Introduction
Hypophosphatasia (HPP) is a rare skeletal systemic
disease that was first described by Rathbun in 1948(1). HPP can be inherited in an autosomal
recessive or autosomal dominant manner (1). A total of 1 in 25 individuals carry
the alkaline phosphatase gene (ALPL) founder mutation and ~1 in
2,500 newborns suffer from lethal HPP among Mennonites in Canada
(2,3). A previous study of ALPL mutation
analysis predicted that the incidence of severe and moderately
severe HPP in Europe was 1/300,000 and 1/6,370, respectively, in
2011(4).
HPP appears as a defective mineralization of the
bone or teeth with low serum activity and bone alkaline phosphatase
(5). Clinical features are varied
and range from the prenatal lethal form without mineralized bone to
adult HPP with pathological fractures of the lower limbs in
adulthood (6,7). A total of six clinical forms,
depending on age at diagnosis and severity of symptoms, are
distinguished (8). Severe perinatal
HPP is characterized by respiratory failure and hypercalcemia
(8,9). Benign perinatal HPP displays prenatal
skeletal anomalies (8,10). Clinical symptoms of infantile HPP
appear between birth and six months of age and include
craniosynostosis and rickets without elevated serum alkaline
phosphatase activity (6,11,12).
Features of childhood HPP mainly comprise low bone mineral density
with unexplained fractures and early loss of primary teeth
(7). Patients with adult HPP
present with stress fractures and pseudofractures of the lower
limbs in middle age, occasionally accompanied by premature loss of
permanent teeth (13-15).
Odontohypophosphatasia presents with premature exfoliation of
primary teeth or severe dental caries (6,8).
Alkaline phosphatases (ALP), also known as
glycoprotein enzymes, catalyze the hydrolysis of phosphoesters to
release inorganic phosphate (16).
There are four different ALP isoenzymes, three tissue-specific ALPs
(placenta, intestine and germ cell) and one tissue-non-specific ALP
(TNAP or TNSALP) (17-20).
TNAP, which is encoded by the ALPL gene, is not only highly
expressed in bone, liver, kidney and intestinal mucosa, but also in
brain, fibroblasts and endothelium (6,21).
The current study presented with a young Chinese
male with HPP caused by a compound heterozygous mutation in ALPL
inherited from his parents. The results of the present study on the
function and potential pathogenic mechanism of HPP suggested that
the compound heterozygous mutations led to diminished expression of
ALPL, which ultimately caused HPP. The present research may provide
a reference for future studies of the potential pathogenical
mechanisms by which mutations in ALPL cause HPP.
Materials and methods
Patients
The patient and his family members were recruited in
Nanfang Hospital. The proband (III-1), aged six, was referred to
the Surgery of Joints and Osteopathy at Nanfang Hospital, China for
consultation associated with skeletal pain and premature loss of
primary teeth between March and April 2016. Physical and imaging
examinations of the proband were performed by an experienced
orthopedic surgeon. Osteoporosis was investigated by X-ray imaging.
A total of 200 healthy controls were randomly recruited from the
Department of Medical Genetics, School of Basic Medical Sciences,
Southern Medical University, China between June and September 2016.
The criteria for inclusion were matched ethnic origin, age range
between 20-50 years and healthy individuals without major
diseases.
Mutation screening
Genomic DNA was isolated from the peripheral blood
of the proband and his family members using standard
phenol/chloroform extraction. A total of 2 ml EDTA-anticoagulated
whole blood was added to a 15 ml tube with 10 ml water for
solvation of erythrocytes and centrifuged for 10 min at 2,580 x g
at room temperature. The supernatant was discarded and the
aforementioned step was repeated. A total of 500 µl sodium
chloride-Tris-EDTA buffer (0.1 M NaCl, 0.01 M Tris-HCl pH 8.0,
0.001 M Na2EDTA pH 8.0), 50 µl 10% SDS and 5 µl 10 mg/ml
proteinase K were added to the tube and the tube was shaken
overnight at 55˚C. The lysate was transferred to a 1.5 ml Eppendorf
tube. A total of 600 µl saturated phenol was added to the tube,
vortexed vigorously and centrifuged for 5 min at 13,524 x g at room
temperature. The aqueous phase was transferred to a new 1.5 ml tube
and 300 µl saturated phenol and 300 µl buffer-chloroform/isoamylol
was added. The tube was vigorously vortexed, centrifuged for 5 min
at 13,524 x g at room temperature and the aqueous phase was
transferred to another new tube followed by addition of 600 µl
buffer-chloroform//isoamylol. The tube was centrifuged for 2 min at
13,524 x g at room temperature and the supernatant was transferred
to a new tube. Subsequently, 60 µl 3 M sodium acetate and 1.2 ml
ethanol was added to the tube, and the tube was stored at -20˚C for
20 min. The tube was then centrifuged for 12 min at 13,524 x g,
left to precipitate and washed with 75% ethanol twice. The
precipitate was dried and resuspended in 50 µl TE buffer (0.1 M
Tris-HCl pH8.0 and 0.01 M Na2EDTA). All exons of ALPL
were amplified by PCR using Taq DNA polymerase (Beijing Dingguo
Changsheng Biotechnology Co., Ltd.). Agarose gel (1.2%)
electrophoresis and Sanger sequencing were used to analyze the PCR
products. Two significant mutations within exons 4 and 6 were
identified. The following primer pairs were used for the PCR: ALPL
exon 4 forward, 5'-CTTACCCCGCCAAGTAACTG-3' and reverse,
5'-ACAGACCTGAACTGGGCTTG-3' and ALPL exon 6 forward,
5'-ACACCCCGATCTGTGGATAA-3' and reverse, 5'-CAACCGCAAATCCCCTAAT-3'.
