Introduction
Osteoarthritis (OA) is a degenerative joint disease
characterized by articular cartilage degeneration and a high
incidence rate in the elderly population (1,2).
Chondrocyte degradation and synthesis imbalance, extracellular
matrix (ECM) and subchondral bones are involved in the degradation
of articular cartilage, which leads to OA progression (3,4). OA is
a disease that affects the quality of life in the elderly worldwide
(5). However, the pathogenesis of
OA has not been fully elucidated. Thus, exploring the pathogenesis
of OA is necessary.
Long non-coding RNAs (lncRNAs) are non-coding RNAs
(ncRNAs) >200 nucleotides (nts) in length that serve crucial
roles in various biological processes, such as tumorigenesis, cell
growth, metastasis and apoptosis (6,7).
lncRNA taurine upregulated gene 1 (TUG1) has been reported to be
highly expressed and involved in the development of human diseases.
For example, Niu et al (8)
demonstrated that TUG1 was upregulated in small cell lung cancer
(SCLC), that TUG1 depletion inhibited SCLC cell growth and
metastasis, and that TUG1 induced apoptosis. Huang et al
(9) reported increased TUG1 levels
in hepatocellular carcinoma (HCC) and demonstrated that TUG1
inhibition repressed HCC progression. While various lncRNAs, such
as HOX transcript antisense RNA (10), maternally expressed gene 3(11) and X-inactive specific transcript
(12) have been demonstrated to
participate in OA regulation, the association between TUG1 and OA
remains unclear. Thus, the current study aimed to investigate the
functional role and potential mechanism of TUG1 in the progression
of OA.
MicroRNAs (miRNAs or miRs) are a series of ncRNAs
19-25 nts in length that modulate gene expression by recognizing
the 3'-untranslated region (3'-UTR) of target genes (13). Numerous studies have reported that
miRs are dysregulated in OA and serve various roles in OA
development. For instance, Huang et al (14) demonstrated that miR-204 and miR-211
maintained joint homeostasis and inhibited OA progression. Meng
et al (15) reported
decreased miR-193b-3p expression in OA, while miR-193b-3p
upregulation promoted cartilage formation by interacting with
histone deacetylase 3. However, Cai et al (16) determined that miR-27a expression was
elevated in OA cartilage tissues and interleukin (IL)-1β-induced
chondrocytes and facilitated chondrocyte apoptosis. Furthermore,
Yang et al (17) reported
elevated miR-145 expression in IL-1β-stimulated chondrocytes,
leading to ECM degradation in OA cartilage. These data suggested
that miRNAs served different roles in OA development.
A previous study reported that miR-17-5p was
associated with OA (10). However,
the specific pathogenesis of miR-17-5p in OA has not been fully
elucidated. As lncRNAs have been reported to alter mRNA expression
as mRNA targets (18), further
research is required to establish whether TUG1 targets
miR-27-5p.
Fucosyltransferases (FUTs) are a group of
glycosylation synthetases (19).
Previous studies have revealed that certain FUT genes may serve
important roles in OA. For example, FUT4 was abnormally expressed
and reportedly involved in IL-1β-mediated ECM regulation,
chondrocyte growth and apoptosis in OA (20). FUT2 expression was also reportedly
elevated in OA and aggravated OA progression by impairing the ECM,
promoting cell apoptosis and inhibiting cell growth (10). To the best of our knowledge, there
are few reports regarding FUT1 in OA and few have assessed whether
miR-17-5p targets FUT1.
In the current study, TUG1, miR-17-5p and FUT1
expression was determined in OA and their role and mechanism of
action in the regulation of ECM degradation, cell viability and
apoptosis.
Materials and methods
Tissue collection
The current study was approved by the Ethics
Committee of Hanyang Hospital affiliated with Wuhan University of
Science and Technology. Written informed consent was obtained from
all participants.
A total of 25 OA cartilage tissue samples (10 male
and 15 female; age, 52-70 years) were collected from patients who
had undergone knee or hip arthroplasty and 25 normal cartilage
tissue samples (12 male and 13 female; age, 35-46 years) were
obtained from patients who underwent traumatic amputations at
Hanyang Hospital between October 2016 and March 2018. OA patients
were diagnosed according to the American College of Rheumatology
criteria (21). The control
individuals had no history of joint disease. At the time of
surgery, the patients had symptomatic disease requiring medical
treatment. None had received intra-articular steroid injections
within 3 months prior to surgery. Samples were stored at -80˚C for
total RNA and protein extraction.
Cell culture
Chondrocytes were collected from OA cartilage
tissues and cut into small sections (<1 mm3). Samples
were pre-treated with 0.25% trypsin (Beijing Solarbio Science &
Technology Co., Ltd.) for 10 min and then digested with 0.2%
collagenase II (Beijing Solarbio Science & Technology Co.,
Ltd.) in DMEM (Nissui Pharmaceutical, Co., Ltd.) supplemented with
10% FBS (Beijing Solarbio Science & Technology Co., Ltd.)
overnight at 37˚C. Undigested samples were removed with a filter
(40 µm; Beijing Solarbio Science & Technology Co., Ltd.).
