated with
beta-thalassemia. Pediatr Hematol Oncol. 18:477–484.
2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ha VH, Jupp J and Tsang RY: Oncology drug
dosing in gilbert syndrome associated with UGT1A1: A summary of the
literature. Pharmacotherapy. 37:956–972. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fang X, Zaman MH, Guo X, Ding H, Xie C,
Zhang X and Deng GM: Role of hepatic deposited immunoglobulin G in
the pathogenesis of liver damage in systemic lupus erythematosus.
Front Immunol. 9(1457)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chowdhary VR, Crowson CS, Poterucha JJ and
Moder KG: Liver involvement in systemic lupus erythematosus: Case
review of 40 patients. J Rheumatol. 35:2159–2164. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Vitek L, Muchova L, Jancova E, Pesickova
S, Tegzova D, Peterova V, Pavelka K, Tesar V and Schwertner H:
Association of systemic lupus erythematosus with low serum
bilirubin levels. Scand J Rheumatol. 39:480–484. 2010.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Petri M, Orbai AM, Alarcon GS, Gordon C,
Merrill JT, Fortin PR, Bruce IN, Isenberg D, Wallace DJ, Nived O,
et al: Derivation and validation of the Systemic Lupus
International Collaborating Clinics classification criteria for
systemic lupus erythematosus. Arthritis Rheum. 64:2677–2686.
2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ehmer U, Kalthoff S, Fakundiny B, Pabst B,
Freiberg N, Naumann R, Manns MP and Strassburg CP: Gilbert syndrome
redefined: A complex genetic haplotype influences the regulation of
glucuronidation. Hepatology. 55:1912–1921. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bombardier C, Gladman DD, Urowitz MB,
Caron D and Chang CH: Derivation of the SLEDAI: A disease activity
index for lupus patients. The Committee on Prognosis Studies in
SLE. Arthritis Rheum. 35:630–640. 1992.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gladman DD, Ibañez D and Urowitz MB:
Systemic lupus erythematosus disease activity index 2000. J
Rheumatol. 29:288–291. 2002.PubMed/NCBI
|
|
18
|
Kumar D, Parakh A and Sharma S: Gilbert
syndrome increasing unconjugated hyperbilirubinemia in a child with
hereditary spherocytosis. J Pediatr Hematol Oncol. 34:54–56.
2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bale G, Avanthi US, Padaki NR, Sharma M,
Duvvur NR and Vishnubhotla VRK: Incidence and risk of gallstone
disease in gilbert's syndrome patients in Indian population. J Clin
Exp Hepatol. 8:362–366. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li XX, Shi J, Huang ZD, Shao YQ, Nie N,
Zhang J, Ge ML, Huang JB and Zheng YZ: Clinical characteristics and
gene mutations of gilbert syndrome complicated with
myeloproliferative neoplasm. Zhongguo Shi Yan Xue Ye Xue Za Zhi.
25:567–571. 2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
21
|
Canu G, Minucci A, Zuppi C and Capoluongo
E: Gilbert and crigler najjar syndromes: An update of the
UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database.
Blood Cells Mol Dis. 50:273–280. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Mendez L, Lagoa M, Quiroga T, Margozzini
P, Azocar L, Molina HR, Vera A, Villarroel L, Arrese M, Hampe J, et
al: Prevalence of Gilbert syndrome and its genetic determinants in
Chile. Rev Med Chil. 141:1266–1274. 2013.PubMed/NCBI View Article : Google Scholar : (In Spanish).
|
|
23
|
Gwee KA, Koay ES and Kang JY: The
prevalence of isolated unconjugated hyperbilirubinaemia (Gilbert's
syndrome) in subjects attending a health screening programme in
Singapore. Singapore Med J. 33:588–589. 1992.PubMed/NCBI
|
|
24
|
Hemmati F, Saki F, Saki N and Haghighat M:
Gilbert syndrome in Iran, fars province. Ann Saudi Med.