The following thermocycling conditions were used for the PCR:
Initial denaturation for 5 min at 95˚C; followed by 35 cycles of
denaturation (30 sec; 95˚C), annealing (30 sec; 60˚C), extension
(45 sec for exon 4; 30 sec for exon 6; 72˚C), and a final extension
for 10 min at 72˚C.
Bioinformatics
The secondary and three-dimensional structures of
wild-type (WT) and mutant ALPL proteins were predicted using the
Self-Optimized Prediction method with the Alignment (SOPMA) method
(22), Iterative Threading ASSEmbly
Refinement (I-TASSER; http://zhanglab.ccmb.med.umich.edu/I-TASSER/) and
SWISS-MODEL (based on P05816) (https://swissmodel.expasy.org/), respectively.
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT
(http://sift.jcvi.org/) and Mutation Taster
(http://www.mutationtaster.org/) were
used to predict the harmfulness of mutations and the University of
California, Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu) was used to analyze the
conservation of ALPL across species.
Cellular localization
The coding sequence of the ALPL gene was cloned into
the BamHI and EcoRI sites of the pcDNA3.1(+) vector
(Invitrogen; Thermo Fisher Scientific, Inc.). The plasmid was used
as a template for generating the mutant plasmids ALPL-pT68M and
ALPL-pE191K. The following primer pairs were used for the PCR:
c.203C>T forward, 5'-GTGTCTCCACAGTGATGGCTGCCCGCATCCT-3' and
reverse, 5'-AGGATGCGGGCAGCCATCACTGTGGAGACAC-3' and c.571G>A
forward, 5'-TGGTACTCAGACAACAAGATGCCCCCTGAGG-3' and reverse,
5'-CCTCAGGGGGCATCTTGTTGTCTGAGTACCA-3'. The following thermocycling
conditions were used for the PCR: Initial denaturation for 5 min at
95˚C; followed by 20 cycles of denaturation (10 sec; 98˚C),
annealing (15 sec; 68˚C), extension (7 min; 72˚C); and a final
extension for 10 min at 72˚C. 293 T cells (Guangzhou Saiku
Biotechnlogy Co., Ltd.) were grown in DMEM (Invitrogen; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) at 37˚C and transiently transfected with
recombinant constructs (3 µg) using Lipofectamine™ 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h.
Immunostaining
Following 48 h of transfection, cells were washed
with PBS (pH 7.4) three times, fixed with ice-cold methanol for 10
min, and dried and washed with PBS. Cells were then permeabilized
for 15 min with PBS-Tween-20 (0.05%) and washed with PBS three
times. Subsequently, cells were blocked with 3% BSA (Beyotime
Institute of Biotechnology) and incubated with mouse anti-TNAP
(1:50; cat. no. sc21708; Santa Cruz Biotechnology, Inc.) at 4˚C for
16 h and stained with FITC-conjugated secondary antibodies (1:200;
cat. no. F2012; Sigma-Aldrich; Merck KGaA) at room temperature for
2 h. Cell nuclei were stained with DAPI (Sigma-Aldrich; Merck KGaA)
for 10 min at room temperature. Fluorescence microscopy (LSM 880;
Carl Zeiss AG) was used to visualize the fluorescence signal of
transfected cells at x100, magnification.
ALPL expression in 293T cells
To determine the expression of WT and mutant ALPL,
recombinant constructs were transfected into 293 T cells using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.).
At 24 or 48 h after transfection, cells were harvested for protein
extraction with radioimmunoprecipitation assay buffer (Cell
Signaling Technology, Inc.) supplemented with protease inhibitor
cocktail (Sigma-Aldrich; Merck KgaA). The lysates were quantified
using a bicinchoninic acid assay (Thermo Fisher Scientific, Inc.)
and then boiled with protein loading buffer (Beyotime Insititute of
Biotechnology). A total of 20 µg protein/lane was separated by
SDS-PAGE with 5% stacking gel and 10% separation gels.
Subsequently, proteins were transferred to polyvinylidene fluoride
membranes (EMD Millipore). The membranes were blocked with 5%
nonfat milk at room temperature for 1 h and then incubated with
mouse anti-TNAP (1:100; cat. no. sc21708; Santa Cruz Biotechnology,
Inc.) overnight at 4˚C and incubated with goat-anti-mouse
immunoglobulin G-horseradish peroxidase (HRP; 1:4,000; cat. no.
RM3001; Beijing Ray Antibody Biotech) at room temperature for 1 h.
Signal detection was performed using the Immobilon Western
Chemiluminescent HRP substrate (EMD Millipore; Merck KGaA).
TNAP relative enzyme activity
test
293T cells were transfected with recombinant
constructs and an alkaline phosphatase detection kit (cat. no.
P0321; Beyotime Institute of Biotechnology) was used to investigate
the relative TNAP enzyme activity of WT and mutant ALPL after
transfection for 24 or 48 h.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 5 (GraphPad Software Inc.). Statistical differences among the
four groups were determined using one-way ANOVA followed by
Bonferroni's correction. Data are presented as the mean ± SEM and
the experiments were repeated three times. *P<0.05
was considered to indicate a statistically significant
difference.
