Introduction
The transmembrane O-mannosyltransferase targeting
cadherins 3 (TMTC3) gene (Online Mendelian Inheritance in
Man no. 617218) is located on chromosome 12q21.32 and encodes a
type of O-mannosyltransferase comprised of 914 amino acids composed
of nine transmembrane domains and 10 tetratricopeptide repeat (TPR)
domains (data obtained from the UniProt database; uniprot.org/uniprot/Q6ZXV5). The most well-known
function of the TMTC3 protein is its action as a positive regulator
of the endoplasmic reticulum (ER) stress response by binding and
interacting with protein disulphide-isomerase A3, which is an ER
protein involved in the folding of glycoproteins and is
overexpressed under conditions of ER stress such as the
accumulation of misfolded proteins (1,2).
Previous studies have reported that biallelic
mutations of the TMTC3 gene result in two different
neurologic defect syndromes in humans. The first one is cobblestone
lissencephaly (COB), reported by Jerber et al (3) in 2016, which is mainly characterized
by moderate to severe psychomotor delay, language development
delay, intellectual disability (ID), truncal hypotonia, intractable
seizure and malformations of the brain (agyria, ventriculomegaly,
hypoplasia of the corpus callosum and hypoplasia and/or dysplasia
of the brainstem and cerebellum). In addition, certain patients
exhibit microcephaly, clubfoot and visual problems (3). The other syndrome was described by
Farhan et al (4) in 2017, in
which the patients were affected with periventricular nodular
heterotopia (PVNH), ID and nocturnal seizures. Though there are
certain overlapping clinical symptoms (ID and seizure) between the
two syndromes, large phenotypic differences are observed,
particularly for psychomotor and language development and the onset
age of seizures (4). These findings
prompted the current study to investigate an additional case to
further examine the TMTC3 gene variation-related
phenotype.
The current study described a novel compound
heterozygous variant of the TMTC3 gene in a 22-month-old
Chinese boy who, to a certain extent, exhibited a COB-like
phenotype. The degree of brain deformity in the patient was between
that characterized by COB and PVNH. Additionally, bilateral single
transverse palmar creases as a novel phenotype due to TMTC3
variation were reported.
Case report
The patient was a 22-month-old Chinese boy [height,
88 cm (+0.2 SD); weight, 11.5 kg (-0.7 SD); head circumference, 45
cm (-2.3 SD)] who was the second child born at full term via
cesarean section to physically healthy and nonconsanguineous