Chondrocytes were collected following centrifugation at 2,000 x g
for 5 min at 37˚C.
All chondrocytes were cultured in DMEM (Nissui
Pharmaceutical, Co., Ltd.) supplemented with 10% FBS (Beijing
Solarbio Science & Technology Co., Ltd.) and 1%
penicillin-streptomycin (Beijing Solarbio Science & Technology
Co., Ltd.) at 37˚C in a humidified atmosphere with 5%
CO2. Chondrocytes at passages 2 and 3 were analyzed.
Cell transfection and IL-1β
treatment
Small interfering RNA (siRNA) targeting TUG1
(si-TUG1; 5'-CCAUCUCACAAGGCUUCAATT-3'), FUT1 (si-FUT1;
5'-UCGAUGUUUUCUUUACACCAC-3') and controls (si-NC;
5'-UUCUCCGAACGUGUCACGUTT-3'); the pcDNA3.1-TUG1 overexpression
vector (pc-TUG1) and corresponding empty vector (vector); miR-17-5p
mimics (miR-17-5p; 5'-CAAAGUGCUUACAGUGCAGGUAG-3') and controls
(miR-NC; 5'-UUCUCCGAACGUGUCACGUTT-3'); and miR-17-5p inhibitors
(anti-miR-17-5p; 5'-CAAAGUGCUUACAGUGCAGGUAG-3') and controls
(anti-miR-NC; 5'-CAGUACUUUUGUGUAGUACAA-3') were purchased from
GeneCopoeia, Inc. Chondrocytes were seeded into 24-well plates at a
density of 1.0x104 cells/well and the oligonucleotides
(50 nM) or vectors (2 µg) were transfected into chondrocytes using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.).
Chondrocytes were stimulated with 10 ng/ml IL-1β
(Beyotime Institute of Biotechnology) for 24 h at 37˚C. Untreated
normal primary chondrocytes were used as controls.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cartilage tissue samples and chondrocytes were lysed
with RNAiso Plus (Takara Biotechnology Co., Ltd.) to extract total
RNA and concentrations were measured using a NanoDrop 2000c
spectrophotometer (Thermo Fisher Scientific, Inc.). RNA was reverse
transcribed into cDNA using the HiScript® II Reverse
Transcriptase kit (Vazyme Biotech Co., Ltd.) under the thermal
conditions of 50˚C for 15 min followed by 85˚C for 5 sec or miRNA
using the 1st Strand cDNA Synthesis kit (Vazyme Biotech Co., Ltd.)
under the thermal conditions of 25˚C for 5 min, 50˚C for 15 min
followed by 85˚C for 5 min. RT-qPCR was performed using a AceQ
Universal SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd.) on an ABI
7500 PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycling conditions of the qPCR reaction were: i)
Initial denaturation at 95˚C for 5 min; ii) 40 cycles of 95˚C for
10 sec and 60˚C for 30 sec; iii) 95˚C for 15 sec, 60˚C for 60 sec
and 95˚C for 15 sec. TUG1, FUT1 and miR-17-5p expression was
calculated using the 2-ΔΔCq method
(22) with GAPDH or U6 as controls.
The primers were as follows: TUG1 forward,
5'-TAGCAGTTCCCCAATCCTTG-3' and reverse, 5'-CACAAATTCCCATCATTCCC-3';
miR-17-5p forward, 5'-TGCGCCAAAGTGCTTACAGTGCA-3' and reverse,
5'-CCAGTGCAGGGTCCGAGGTATT-3'; FUT1 forward,
5'-AAAGCGGACTGTGGATCT-3' and reverse, 5'-GGACACAGGATCGACAGG-3';
GAPDH forward, 5'-TGTTCGTCATGGGTGTGAAC-3' and reverse,
5'-ATGGCATGGACTGTGGTCAT-3'; U6 forward, 5'-CTTCGGCAGCACATATACT-3'
and reverse, 5'-AAAATATGGAACGCTTCACG-3'.
Cell counting kit-8 (CCK-8) assay
Chondrocyte viability was assessed using a CCK-8
assay following transfection and treatment. Collected chondrocytes
(5.0x103 cells/well) were seeded into 96-well plates and
10 µl CCK-8 reagent (Beyotime Institute of Biotechnology) was added
to each well at 48 h. After incubation for 4 h at 37˚C and 5%
CO2, absorbance at 450 nm was determined using a
microplate reader (BioTek Instruments, Inc.).
Flow cytometry analysis
Chondrocyte apoptosis was evaluated using an Annexin
FITC/PI Apoptosis Detection kit (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions
following transfection and treatment. Chondrocytes were harvested,
washed with PBS (Beijing Solarbio Science & Technology Co.,
Ltd.), resuspended at a concentration of 1.0x106
cells/ml and incubated with 5 µl Annexin V-FITC and 10 µl PI for 10
min at room temperature in the dark. Chondrocyte apoptotic rate was
analyzed using a FACScan® flow cytometer (BD
Biosciences) for 1 h. The level of apoptotic cells were analyzed
using FlowJo 7.6.1. (FlowJo LLC). Apoptosis rate was calculated as
the sum of the early apoptosis rate (the lower right quadrant) and
the late apoptosis rate (the upper right quadrant).