Results
Clinical features
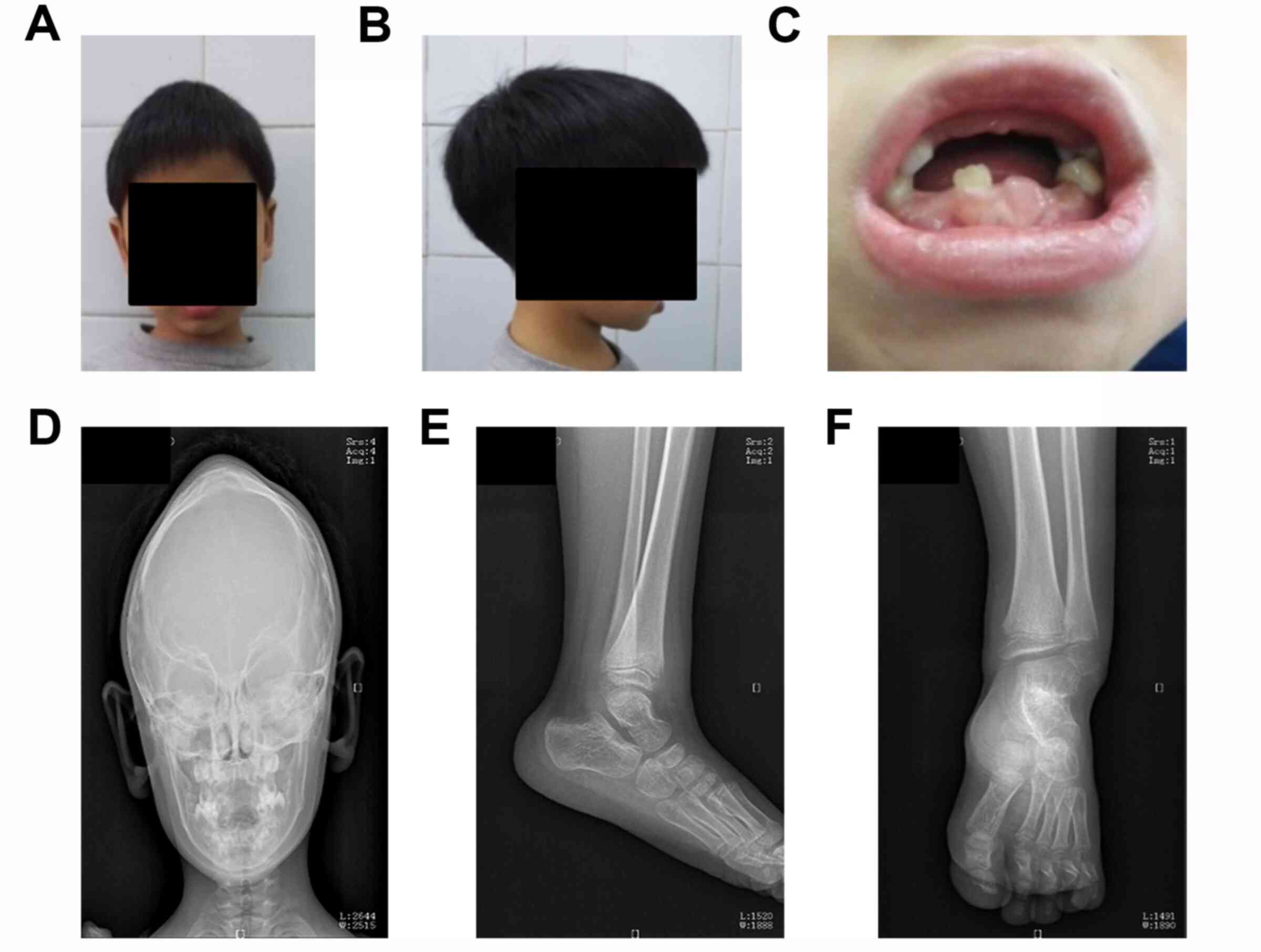
The results of the physical examination indicated
that the proband exhibited cerebral abnormalities, premature loss
of primary teeth (Fig. 1A-C), and
mild pigeon breast. Head CT scan results of the proband when he was
1 year of age revealed that the anteroposterior diameter of the
cranial cavity was larger and biparietal diameter was shorter than
normal. In addition, the patient's forehead was extremely
prominent, while the sagittal suture was closed and the coronal
seam and lambdoidal suture were present, which indicated that
scaphocephaly resulted from premature fusion of cranial sutures
(Fig. 1D). X-ray findings of the
left ankle indicated decreased bone density and loosened bone
trabeculae. In addition, the distal tibiofibula was slightly
enlarged so that it looked like a brush and the epiphysis of the
distal fibula was less regular with soft tissue around it, causing
left ankle joint swelling (Fig. 1E
and F). Serum alkaline phosphatase
activity of the proband (III-3) was below the normal reference
range, while that of his mother (II-4) was near the threshold, and
that of his father (II-3) was normal (Table I).
 | Table IClinical data and laboratory
parameters of the HPP proband (III-1) and his parents (II-3 and
II-4). |
Table I
Clinical data and laboratory
parameters of the HPP proband (III-1) and his parents (II-3 and
II-4).