Chinese parents. He was admitted to Department of Pediatric
Internal Medicine of Fujian Maternity and Child Health Hospital
(Fuzhou, China) for a comprehensive examination in March 2019. The
patient presented with recurrent epileptic spasms at a frequency of
1-3 times/day for 2-3 days/week since he was 1 year old. Coronal
magnetic resonance imaging from Department of Pediatrics, Nanping
People's Hospital (Nanping, Fujian) revealed white matter plaques,
a small frontal lobe and myelin dysplasia. Physical examination
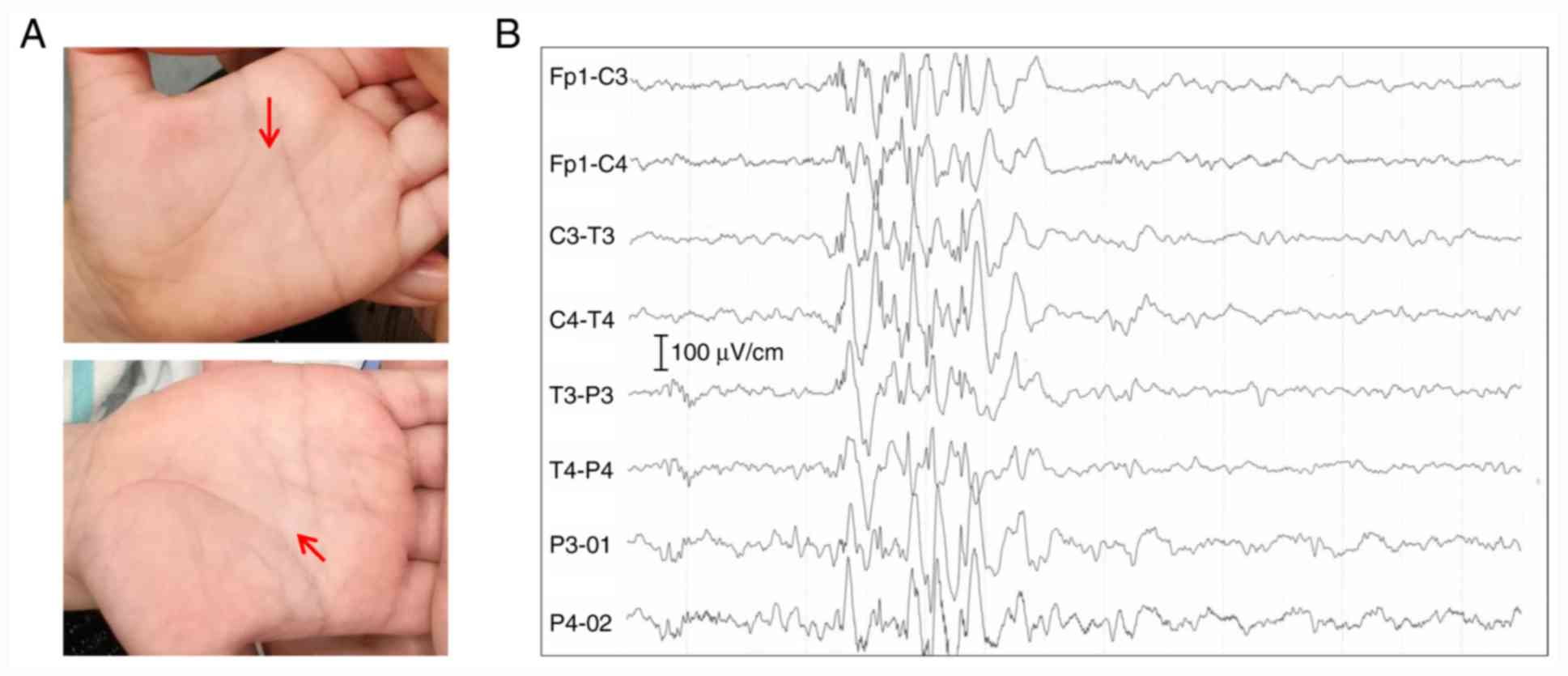
identified truncal hypotonia and bilateral single transverse palmar
creases (Fig. 1A), and the
electroencephalogram reported typical epileptoid discharge
(Fig. 1B). The patient was
diagnosed with intractable epilepsy and infantile spasm. The gross
and fine motor development of the patient was significantly delayed
compared with his peers. The patient could sit with support;
however, he was unable to stand alone or speak. Infections by the
Epstein-Barr virus or cytomegalovirus were ruled out. No
abnormalities were reported using abdominal ultrasound or
ophthalmic examination. The 12-year-old sister of the patient did
not exhibit the aforementioned features.
The patient was suspected of having a genetic
central nervous system syndrome. Therefore, whole-exome sequencing
(WES) was performed to identify the genetic makeup of the patient,
using a previously described experimental procedure, according to
the manufacturer's protocols (5,6).
Briefly, a total of 3 µg DNA from the patient was sheared to
segments sized 150-200 bp using a Covarias® M220
Ultrasonicator system (Covaris, Inc.). DNA integrity was verified
by electrophoresis on a 1% agarose gel. The adapter-ligated library
was generated using the Agilent SureSelect Target Enrichment system
(Agilent Technologies, Inc.) and the capture library, including
both coding exons and flanking intronic regions, was produced with
a SureSelect XT Human All Exon V6 reagent kit (cat. no. 5190-8863;
Agilent Technologies, Inc.). Following this, clusters were
generated via isothermal bridge amplification using an Illumina
cBot station (Illumina, Inc.). Mass concentrations were measured
with a Qubit dsDNA HS Assay kit (cat. no. Q32851; Thermo Fisher
Scientific, Inc.). The library peak size was detected using the
Bioanalyzer 2100 system (Agilent Technologies, Inc.). The average
size value from the Bioanalyzer 2100 was used as the library size
for conversion of mass concentration into molar concentration.