Western blot assay
Total protein was extracted by lysing cartilage
tissues and chondrocytes with RIPA buffer (Beyotime Institute of
Biotechnology) and quantified using a NanoDrop 2000c
spectrophotometer (Thermo Fisher Scientific, Inc.). Protein samples
(30 µg/lane) were subjected to 10% SDS gel (Beijing Solarbio
Science & Technology Co., Ltd.) and transferred onto
polyvinylidene difluoride membranes (Pall Life Sciences). Membranes
were blocked with 5% non-fat milk for 1 h at room temperature and
then incubated with primary antibodies against FUT1 (cat. no.
bs-7636R; BIOSS; 1:200), matrix metalloprotein 13 (MMP13; cat. no.
bs-10250R; BIOSS; 1:300), collagen II (cat. no. bs-11929R; BIOSS;
1:1,000), aggrecan (cat. no. ab3778; Abcam; 1:100) or GAPDH (cat.
no, ab9485; Abcam; 1:2,000) overnight at 4˚C. Samples were then
incubated with horseradish peroxidase-conjugated secondary
antibodies (cat. no. bs-40296G; BIOSS; 1:5,000) for 1 h at room
temperature. Bands were detected using an enhanced
chemiluminescence kit (Beyotime Institute of Biotechnology)
according to the instructions of manufacturer and analyzed using
ImageJ v1.8.0 (National Institutes of Health).
Dual-luciferase reporter assay
Potential binding sites between TUG1 and miR-17-5p,
and miR-17-5p and FUT1 were predicted by online software MIRcode 11
(http://www.mircode.org/) and starBase2.0
(http://starbase.sysu.edu.cn/index.php) and then
verified using a dual-luciferase reporter assay. TUG1 and FUT1 wild
type (WT) 3'-UTR sequences containing potential miR-17-5p binding
sites and the sequences of TUG1 and FUT1 mutant (MUT) 3'-UTR, with
the binding site removed, were cloned into a pGL3 vector (Promega
Corporation) to generate TUG1 WT, FUT1 WT, TUG1 MUT and FUT1 MUT
luciferase reporter vectors. Chondrocytes were then seeded into
24-well plates (5.0x104 cells/well) and co-transfected
with indicated vectors (100 ng) and miR-17-5p or miR-NC (50 nM)
utilizing Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Cells were gathered after 48 h and a
dual-luciferase reporter assay kit (Promega Corporation) was used
to detect luciferase activity according to the instructions of
manufacturer. Renilla luciferase activity was normalized to
firefly luciferase activity.
RNA immunoprecipitation (RIP)
assay
An RIP assay was performed to determine whether TUG1
was expressed in the RNA-induced silencing complex using a Magna
RNA-binding protein immunoprecipitation kit (EMD Millipore).
Chondrocytes (2.0x105 cells) were collected, lysed in
RIP buffer (Beijing Solarbio Science & Technology Co., Ltd.)
and incubated with magnetic beads (cat. no. 88847; Thermo Fisher
Scientific, Inc.) conjugated with anti-argonaute-2 (anti-Ago2; cat.
no. ab32381; Abcam; 2 µg/ml) or anti-immunoglobulin G (IgG; cat.
no. ab133470; Abcam; 1:5,000) overnight at 4˚C following incubation
with Proteinase K (Beijing Solarbio Science & Technology Co.,
Ltd.) at 55˚C for 30 min. Purified RNA was isolated from the
magnetic beads using RNAiso Plus (Takara Biotechnology Co., Ltd.)
and analyzed via the aforementioned RT-qPCR assay to determine TUG1
and miR-17-5p enrichment.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software (version 7; GraphPad Software, Inc.). All
experiments were performed in triplicate and data were presented as
the mean ± standard deviation. Paired Student's t-test or one-way
ANOVA followed by Tukey's test was used for analysis between
groups. Spearman's correlation analysis was used to analyze the
association between TUG1, miR-17-5p and FUT1 in OA cartilage
tissue. P<0.05 was considered to indicate a statistically
significant difference.