| | | | Serum | Urine |
|---|
| Name | Sex | Age | Ca (mmol/l) | P (mmol/l) | ALP (IU/l) | 24 h Ca (mmol) | 24 h P (mmol) |
|---|
| II-3 | M | 39 | 2.34 | 0.99 | 45 | 5.3 | 18.43 |
| II-4 | F | 34 | 2.34 | 1.16 | 29 | 2.2 | 18.01 |
| III-1 | M | 6 | 2.35 | 1.65 | 41 | 1.55 | 6.58 |
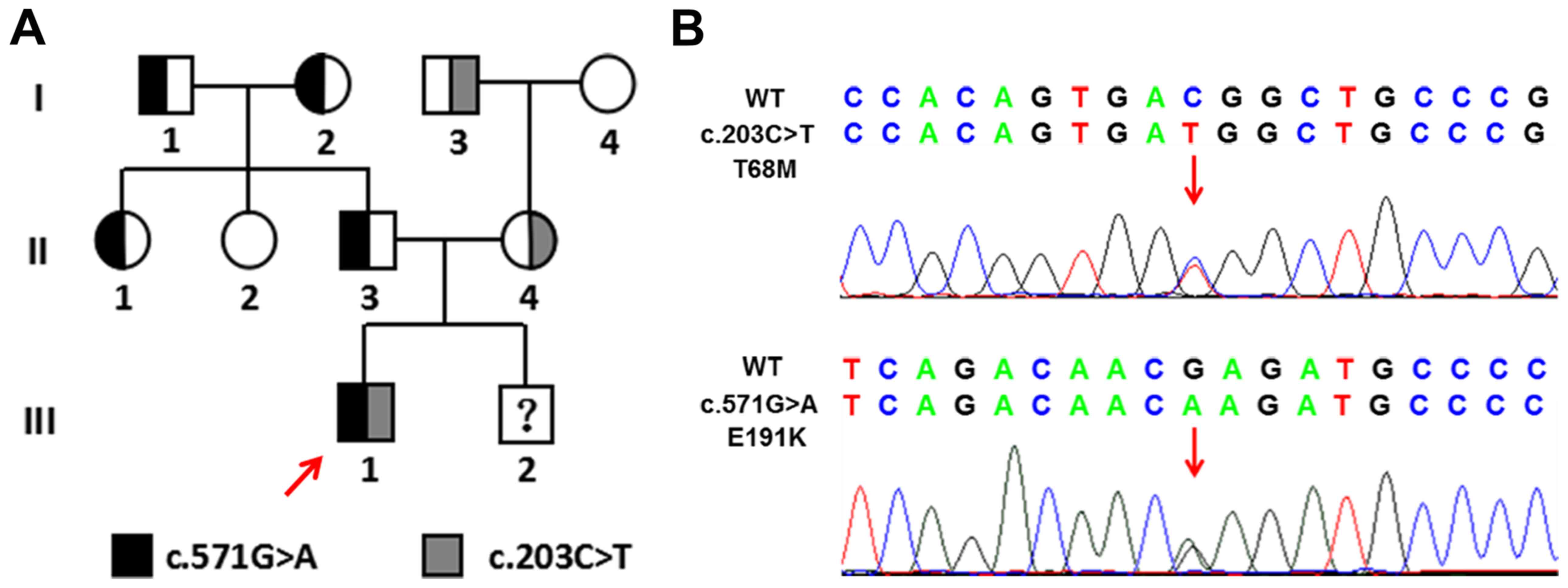
Identification of ALPL mutations
Genetic analysis of the ALPL gene from all family
members was performed, and two variants that were not indicated in
the 200 control samples were identified. The c.571G>A mutation
[GenBank accession no. NM_000478.5, p.Glu191Lys; Human Gene
Mutation Database (HGMD) accession no. CM920019] within exon 6 of
ALPL is a common pathogenic mutation which has been previously
identified (Fig. 2) (23). The c.203C>T mutation (GenBank
accession no. NM_000478.5, p.Thr68Met) within exon 4 was revealed
to be an allele associated with severe childhood HPP and is
considered to be pathogenic (HGMD accession no. CM012046) (24). The frequency of ALPL 571A allele
(ClinVar accession no. rs121918007) is 0.00247 in Genome
Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/) and 0.00258 in
Exome Aggregation Consortium (ExAC; https://gnomad.broadinstitute.org/) However, the
frequency of ALPL 203T allele (chr1:21887611) was not able to be
indicated in ExAC or gnomAD. Sanger sequencing indicated that the
proband presented both heterozygous mutations in ALPL of which the
c.571G>A in exon 6 was from his father and the c.203C>T in
exon 4 was from his mother.