Molar concentration=(Ax1,000,000)/(Sx650), where A is the mass
concentration (ng/µl) and S is the library size (bp). The final
loading concentration was 0.7 nM. Paired-end sequencing with a read
length of 150 bp was performed using a NovaSeq 6000 S4 reagent kit
(cat. no. 20012866; Illumina, Inc.) on an Illumina NovaSeq 6000
System (Illumina, Inc.).
After sequencing, the image files in binary base
cell format were generated. CASAVA software (version no. 1.8;
Illumina, Inc.) was used to perform base calling and demultiplexing
to generate raw fastq files (primary analysis). Raw reads were
trimmed using Skewer (version no. 0.2.2; https://sourceforge.net/projects/skewer/) to remove
adapter sequences and low-quality reads. Subsequently, the trimmed
data were aligned against a reference human genome (GRCh37/hg19;
single nucleotide polymorphism; 153) using NextGENe®
software (version no. 2.4.2; softgenetics.com/NextGENe_011.php; SoftGenetics, LLC).
All single nucleotide variants and indels were presented in variant
cell format and were uploaded to Ingenuity® Variant
Analysis™ (version no. 2.11; qiagenbioinformatics.com/products/ingenuity-variant-analysis;
Qiagen, Inc.) for bioinformatics analysis and interpretation. WES
raw data was deposited into the Mendeley Data online database
(dx.doi.org/10.17632/69j4nzggdx.1).
Common variants with allele frequencies (AF) >1%
in the gnomAD database (version no. 2.1.1; gnomad.broadinstitute.org) and benign variants,
including synonymous, harmless missense variants predicted using
PolyPhen-2 (version no. 2.2; genetics.bwh.harvard.edu/pph2/) and MutationTaster
(https://mutationtaster.org/) (7) software and those predicted to have no
impact on splicing using MaxEntScan software (hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html)
(8), were initially excluded.
Subsequently, clinical symptoms of global developmental delay and
epilepsy served as filtering indexes to analyze candidate variants.
Finally, a compound heterozygous variant in the TMTC3 gene
was identified in the patient (Fig.
S1). In total, one was a missense variant with an extremely low
AF (0.00042%; gnomAD database; GenBank accession no. NM_181783.3)
in exon 8 that generates an amino acid conversion (c.1123G>A,