30(84)2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bosma PJ, Chowdhury JR, Bakker C, Gantla
S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude
Elferink RP, et al: The genetic basis of the reduced expression of
bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N
Engl J Med. 333:1171–1175. 1995.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sato H, Adachi Y and Koiwai O: The genetic
basis of Gilbert's syndrome. Lancet. 347:557–558. 1996.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Burchell B and Hume R: Molecular genetic
basis of Gilbert's syndrome. J Gastroenterol Hepatol. 14:960–966.
1999.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hsieh TY, Shiu TY, Huang SM, Lin HH, Lee
TC, Chen PJ, Chu HC, Chang WK, Jeng KS, Lai MM and Chao YC:
Molecular pathogenesis of Gilbert's syndrome: Decreased
TATA-binding protein binding affinity of UGT1A1 gene promoter.
Pharmacogenet Genomics. 17:229–236. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Radoi VE, Ursu RI, Poenaru E, Arsene C,
Bohiltea CL and Bohiltea R: Frequency of the UGT1A1*28 polymorphism
in a Romanian Cohort of gilbert syndrome individuals. J
Gastrointestin Liver Dis. 26:25–28. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sun L, Li M, Zhang L, Teng X, Chen X, Zhou
X, Ma Z, Qi L and Wang P: Differences in UGT1A1 gene mutations and
pathological liver changes between Chinese patients with Gilbert
syndrome and Crigler-Najjar syndrome type II. Medicine (Baltimore).
96(e8620)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Nakagawa T, Mure T, Yusoff S, Ono E,
Kusuma Harahap IS, Morikawa S, Morioka I, Takeshima Y, Nishio H and
Matsuo M: A homozygous mutation in UGT1A1 exon 5 may be responsible
for persistent hyperbilirubinemia in a Japanese girl with Gilbert's
syndrome. Kobe J Med Sci. 57:E26–E31. 2011.PubMed/NCBI
|
|
32
|
Court MH, Duan SX, von Moltke LL,
Greenblatt DJ, Patten CJ, Miners JO and Mackenzie PI:
Interindividual variability in acetaminophen glucuronidation by
human liver microsomes: Identification of relevant acetaminophen
UDP-glucuronosyltransferase isoforms. J Pharmacol Exp Ther.
299:998–1006. 2001.PubMed/NCBI
|
|
33
|
Gelotte CK, Auiler JF, Lynch JM, Temple AR
and Slattery JT: Disposition of acetaminophen at 4, 6, and 8 g/day
for 3 days in healthy young adults. Clin Pharmacol Ther.
81:840–848. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lester R and Schmid R: Bilirubin
Metabolism. N Engl J Med. 270:779–786. 1964.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Watchko JF and Tiribelli C:
Bilirubin-induced neurologic damage-mechanisms and management
approaches. N Engl J Med. 369:2021–2030. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Vitek L, Jirsa M, Brodanova M, Kalab M,
Marecek Z, Danzig V, Novotny L and Kotal P: Gilbert syndrome and
ischemic heart disease: A protective effect of elevated bilirubin
levels. Atherosclerosis. 160:449–456. 2002.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ilzecka J and Stelmasiak Z: Serum
bilirubin concentration in patients with amyotrophic lateral
sclerosis. Clin Neurol Neurosurg. 105:237–240. 2003.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yuce S, Cure E, Cicek Y, Cumhur Cure M,
Yilmaz A and Kizilkaya B: Evaluation of aortic stiffness in Gilbert
syndrome patients: A protective effect of elevated bilirubin
levels. Turk Kardiyol Dern Ars. 43:599–606. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
McCarty MF: ‘Iatrogenic Gilbert
syndrome’-a strategy for reducing vascular and cancer risk by
increasing plasma unconjugated bilirubin. Med Hypotheses.
69:974–994. 2007.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Maruhashi T, Soga J, Fujimura N, Idei N,
Mikami S, Iwamoto Y, Kajikawa M, Matsumoto T, Kihara Y, Chayama K,
et al: Hyperbilirubinemia, augmentation of endothelial function,
and decrease in oxidative stress in Gilbert syndrome. Circulation.