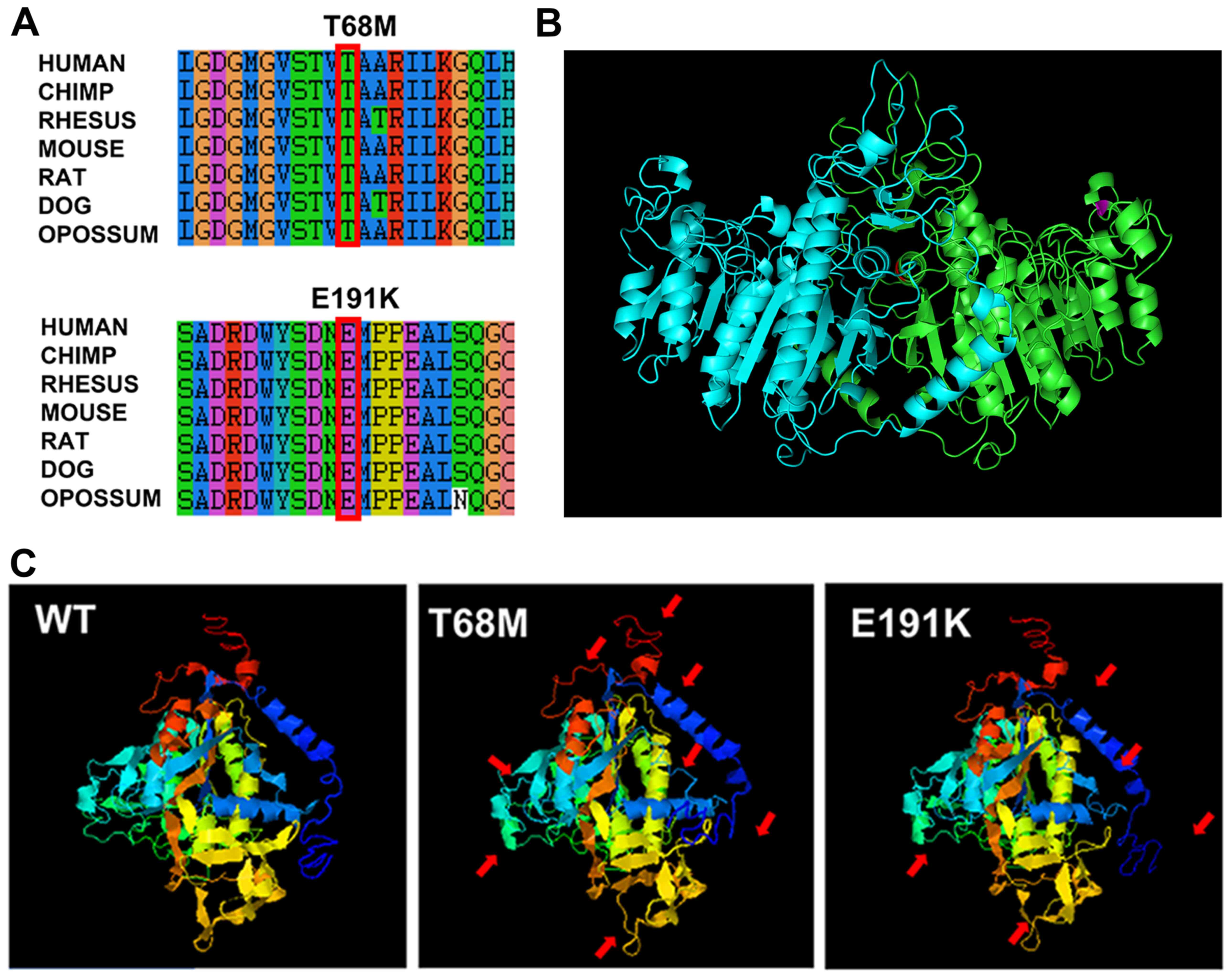
Bioinformatics analysis was subsequently performed.
Both variants are located in evolutionarily conserved regions, as
indicated by UCSC Genome Browser (Fig.
3A). These variants were predicted to be harmful to humans
according to PolyPhen-2 software and Mutation Taster analysis (data
not shown). In addition, SIFT predicted that the c.203C>T is
likely to be harmful, while c.571G>A is tolerated (data not
shown). SOPMA predicted that c.203C>T could result in alteration
of the protein secondary structure, while c.571G>A almost could
not (data not shown). The three-dimensional structure of ALPL
protein monomers predicted by I-TASSER showed a number of changes
in the mutants (red arrows; Fig.
3C). The positions of these variants were marked by red (T68M)
and purple (E191K) in the structure of the protein dimer predicted
by SWISS-MODEL, based on the homology to the placental isozyme
(Fig. 3B). The E191K residue is
located at the surface of the molecule and the T68M residue is
located at the homodimer interface.