p.Glu375Lys; rs750602559). The other was a deletion of four bases
(c.1126_1129delCGAG) in exon 8, which was absent in the gnomAD
database and was predicted to lead to a frameshift mutation
resulting in a premature stop codon (p.Arg376Tyrfs*13). To the best
of our knowledge, neither variant has been previously reported.
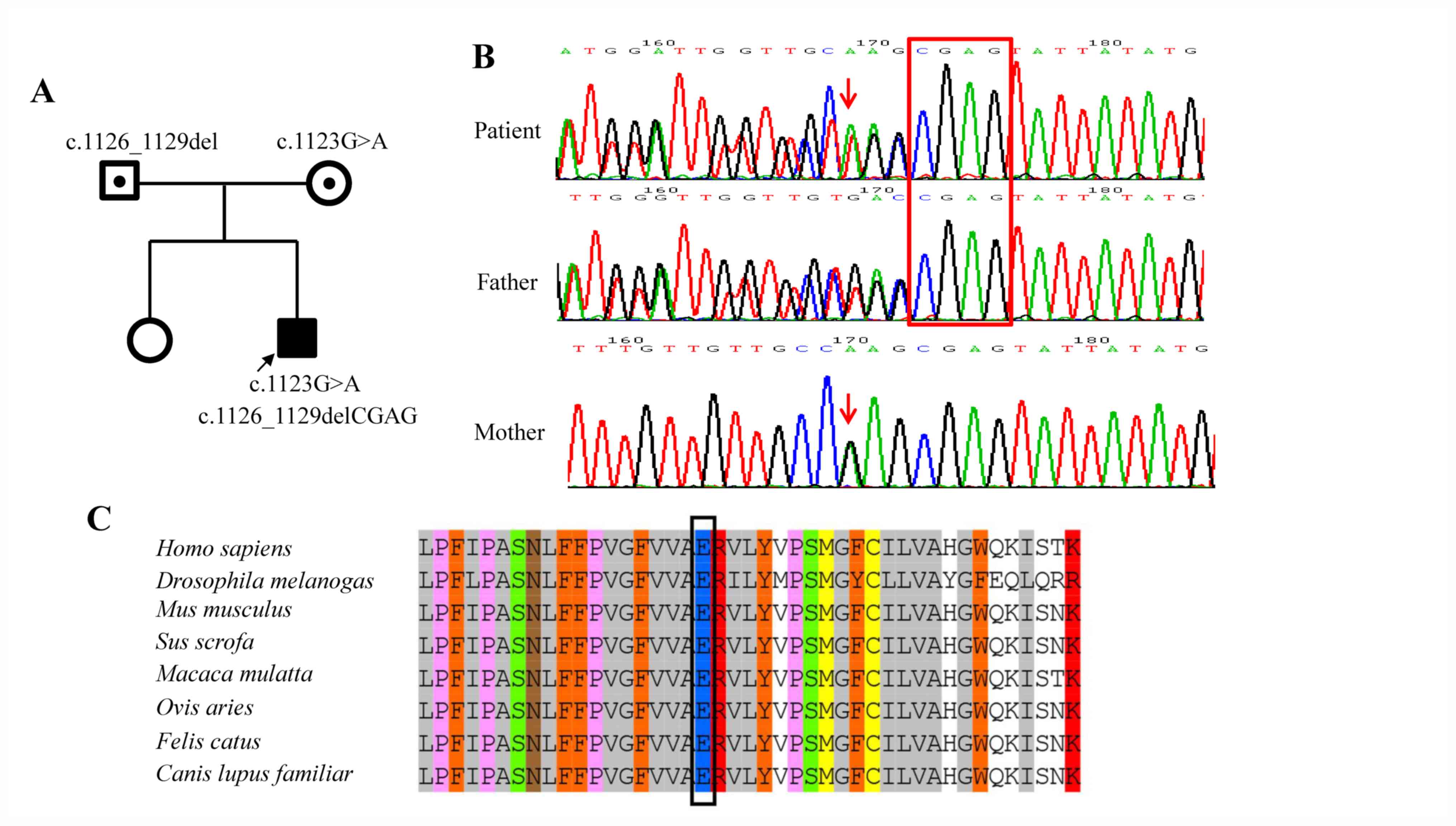
The identified TMTC3 variants were confirmed
in the patient and his parents using Sanger sequencing (Fig. 2A and B). The primers for amplification of the
TMTC3 gene were designed using Prime 3 online software
(version no. 4.1.0; primer3.ut.ee). The primers designed for exon 8
were as follows: Forward, 5'-GGATTCAAGTATCAGATGCCCA-3' and reverse,
5'-AGTAGGTGCCATGGAGCTTT-3'. Both exons and exon-intron boundaries
were amplified by PCR. The reaction mixture for each amplification
contained 1X Premix Taq (Ex Taq™ version 2.0; cat. no. RR003;
Takara Biotechnology Co., Ltd.), 100 ng genomic DNA and 1 pmol
forward and reverse primers to a final volume of 25 µl. The
reaction was performed under the following PCR conditions: Initial
denaturation at 95˚C for 5 min, followed by 19 cycles of 95˚C for
30 sec, 65˚C for 30 sec and 72˚C for 45 sec; 14 cycles of 95˚C for
30 sec, 55˚C for 30 sec and 72˚C for 45 sec; and a final elongation
step at 72˚C for 5 min using a C1000TM Thermal Cycler (Bio-Rad
Laboratories, Inc.). PCR products were separated by 1% agarose gel
(Sangon Biotech Co., Ltd.) with SYBR™ Safe DNA Gel Stain (cat. no.
S33102; Thermo Fisher Scientific; Inc.) and then purified using a
QIAquick Gel Extraction kit (Qiagen GmbH). The purified DNA was
sequenced using an ABI3730XL sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with reverse primers. The sequence data
were analyzed with Mutation Surveyor DNA Variant Analysis software
(version no. 4.0.4; SoftGenetics, LLC). Sanger sequencing revealed
that the missense variant was inherited from the mother of the
patient and the frameshift variant from his father. Additionally,
copy number variation (CNV) analysis was performed by comparing the
sequence depth with the WES data from the other 20 samples of the
same batch, using the NextGENe® software (SoftGenetics
LLC). No clinically significant CNVs were found.