Results
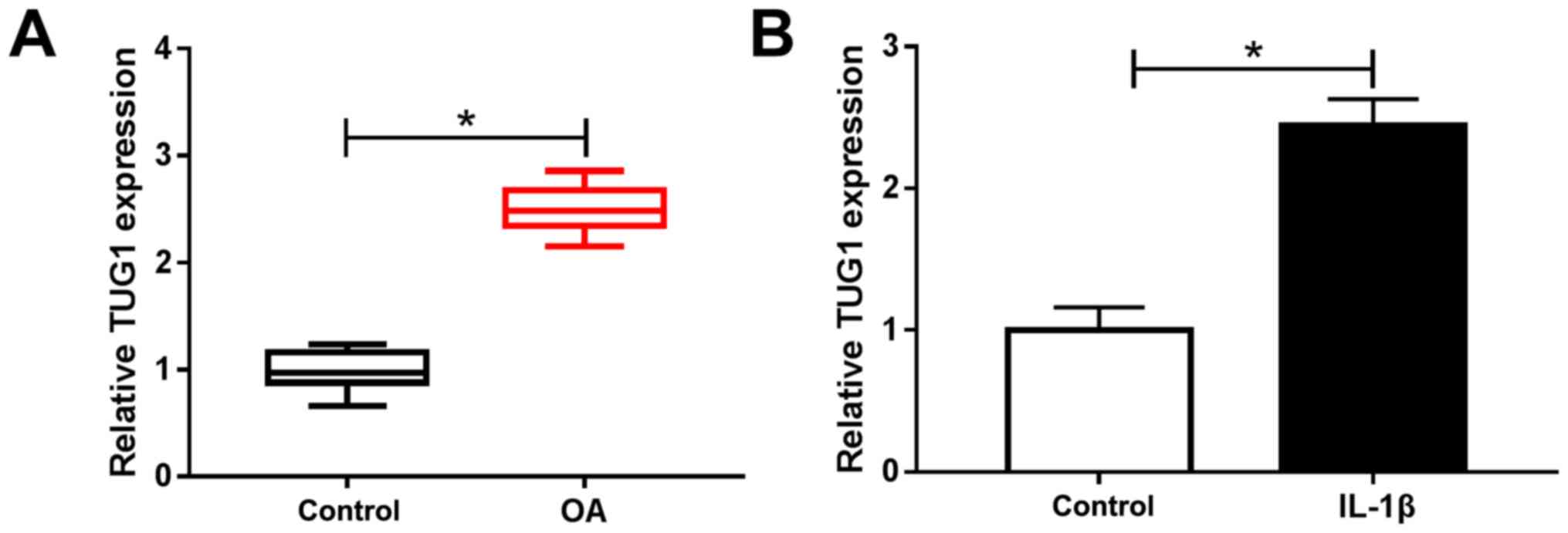
TUG1 is highly expressed in OA
cartilage tissue and IL-1β-induced chondrocytes
RT-qPCR was performed to analyze TUG1 expression in
OA cartilage tissue and normal cartilage tissue (control) and to
determine the potential role of TUG1 in the development of OA. The
results demonstrated that TUG1 expression was significantly
increased in OA cartilage tissue and IL-1β-activated chondrocytes
compared with controls (Fig. 1A and
B). These data indicated that
abnormal TUG1 expression may be involved in OA development.
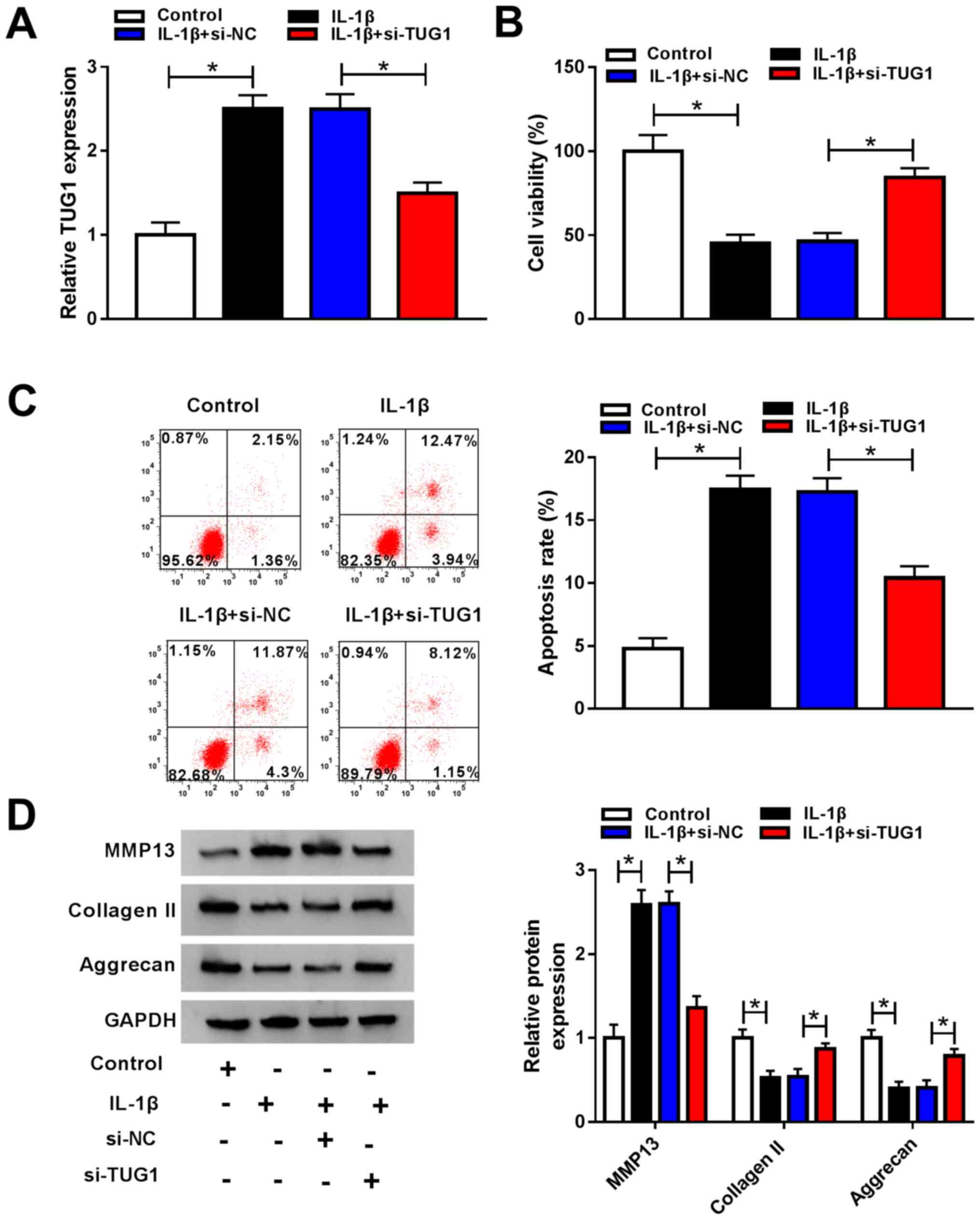
TUG1 knockdown promotes IL-1β-induced
cell viability and inhibits cell apoptosis and ECM degradation in
chondrocytes
Chondrocytes were transfected with si-TUG1 or si-NC
and treated with IL-1β to investigate the functional role of TUG1
in cell viability, apoptosis and ECM degradation in OA. RT-qPCR
demonstrated that IL-1β-induced TUG1 upregulation was markedly
decreased in chondrocytes transfected with si-TUG1 compared with
the si-NC transfected group (Fig.
2A). Chondrocyte viability was significantly inhibited and cell
apoptosis was promoted by IL-1β treatment in comparison with
control, with TUG1 knockdown partially reversing these effects, as
demonstrated by the CCK-8 and flow cytometry assays, respectively
(Fig. 2B and C). Furthermore, western blotting was
performed to determine MMP13, collagen II and aggrecan expression.
The results demonstrated that MMP13 expression was significantly
increased following IL-1β treatment, while collagen II and aggrecan
were significantly decreased, with TUG1 knockdown partially
reversing these effects (Fig. 2D).