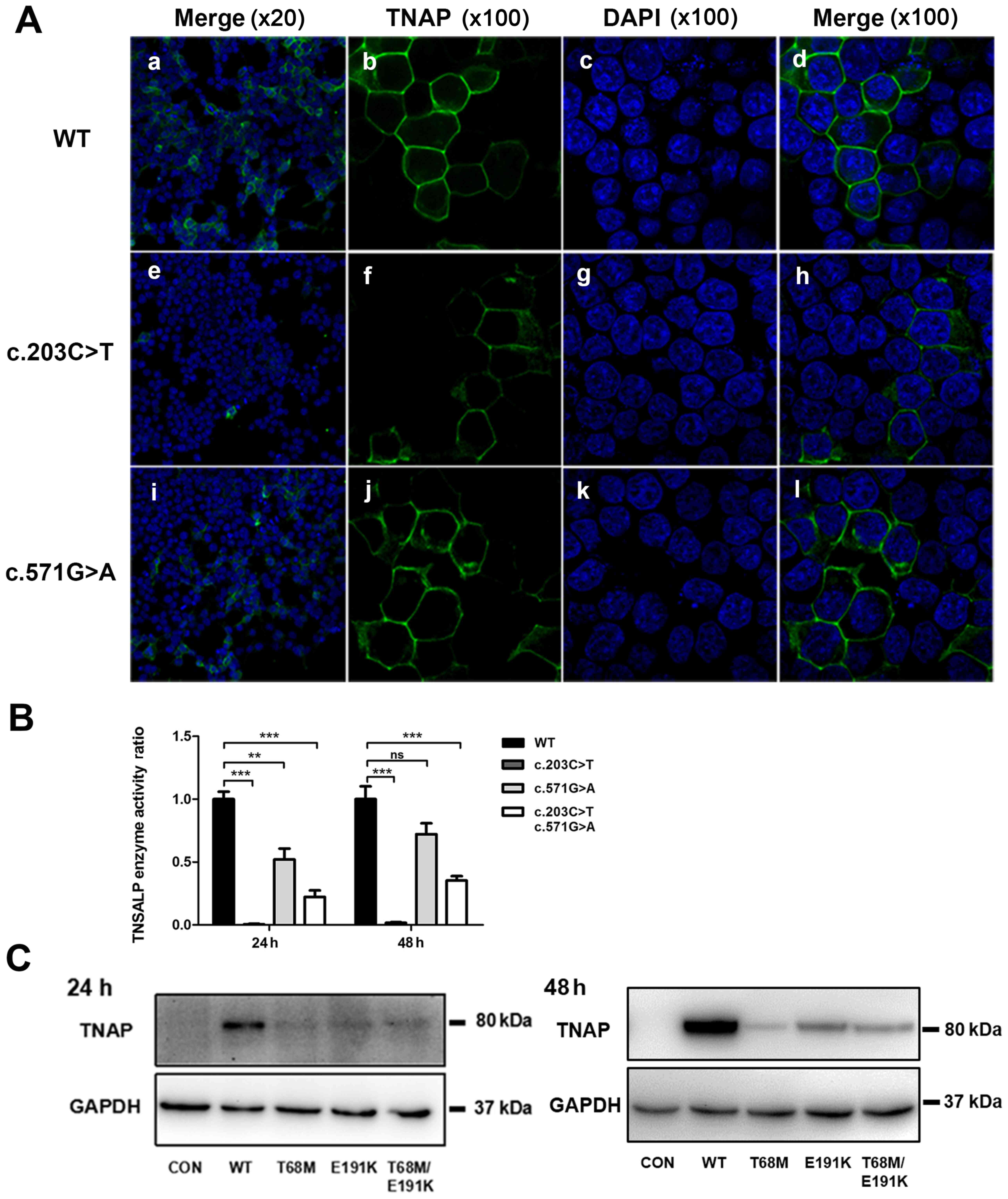
Immunofluorescence
Immunocytochemical staining of 293T cells
transfected with anti-TNAP indicated the subcellular localization
of the WT and mutated TNAP (Fig.
4). After transfection for 48 h, the WT TNAP anchored to the
surface of cells in general, which was shown by the clear
fluorescence of cell membranes. Fluorescence in cells transfected
with the ALPL-pE191K mutant was mainly located in the cell
membrane. However, staining in the cytoplasm was increased relative
to the WT. Almost no fluorescence was identified in 293T cells
transfected with the ALPL-pT68M mutant construct.
Analysis of protein expression
Western blot analysis revealed identically sized
bands of ~80 kDa in transfected cells expressing WT or mutant TNAP.
However, the expression level of mutant ALPL was markedly lower
compared with the WT, especially in cells transfected with the
ALPL-pT68M mutant construct (Fig.
4).
Enzyme activity of ALP in WT and
mutants
ALP activity in 293T cells transfected with ALPL
c.203C>T was 0.64% of WT ALPL enzyme activity after 24 h, and
1.77% after 48 h (Fig. 4). In
addition, the enzyme activity in 293T cells transfected with ALPL
c.571G>A was 52.83% after 24 h and 72.05% after 48 h. The
results suggested that enzyme activity of p.T68M was almost
completely lost and that of p.E191K was partially inactivated.
Discussion
HPP is an inborn error of metabolism characterized
by low serum ALP activity (11).
ALPL has been extensively studied as a pathogenic gene of HPP and
many mutations in the ALPL gene are related to HPP (25). More than 75% are missense mutations
and >20% are splicing mutations, nonsense mutations, small
deletions or insertions (26,27).
These loss-of-function mutations may affect the synthesis and
trafficking of TNAP, and result in diminished expression and/or
altered localization of TNAP (28-33).
The present article described a child suffering from childhood HPP,
with an onset at 1 year of age, who carried two heterozygous
mutations c.203C>T and c.571G>A in the ALPL gene. The serum
TNAP activity of the proband's mother, who carried heterozygous
c.203C>T, was near the threshold, while that of his father, who
carried heterozygous c.571G>A, was normal. The c.571G>A
mutation is widely believed to be associated with childhood
HPP.
The TNAP protein is synthesized as a 66 kDa immature
form; it is converted to an 80 kDa mature form as it migrates from
the endoplasmic reticulum to the Golgi apparatus and is finally
fixed on the cell surface as a
glycosylphosphatidylinositol-anchored protein (33,34).
The fluorescence intensity was increased in the cytoplasm of both
mutants, which showed that partial protein was not anchored to the
cytomembrane, and seemed to be more severe in T68M. A previous
study suggested that transport of E191K (c.571G>A) was delayed,
and the fluorescence signal of the E191K protein was observed in
the cytoplasm and cytomembrane after 24 h of transfection in COS-7
cells (31). The localization of
E191K protein in 24 h-transfected cells in the present study was
consistent with this observation.
Western blotting demonstrated that the mature
protein was decreased in E191K and T68M mutants compared with WT.