After sequencing, several types of in silico
tools were applied to assess the pathogenicity of the Glu375Lys
variant. The functional prediction of the identified variant was
analyzed with MultAlin online software (multalin.toulouse.inra.fr/multalin) (9), CADD online software (version no. 1.6;
cadd.gs.washington.edu), PROVEAN
(version no. 1.1; provean.jcvi.org), MutationTaster online software and
PolyPhen-2 online software (version no. 2; genetics.bwh.harvard.edu/pph2).
In silico analysis from the MultAlin online
software revealed that the Glu375 amino acid residue of
TMTC3 was highly conserved in multiple species (Fig. 2C). Functional prediction of the
Glu375Lys variant demonstrated a harmful effect on the TMTC3
protein resulting from PolyPhen-2 (probably damaging; score, 1),
PROVEAN (damaging; score, -3.9), MutationTaster (disease causing;
score, 1) and CADD (damaging; score, 34).
Discussion
TMTC3 belongs to a putative family of
O-mannosyltransferases consisting of four TPR-containing proteins
(TMTC1-TMTC4) (3).
TPR domains are critical for protein-protein interactions involved
in various biological processes, including biomineralization,
synaptic vesicle fusion, protein folding, organelle targeting and
protein import (10,11). The human TMTC3 protein was initially
identified in the context of renal transplant surgeries and was
revealed to be upregulated in the blood of operationally tolerant
individuals (12). In addition to
its role in the ER stress response, TMTC3 was recently found
to contribute to the O-mannosylation of E-cadherin, which is
crucial for E-cadherin-mediated cell-cell adhesion (13-15).
Furthermore, mSmile, the murine homolog of TMTC3, is
necessary for bronchial smooth muscle and alveolar myofibroblast
development, and deficiency results in early neonatal lethality in
mice due to airway branching morphogenesis defects during fetal
lung development and alveolarization defects after birth (16).
COB is a severe brain malformation resulting from
the overmigration of neuronal cells, whereas PVNH is characterized
as a common brain malformation due to neurons failing to migrate
from the ventricles (4). However,
the role of TMTC3 in the development of the nervous system
is poorly understood. Farhan et al (4) revealed that TMTC3 is localized
at presynaptic terminals in rat brains via colocalization with the
vesicular γ aminobutyric acid transporter. Specific knockdown of
Drosophila neuronal TMTC3 has been reported to cause
seizure susceptibility, which can be recovered by human
TMTC3 (13). Furthermore,
knockout of the TMTC3 gene in 293 cells was previously
reported to lead to cellular adhesion defects via markedly reduced
binding to the extracellular region of E-cadherin, which was also
hypothesized to be a possible molecular mechanism for neuron
migration defects (14). While the
present study hypothesized that TMTC3 may serve a role in
the development of the nervous system, the precise molecular
mechanisms are yet to be fully elucidated. Further studies should
consider using animal models with a brain tissue-specific
TMTC3 knockout.
The current study presented a male Chinese patient
who exhibited white matter plaques, a small frontal lobe, myelin
dysplasia, microcephaly, psychomotor delay, language development
delay, truncal hypotonia, intractable epilepsy, infantile spasm and
bilateral single transverse palmar creases. DNA sequencing
demonstrated that the patient harbored compound heterozygous
variants for c.1123G>A (p.Glu375Lys) and c.1126_1129delCGAG
(p.Arg376Tyrfs*13) in the TMTC3 gene. According to the
guidelines developed by the American College of Medical Genetics
and Genomics/Association for Molecular Pathology
variant-interpretation (17), the
p.Arg376Tyrfs*13 variant is classified as pathogenic (PVS1+PM2+PP4)
and the p.Glu375Lys variant is likely pathogenic (PM2+PM3+PP3+PP4).