These data suggested that IL-1β treatment decreased cell viability
and promoted cell apoptosis and ECM degradation, and that TUG1
knockdown abrogated these effects.
TUG1 negatively modulates miR-17-5p
expression via direct interaction in chondrocytes
MIRcode11 online software was used to determine
potential TUG1 binding sites to investigate the underlying
mechanism of TUG1 in OA. The results indicated that miR-17-5p was a
predicted target of TUG1 (Fig. 3A)
and that miR-17-5p transfection led to a significant increase in
miR-17-5p expression, indicating that miR-17-5p was successfully
transfected (Fig. 3B). Furthermore,
the dual-luciferase reporter assay demonstrated that luciferase
activity in TUG1 WT and miR-17-5p co-transfected chondrocytes was
significantly decreased compared with TUG1 WT and miR-NC. Activity
was unaltered in the TUG1 MUT group (Fig. 3C). The RIP assay indicated that TUG1
and miR-17-5p were significantly expressed in anti-Ago2
immunoprecipitates compared with anti-IgG (Fig. 3D).
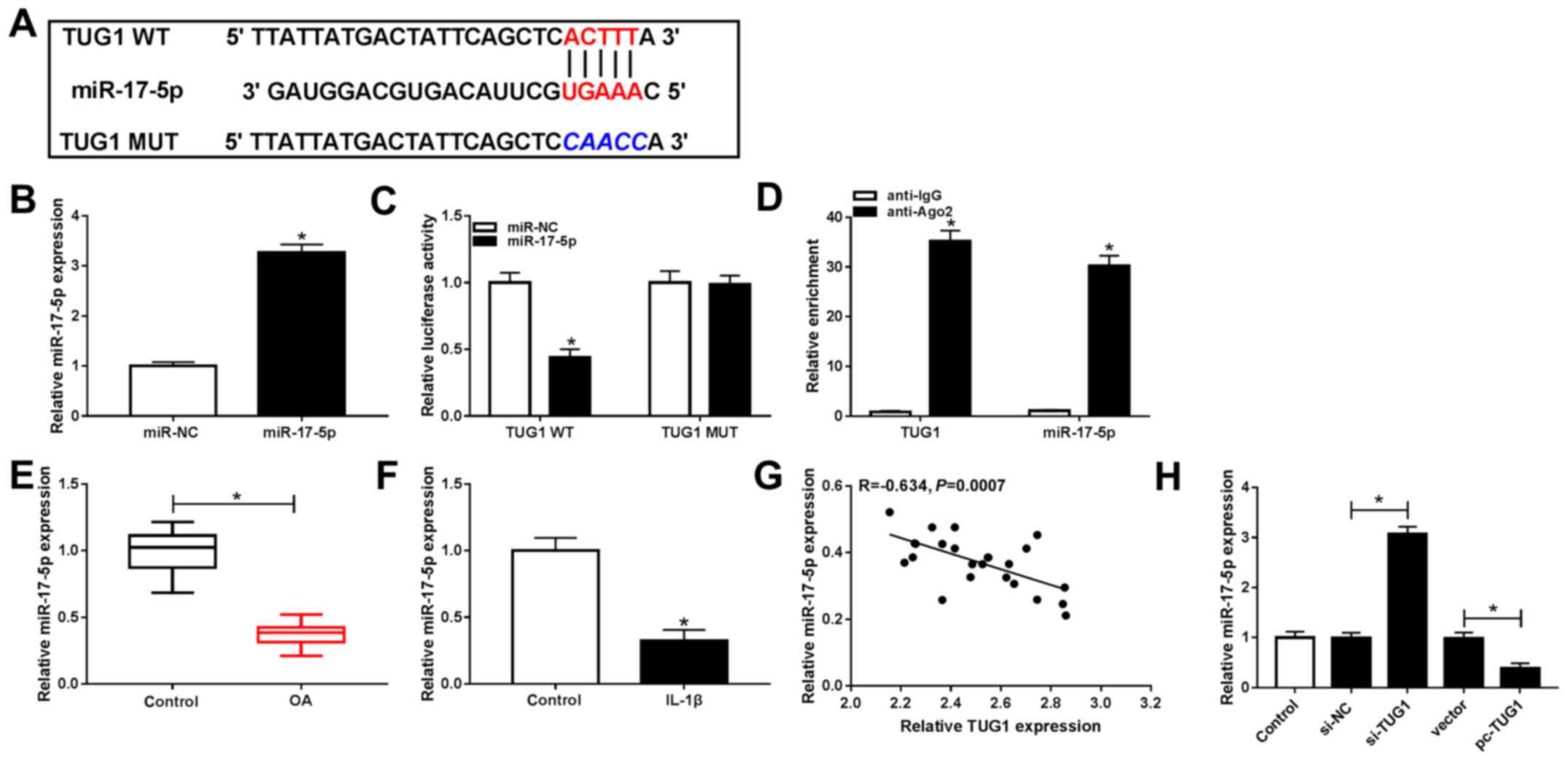 | Figure 3TUG1 negatively regulated miR-17-5p
expression via direct targeting in chondrocytes. (A) Potential
binding sites between TUG1 and miR-17-5p. (B) miR-17-5p expression
following transfection with miR-17-5p or miR-NC was determined via
RT-qPCR. (C) Luciferase activity in TUG1 WT or TUG1 MUT and
miR-17-5p and miR-NC co-transfected chondrocytes was analyzed using
a dual-luciferase reporter assay. (D) An RNA immunoprecipitation
assay was performed to assess the association between TUG1 and
miR-17-5p, and expressions were determined via RT-qPCR. (E)
miR-17-5p expression in OA cartilage tissues and controls was