126:598–603. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Duguay Y, McGrath M, Lépine J, Gagné JF,
Hankinson SE, Colditz GA, Hunter DJ, Plante M, Têtu B, Bélanger A,
et al: The functional UGT1A1 promoter polymorphism decreases
endometrial cancer risk. Cancer Res. 64:1202–1207. 2004.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Strassburg CP: Hyperbilirubinemia
syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and
Rotor syndrome). Best Pract Res Clin Gastroenterol. 24:555–571.
2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ribrag V, Koscielny S, Casasnovas O,
Cazeneuve C, Brice P, Morschhauser F, Gabarre J, Stamatoullas A,
Lenoir G and Salles G: Groupe d'Etude des Lymphomes agressifs
group, Laboratoire de Génétique et de recherche translationnelle,
Institut Gustave Roussy. Pharmacogenetic study in Hodgkin lymphomas
reveals the impact of UGT1A1 polymorphisms on patient prognosis.
Blood. 113:3307–3313. 2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
dos Santos BH, de R Almeida CM and Skare
TL: Systemic Lupus Erythematosus activity and serum bilirubins.
Acta Reumatol Port. 38:242–246. 2013.PubMed/NCBI
|
|
45
|
Lozovoy MA, Simao AN, Panis C, Rotter MA,
Reiche EM, Morimoto HK, Lavado E, Cecchini R and Dichi I: Oxidative
stress is associated with liver damage, inflammatory status, and
corticosteroid therapy in patients with systemic lupus
erythematosus. Lupus. 20:1250–1259. 2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Jamwal M, Aggarwal A, Kumar V, Sharma P,
Sachdeva MU, Bansal D, Malhotra P and Das R: Disease-modifying
influences of coexistent G6PD-deficiency, Gilbert syndrome and
deletional alpha thalassemia in hereditary spherocytosis: A report
of three cases. Clin Chim Acta. 458:51–54. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Singer S, Pilpel N and Pinhas-Hamiel O:
Gilbert syndrome in patients with type 1 diabetes-prevalence,
glycemic control, and microalbuminuria. Pediatr Diabetes.
18:803–807. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Moyer AM, Skierka JM, Kotzer KE, Kluge ML,
Black JL and Baudhuin LM: Clinical UGT1A1 genetic analysis in
pediatric patients: Experience of a reference laboratory. Mol Diagn
Ther. 21:327–335. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Aiso M, Yagi M, Tanaka A, Miura K, Miura
R, Arizumi T, Takamori Y, Nakahara S, Maruo Y and Takikawa H:
Gilbert Syndrome with concomitant hereditary spherocytosis
presenting with moderate unconjugated hyperbilirubinemia. Intern
Med. 56:661–664. 2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Pasha YZ, Kacho MA, Niaki HA, Tarighati M
and Alaee E: The association between prolonged jaundice and TATA
box dinucleotide repeats in Gilbert's syndrome. J Clin Diagn Res.
11:GC05–GC07. 2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Haddad F, Trabelsi N, Chaouch L, Darragi
I, Oueslati M, Boudriga I, Chaouachi D, El-Borgi W, Hafsia R, Abbes
S and Ouragini H: Homozygous mutation on the β-globin
polyadenylation signal in a Tunisian patient with β-Thalassemia
intermedia and coinheritance of Gilbert's syndrome. Hemoglobin.
41:147–150. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Qian JD, Hou FQ, Wang TL, Shao C and Wang
GQ: Gilbert syndrome combined with prolonged jaundice caused by
contrast agent: Case report. World J Gastroenterol. 24:1486–1490.
2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kamal S, Abdelhakam S, Ghoraba D, Massoud
Y, Aziz KA, Hassan H, Hafez T and Abdel Sallam A: The frequency,
clinical course, and health related quality of life in adults with
Gilbert's syndrome: A longitudinal study. BMC Gastroenterol.
19(22)2019.PubMed/NCBI View Article : Google Scholar
|