Most of the characteristics of the patient were consistent with
COB, including microcephaly, psychomotor delay, language
development delay, truncal hypotonia, intractable epilepsy and
infantile spasm (Table I). However,
the brain deformities presented in the patient were more severe
compared with those generally exhibited by PVNH, but less severe
compared with those presented by COB. Moreover, to the best of our
knowledge, the bilateral single transverse palmar creases in the
patient have not been described in previously reported patients,
indicating that this may be a novel phenotype resulting from
TMTC3 variation. The patient was diagnosed with TMTC3
variation-related COB-like syndrome by comparing the phenotype with
two previously reported groups of patients (3,4).
 | Table IPhenotypic comparison of the patient
in the current study and previously reported patients. |
Table I
Phenotypic comparison of the patient
in the current study and previously reported patients.
| Clinical feature | Patients with COB
(n=9) reported by Jerber et al (3) | Patients with PVNH
(n=4) reported by Farhan et al (4) | Patient (n=1) in the
current study |
|---|
| Psychomotor
development | | | |
|
Motor
skills | Delayed (9/9) | Normal | Delayed |
|
Language | Delayed (5/9), absent
(4/9) | Delayed (1/4) | Delayed |
| Seizures | | | |
|
Type | Intractable and
infantile-onset epilepsy | Nocturnal
seizures | Intractable and
infantile-onset epilepsy |
|
Age of
onset | 4-8 months | 2-5 years | 1 year |
|
Frequency | Daily to weekly | ≤4 times/night, ≤4-5
days/week | ≤3 times/day, ≤2-3
days a week |
| Neurological
abnormalities | | | |
|
Hypotonia | 9/9 | None | Yes |
|
Intellectual
disability | 9/9 | 4/4 | Too young for
assessment |
|
Microcephaly | 3/7 | None | Yes |
| MRI findings | | | |
|
COB | 7/9 | None | White matter plaques,
small frontal lobe and myelin dysplasia |
|
Ventriculomegaly | 7/9 | None | |
|
Corpus
callosum hypoplasia | 5/9 | None | |
|
Brainstem
hypoplasia | 6/9 | None | |
|
Cerebellum
hypoplasia | 6/9 | None | |
|
Encephalocele | 2/9 | None | |
|
Bilateral
periventricular heterotopias | None | 3/4 | |
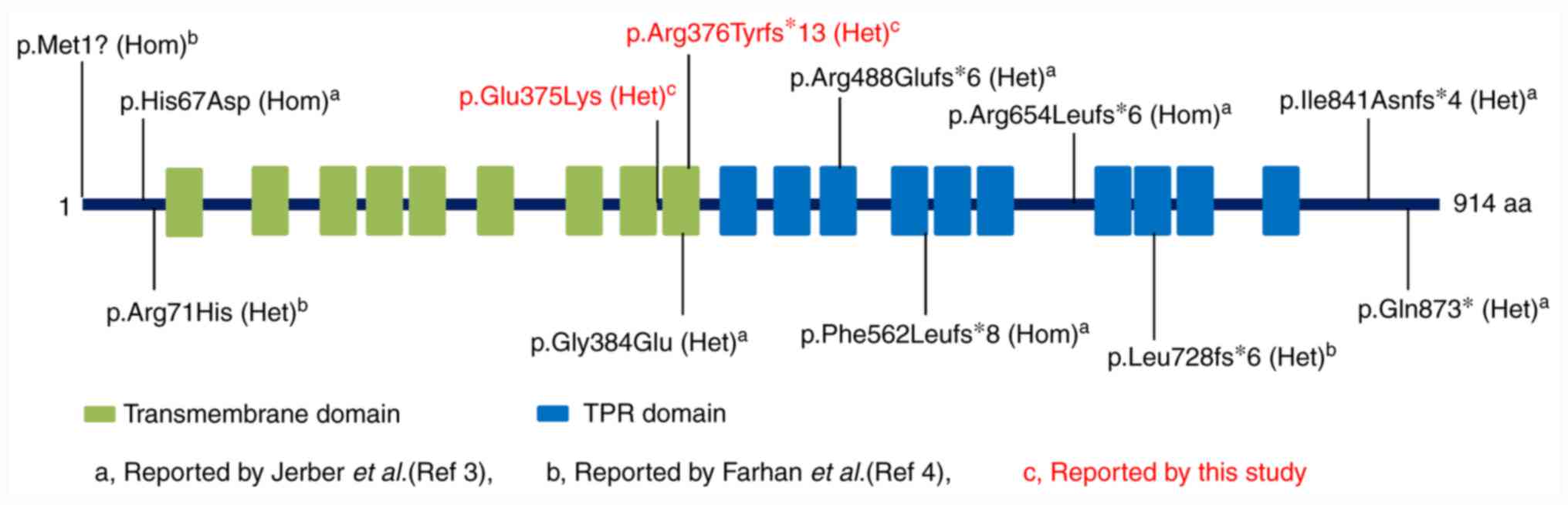
To date, a total of 10 variants of the TMTC3
gene have been reported by two groups. Jerber et al
(3) described four homozygous
(p.Met1?, p.His67Asp, p.Arg654Leufs*6 and p.Phe562Leufs*8) and two
compound heterozygous variants (p.Arg488Glufs*6/p.Gln873* and
p.Gly384Glu/p.Ile841Asnfs*4) in six patients with COB. Farhan et
al (4) identified a compound
heterozygous variant (p.Arg71His/p.Leu728fs*6) from four siblings
in a pedigree where the patients had PVNH with nocturnal seizures
and ID. Unlike patients with COB, patients with PVNH tended to have
normal psychomotor and language development, less severe brain
lesions and a much later onset age of seizures (Table I). However, in addition to the two
variants, the 12 variants, including four missense and eight null
variants, were evenly distributed on the TMTC3 protein (Fig. 3). It is difficult to compare the
phenotypic relationship of the two types of diseases according to
the variant distribution, or the variant type or composition.
Functionally, knockdown of TMTC3 has been reported to result