measured via RT-qPCR. (F) miR-17-5p expression in IL-1β-stimulated
chondrocytes and controls was examined via RT-qPCR. (G) Correlation
between the expression of TUG1 and miR-17-5p in OA cartilage
tissues was analyzed via Spearman's correlation analysis. (H)
miR-17-5p expression in untransfected or transfected chondrocytes
with si-NC, si-TUG1, vector or pc-TUG1 was determined via RT-qPCR.
*P<0.05 vs. the miR-NC group, *P<0.05
vs. the anti-IgG group, *P<0.05 vs. the Control
groups, *P<0.05 vs. the si-NC groups,
*P<0.05 vs. the Vector groups. TUG1, taurine
upregulated gene 1; miR, microRNA; miR-NC, microRNA negative
control; RT-qPCR, reverse transcription quantitative-PCR; WT, wild
type; MUT, mutant; OA, osteoarthritis; IL, interleukin; siRNA,
small interfering RNA;. si-NC, small interfering RNA negative
control; si-TUG1, small interfering RNA targeting TUG1. |
Furthermore, miR-17-5p expression in OA cartilage
tissues and IL-1β-stimulated chondrocytes was assessed using
RT-qPCR. It was revealed that miR-17-5p expression was
significantly decreased in OA cartilage tissues and
IL-1β-stimulated chondrocytes compared with controls (Fig. E and
F). miR-17-5p was also revealed to be negatively correlated with
TUG1 in OA cartilage tissues, as demonstrated by Spearman's
correlation analysis (Fig. 3G).
Additionally, TUG1 knockdown resulted in significantly increased
miR-17-5p expression, while TUG1 overexpression resulted in the
significant inhibition of miR-17-5p expression in chondrocytes
(Fig. 3H). The results revealed
that TUG1 directly targeted miR-17-5p and negatively modulated
miR-17-5p expression in chondrocytes.