Levels of enzyme activity of mutant proteins expressed in 293T
cells also decreased compared with the WT. The T68M mutant
indicated almost no serum activity after 24 h and 1.77% serum
activity after 48 h. A previous study also demonstrated very low
enzyme activity (~4.5%) in the T68M mutant transiently expressed in
COS-1 cells after 48 h (24). The
results are consistent with the phenotype of family members in this
pedigree. 3D model analysis suggested that the E191K residue is
located at the surface of the molecule and may be associated with
substrate approach and stabilization. The T68M residue is located
at the homodimer interface and may serve an important role in
maintaining the correct fold to allow homodimer interactions
(35). A change in these positions
may repress the process of post-translation and cause unsuccessful
or delayed modification of the immature protein, which may then be
degraded. This might be the reason for decreased enzyme activity in
the mutants. According to previous reports, decreased TNAP activity
can cause the accumulation of inorganic phosphate, which inhibits
the formation of hydroxyapatite, thus preventing biomineralization
(16,19).
Herein, the current presented a molecular
cytogenetic characterization of the compound heterozygous
c.571G>A and c.203C>T mutations within ALPL that resulted in
decreased protein expression and loss of enzyme activity with TNAP,
leading to the childhood form of HPP in a six-year-old male
patient. Potential genotype-phenotype correlations were discussed
in the present case. The present data expanded the known ALPL
compound heterozygous mutation spectrum and may potentially provide
insight into the development of HPP.
Acknowledgements
Not applicable.
Funding
This study and publication costs were financially
supported from the National Natural Science Foundation of China
(grant no. 31970558), Natural Science Foundation of Guangdong
Province (grant no. 2018B030311033), Science and Technology
Planning Project of Guangdong (grant no. 2019A030317019) and
Science and Technology Planning Project of Guangzhou (grant no.
201707010301).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW participated in clinical sampling and data
collection. FX and XX designed the experiments. YL, XZ and XW
performed genomic DNA isolation and mutation screening. HH, DG and
HS performed the functional studies. HH and FX wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Southern Medical
University Ethics Committee. Written informed consent was obtained
from the participants and the parents of the child.
Patient consent for publication
Written informed consent was obtained from the
patient's legal guardian and his family for genetic analysis,
publication of the research and the patient's images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rathbun JC: Hypophosphatasia; a new
developmental anomaly. Am J Dis Child. 75:822–831. 1948.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Orton NC, Innes AM, Chudley AE and
Bech-Hansen NT: Unique disease heritage of the dutch-german
mennonite population. Am J Med Genet A. 146A:1072–1087.
2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Greenberg CR, Evans JA, McKendry-Smith S,
Redekopp S, Haworth JC, Mulivor R and Chodirker BN: Infantile
hypophosphatasia: Localization within chromosome region 1p36.1-34
and prenatal diagnosis using linked DNA markers. Am J Hum Genet.
46:286–292. 1990.PubMed/NCBI
|
|
4
|
Mornet E, Yvard A, Taillandier A, Fauvert
D and Simon-Bouy B: A molecular-based estimation of the prevalence
of hypophosphatasia in the European population. Ann Hum Genet.
75:439–445. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mornet E: Hypophosphatasia. Orphanet J
Rare Dis. 2(40)2007.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Whyte MP: Genetics of Bone Biology and
Skeletal Disease. Thakker RV, Whyte MP, Eisman J, Igarashi T
(eds.). 1st edition.Academic Press pp337-360, 2012.
|
|
7
|
Whyte MP, Zhang F, Wenkert D, McAlister
WH, Mack KE, Benigno MC, Coburn SP, Wagy S, Griffin DM, Ericson KL
and Mumm S: Hypophosphatasia: Validation and expansion of the
clinical nosology for children from 25 years experience with 173
pediatric patients. Bone. 75:229–239. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mornet E and Nunes ME: Hypophosphatasia.
In: GeneReviews®. Adam MP, Ardinger HH, Pagon RA, et
al (eds.). Seattle (WA): University of Washington, Seattle,
1993-2020.
|
|
9
|
Silver MM, Vilos GA and Milne KJ:
Pulmonary hypoplasia in neonatal hypophosphatasia. Pediatr Pathol.
8:483–493. 1988.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wenkert D, McAlister WH, Coburn SP, Zerega
JA, Ryan LM, Ericson KL, Hersh JH, Mumm S and Whyte MP:
Hypophosphatasia: Non-lethal disease despite skeletal presentation
in utero (17 new cases and literature review). J Bone Miner Res.
26:2389–2398. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Fraser D: Hypophosphatasia. Am J Med.
22:730–746. 1957.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Whyte MP, Greenberg CR, Salman NJ, Bober
MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS,
Bauer ML, et al: Enzyme-replacement therapy in life-threatening
hypophosphatasia. N Engl J Med. 366:904–913. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Whyte MP, Teitelbaum SL, Murphy WA,
Bergfeld MA and Avioli LV: Adult hypophosphatasia: Clinical,
laboratory, and genetic investigation of a large kindred with
review of the literature. Medicine (Baltimore). 58:329–347.
1979.PubMed/NCBI
|
|
14
|
Sutton RAL, Mumm S, Coburn SP, Ericson KL
and Whyte MP: Atypical femoral fractures' during bisphosphonate
exposure in adult hypophosphatasia. J Bone Miner Res. 27:987–994.