in delayed gastrulation in Xenopus laevis, which may be
rescued by complementation of wild-type TMTC3. The three
disease-causing missense variants (p.His67Asp, p.Arg71His and
p.Gly384Glu) failed to rescue the delayed gastrulation phenotype,
further supporting the deleterious impact on the TMTC3 protein
(14). The in vitro study
using mutant plasmids revealed that the PVNH-related TMTC3 mutant
protein (p.Arg71His) had a similar half-life compared with
wild-type TMTC3, whereas COB-associated TMTC3
variants (p.Gly384Glu, p.Arg488Glufs*6 and p.Phe562Leufs*8) led to
significantly reduced half-lives (14). This increased instability may be an
explanation for the severe phenotype of patients with COB. However,
further functional investigations should be performed to directly
study how the variants affect the development of the nervous
system, as no half-life alterations were observed from the other
four COB-associated TMTC3 variants (p.His67Asp
p.Arg654Leufs*6 p.Ile841Asnfs*4 p.Gln873*) (14).
In conclusion, the current study identified a novel
compound heterozygous variant in the TMTC3 gene that caused
severe neurological defects in a pediatric Chinese patient. The
results further support the observation that variation in the
TMTC3 gene leads to a recessive COB phenotype. Moreover, to
the best of our knowledge, the current study was the first to
indicate the feature of bilateral single transverse palmar creases
in patients with TMTC3 variation.
Supplementary Material
Analysis of the whole-exome sequencing
raw data (variant cell format files) using NextGENe®
software. Compared with the reference sequence, the WES data
suggest that there are two close variants in TMTC3, one is a
missense variant (c.1123G>A) and the other is a four
bases-deletion variant (c.1126_1129delCGAG). TMTC3, transmembrane
O-mannosyltransferase targeting cadherins 3.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Guiding
Project of Fujian Province Health Commission (grant no. 2019Y0057)
and the Project of Science and Technology Innovation, Fujian
Province Health Commission [grant no. 2017(804)].
Availability of data and materials
The WES raw data (named ‘VCF file of WES data for a
TMTC3 variation patient’) is available from the Mendeley Data
online database (http://dx.doi.org/10.17632/69j4nzggdx.1).
Authors' contributions
GL, HY and JW conceptualized and designed the
current study, drafted the initial manuscript and reviewed and
revised the manuscript. NL and JW were responsible for the genetic
diagnosis and interpretation of genetic findings. QZ and HL were
responsible for patient treatment and medical history data
collection. The authors agreed to be accountable for all aspects of
the current work. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures followed were in accordance with the
ethical standards of the Fujian Maternity and Child Health Hospital
(Fuzhou, China) on human experimentation and with the Helsinki
Declaration of 1975 (revised in 2000), and the protocol was
approved by the Ethics Committee of Fujian Maternity and Child
Health Hospital, Fuzhou, China (approval no. FMCHHIRB-2019019). The
patient was enrolled from the Fujian Maternity and Child Health
Hospital, and written informed consent was obtained from the
patient's family.