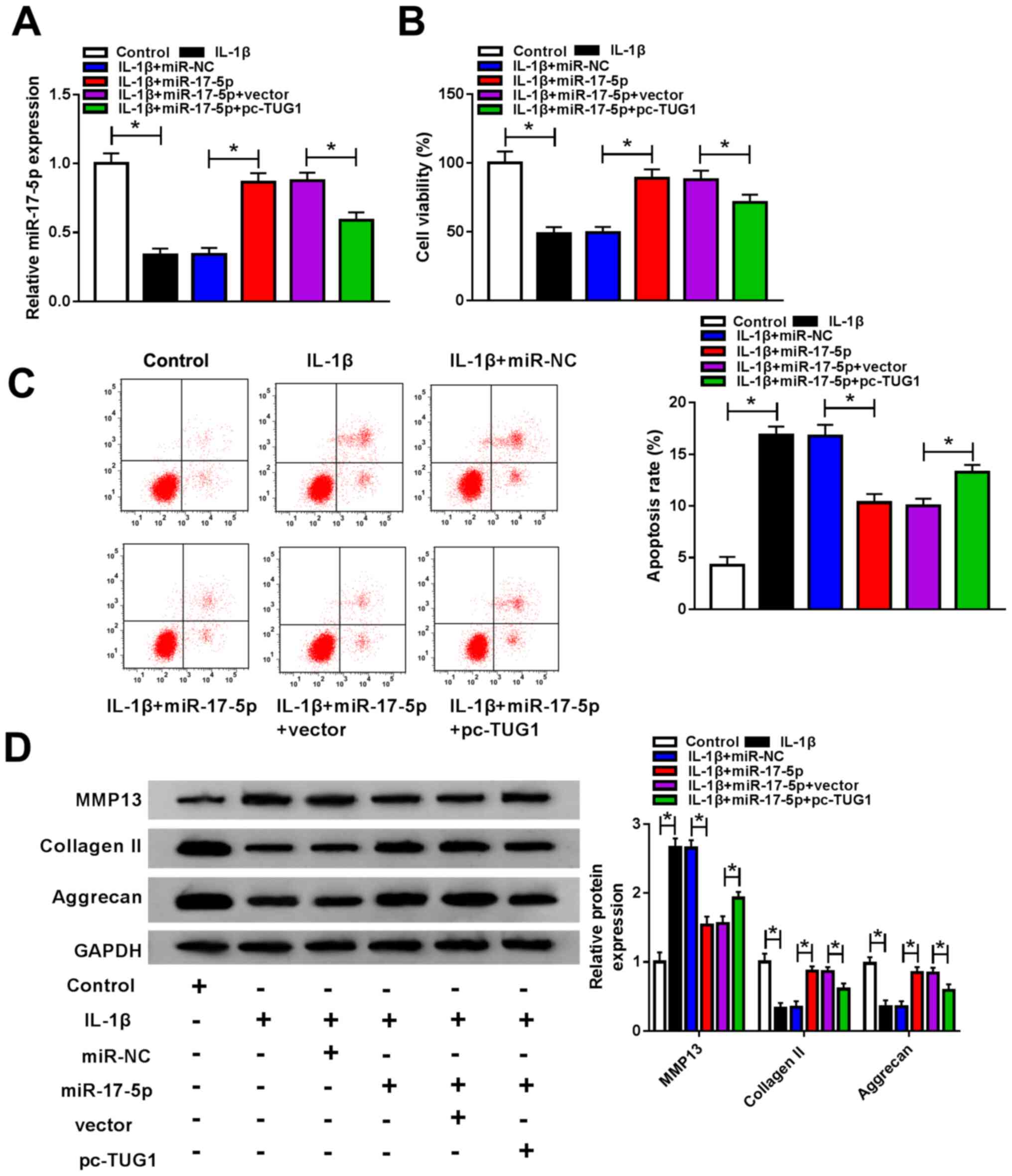
Upregulation of TUG1 attenuates the
effects of miR-17-5p overexpression on cell viability, cell
apoptosis and ECM degradation in IL-1β-stimulated chondrocytes
Chondrocytes were untransfected or transfected with
IL-1β, IL-1β + miR-NC, IL-1β + miR-17-5p, IL-1β + miR-17-5p +
vector or IL-1β + miR-17-5p + pc-TUG1 and miR-17-5p expression was
determined to investigate whether TUG1 regulated cell viability,
apoptosis and ECM degradation by targeting miR-17-5p in
IL-1β-stimulated chondrocytes. The results revealed that
IL-1β-mediated miR-17-5p downregulation was reversed by miR-17-5p
transfection and that TUG1 overexpression inhibited this effect
(Fig. 4A). IL-1β-mediated cell
viability suppression was significantly increased following
miR-17-5p transfection as demonstrated by a CCK-8 assay. However,
this increase was partly abrogated by TUG1 overexpression (Fig. 4B). Flow cytometry analysis revealed
that IL-1β increased cell apoptosis and that this was significantly
decreased by miR-17-5p transfection. However, TUG1 overexpression
reversed this effect (Fig. 4C).
Western blotting demonstrated increased MMP13 and decreased
collagen II and aggrecan expression following IL-1β treatment.
These levels were subsequently abolished by miR-17-5p and restored
by TUG1 overexpression (Fig. 4D).
In summary, miR-17-5p overexpression reversed the effect of IL-1β
on cell viability, apoptosis and ECM degradation, and these effects
were reversed by TUG1.
miR-17-5p inhibits FUT1 expression in
chondrocytes
The online software, starBase2.0, was used to
identify FUT1 as a target gene of miR-17-5p. Their potential
binding sites were established (Fig.