2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Khandwala HM, Mumm S and Whyte MP: Low
serum alkaline phosphatase activity and pathologic fracture: Case
report and brief review of hypophosphatasia diagnosed in adulthood.
Endocr Pract. 12:676–681. 2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Millan JL: The role of phosphatases in the
initiation of skeletal mineralization. Calcif Tissue Int.
93:299–306. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Caswell AM, Whyte MP and Russell RG:
Hypophosphatasia and the extracellular metabolism of inorganic
pyrophosphate: Clinical and laboratory aspects. Crit Rev Clin Lab
Sci. 28:175–232. 1991.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Mornet E: Hypophosphatasia. Best Pract Res
Clin Rheumatol. 22:113–127. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Whyte MP: Physiological role of alkaline
phosphatase explored in hypophosphatasia. Ann N Y Acad Sci.
1192:190–200. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Saraff V, Narayanan VK, Lawson AJ, Shaw
NJ, Preece MA and Högler W: A diagnostic algorithm for children
with low alkaline phosphatase activities: Lessons learned from
laboratory screening for hypophosphatasia. J Pediatr.
172:181–186.e1. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cole DE: Hypophosphatasia update: Recent
advances in diagnosis and treatment. Clin Genet. 73:232–235.
2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Combet C, Blanchet C, Geourjon C and
Deléage G: NPS@: Network protein sequence analysis. Trends Biochem
Sci. 25:147–150. 2000.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Henthorn PS, Raducha M, Fedde KN, Lafferty
MA and Whyte MP: Different missense mutations at the
tissue-nonspecific alkaline phosphatase gene locus in autosomal
recessively inherited forms of mild and severe hypophosphatasia.
Proc Natl Acad Sci USA. 89:9924–9928. 1992.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Orimo H, Girschick HJ, Goseki-Sone M, Ito
M, Oda K and Shimada T: Mutational analysis and functional
correlation with phenotype in german patients with childhood-type
hypophosphatasia. J Bone Miner Res. 16:2313–2319. 2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Weiss MJ, Cole DE, Ray K, Whyte MP,
Lafferty MA, Mulivor RA and Harris H: A missense mutation in the
human liver/bone/kidney alkaline phosphatase gene causing a lethal
form of hypophosphatasia. Proc Natl Acad Sci USA. 85:7666–7669.
1988.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mornet E, Hofmann C, Bloch-Zupan A,
Girschick H and Merrer ML: Clinical utility gene card for:
Hypophosphatasia-update 2013. Eur J Hum Genet. 22:2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Silvent J, Gasse B, Mornet E and Sire JY:
Molecular evolution of the tissue-nonspecific alkaline phosphatase
allows prediction and validation of missense mutations responsible
for hypophosphatasia. J Biol Chem. 289:24168–24179. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fukushi-Irie M, Ito M, Amaya Y, Amizuka N,
Ozawa H, Omura S, Ikehara Y and Oda K: Possible interference
between tissue-non-specific alkaline phosphatase with an
Arg54->Cys substitution and acounterpart with an Asp277->Ala
substitution found in a compound heterozygote associated with
severe hypophosphatasia. Biochem J. 348 (Pt 3):633–642.
2000.PubMed/NCBI
|
|
29
|
Ito M, Amizuka N, Ozawa H and Oda K:
Retention at the cis-Golgi and delayed degradation of
tissue-non-specific alkaline phosphatase with an Asn153->Asp
substitution, a cause of perinatal hypophosphatasia. Biochem J.
361(Pt 3):473–480. 2002.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ishida Y, Komaru K, Ito M, Amaya Y, Kohno
S and Oda K: Tissue-nonspecific alkaline phosphatase with an
Asp(289)->Val mutation fails to reach the cell surface and
undergoes proteasome-mediated degradation. J Biochem. 134:63–70.
2003.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Brun-Heath I, Lia-Baldini AS, Maillard S,
Taillandier A, Utsch B, Nunes ME, Serre JL and Mornet E: Delayed
transport of tissue-nonspecific alkaline phosphatase with missense
mutations causing hypophosphatasia. Eur J Med Genet. 50:367–378.
2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yang H, Wang L, Geng J, Yu T, Yao RE, Shen
Y, Yin L, Ying D, Huang R, Zhou Y, et al: Characterization of six
missense mutations in the tissue-nonspecific alkaline phosphatase
(TNSALP) gene in Chinese children with hypophosphatasia. Cell
Physiol Biochem. 32:635–644. 2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Satou Y, Al-Shawafi HA, Sultana S, Makita
S, Sohda M and Oda K: Disulfide bonds are critical for
tissue-nonspecific alkaline phosphatase function revealed by
analysis of mutant proteins bearing aC(201)-Y or C(489)-S
substitution associated with severe hypophosphatasia. Biochim
Biophys Acta. 1822:581–588. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Nasu M, Ito M, Ishida Y, Numa N, Komaru K,
Nomura S and Oda K: Aberrant interchain disulfide bridge of
tissue-nonspecific alkaline phosphatase with an Arg433->Cys
substitution associated with severe hypophosphatasia. FEBS J.
273:5612–5624. 2006.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mornet E, Stura E, Lia-Baldini AS,
Stigbrand T, Ménez A and Le Du MH: Structural evidence for a
functional role of human tissue nonspecific alkaline phosphatase in
bone mineralization. J Biol Chem. 276:31171–31178. 2001.PubMed/NCBI View Article : Google Scholar
|