Patient consent for publication
Written informed consent was obtained from the
patient's family.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Racapé M, Duong Van Huyen JP, Danger R,
Giral M, Bleicher F, Foucher Y, Pallier A, Pilet P, Tafelmeyer P,
Ashton-Chess J, et al: The involvement of SMILE/TMTC3 in
endoplasmic reticulum stress response. PLoS One.
6(e19321)2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Parakh S and Atkin JD: Novel roles for
protein disulphide isomerase in disease states: A double edged
sword? Front Cell Dev Biol. 3(30)2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jerber J, Zaki MS, Al-Aama JY, Rosti RO,
Ben-Omran T, Dikoglu E, Silhavy JL, Caglar C, Musaev D, Albrecht B,
et al: Biallelic mutations in TMTC3, encoding a transmembrane and
TPR-containing protein, lead to cobblestone lissencephaly. Am J Hum
Genet. 99:1181–1189. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Farhan SMK, Nixon KCJ, Everest M, Edwards
TN, Long S, Segal D, Knip MJ, Arts HH, Chakrabarti R, Wang J, et
al: Identification of a novel synaptic protein, TMTC3, involved in
periventricular nodular heterotopia with intellectual disability
and epilepsy. Hum Mol Genet. 26:4278–4289. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yu T, Li J, Li N, Liu R, Ding Y, Chang G,
Chen Y, Shen Y, Wang X and Wang J: Obesity and developmental delay
in a patient with uniparental disomy of chromosome 2. Int J Obes
(Lond). 40:1935–1941. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li N, Xu Y, Zhang Y, Li G, Yu T, Yao R,
Zhou Y, Shen Y, Yin L, Wang X and Wang J: Biallelic ERBB3
loss-of-function variants are associated with a novel multisystem
syndrome without congenital contracture. Orphanet J Rare Dis.
14(265)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yeo G and Burge CB: Maximum entropy
modeling of short sequence motifs with applications to RNA splicing
signals. J Comput Biol. 11:377–394. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Corpet F: Multiple sequence alignment with
hierarchical clustering. Nucleic Acids Res. 16:10881–10890.
1988.PubMed/NCBI View Article : Google Scholar
|
|
10
|
D'Andrea LD and Regan L: TPR proteins: The
versatile helix. Trends Biochem Sci. 28:655–662. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zeytuni N and Zarivach R: Structural and
functional discussion of the tetra-trico-peptide repeat, a protein
interaction module. Structure. 20:397–405. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Brouard S, Mansfield E, Braud C, Li L,
Giral M, Hsieh SC, Baeten D, Zhang M, Ashton-Chess J, Braudeau C,
et al: Identification of a peripheral blood transcriptional
biomarker panel associated with operational renal allograft
tolerance. Proc Natl Acad Sci USA. 104:15448–15453. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Larsen ISB, Narimatsu Y, Joshi HJ,
Siukstaite L, Harrison OJ, Brasch J, Goodman KM, Hansen L, Shapiro
L, Honig B, et al: Discovery of an O-mannosylation pathway
selectively serving cadherins and protocadherins. Proc Natl Acad
Sci USA. 114:11163–11168. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Graham JB, Sunryd JC, Mathavan K, Weir E,
Larsen ISB, Halim A, Clausen H, Cousin H, Alfandari D and Hebert
DN: ER transmembrane protein TMTC3 contributes to O-mannosylation
of E-cadherin, Cellular Adherence and Embryonic Gastrulation. Mol
Biol Cell. 31:167–183. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Larsen ISB, Narimatsu Y, Clausen H, Joshi
HJ and Halim A: Multiple distinct O-Mannosylation pathways in
eukaryotes. Curr Opin Struct Biol. 56:171–178. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yun EJ and Vu TH: mSmile is necessary for
bronchial smooth muscle and alveolar myofibroblast development.
Anat Rec (Hoboken). 295:167–176. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|