5A). si-FUT1 transfection resulted in significant reduction in
FUT1 mRNA and protein levels (Fig.
5B and C). The dual-luciferase
reporter assay revealed that FUT1 WT and miR-17-5p cotransfection
resulted in a significant decrease in luciferase activity, while
FUT1 MUT and miR-17-5p or miR-NC cotransfection had no effect
(Fig. 5D). FUT1 mRNA and protein OA
cartilage tissues were significantly elevated compared with
controls, as demonstrated by RT-qPCR and western blotting,
respectively (Fig. 5E and F). Furthermore, the results revealed an
inverse correlation between FUT1 and miR-17-5p in OA cartilage
tissues (Fig. 5G).
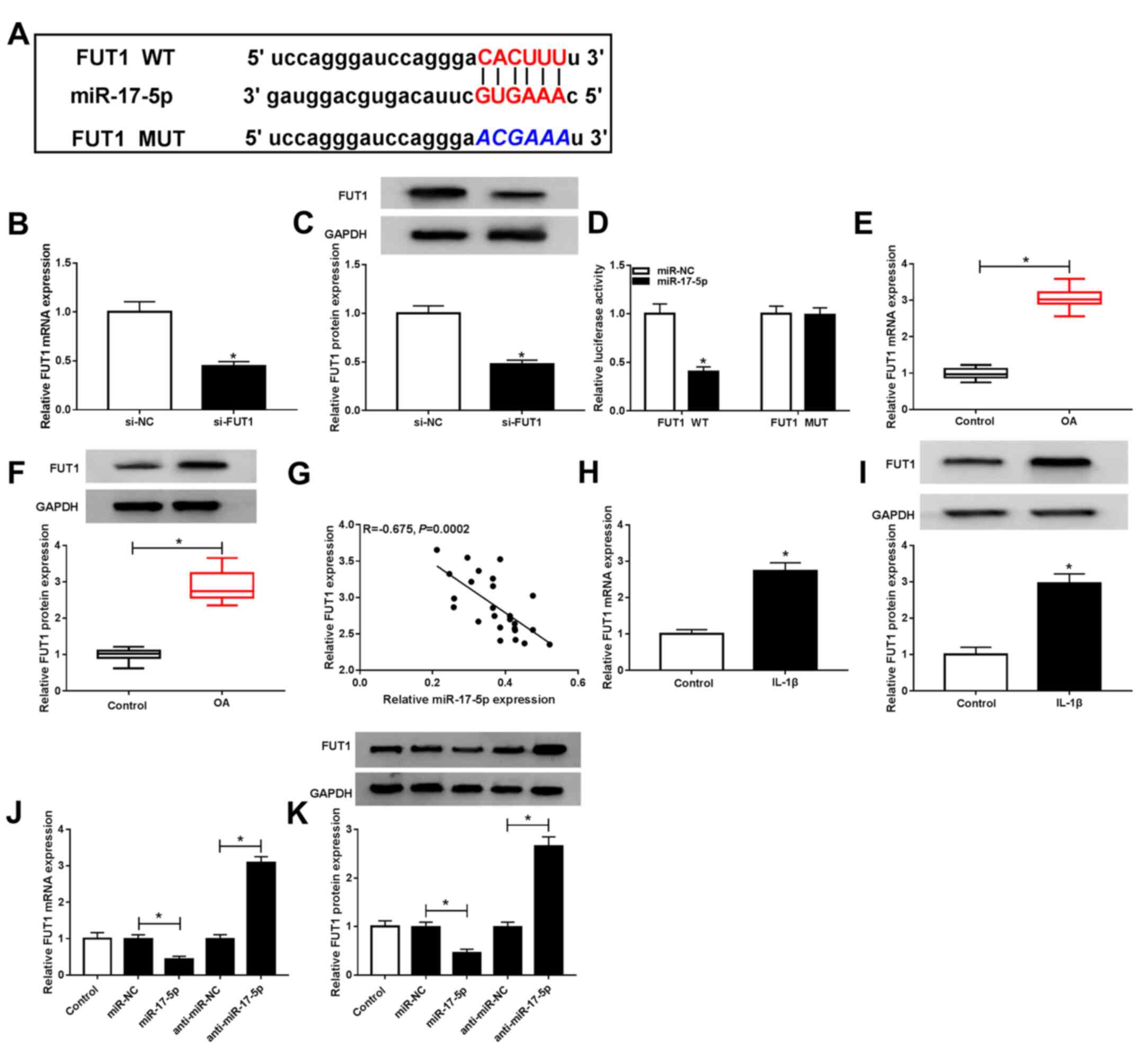 | Figure 5miR-17-5p inhibited FUT1 expression
in chondrocytes. (A) Potential binding sites between miR-17-5p and
FUT1 were predicted via starBase. si-NC or si-FUT1 was transfected
into chondrocytes and FUT1 (B) mRNA and (C) protein expressions
were determined via RT-qPCR and western blotting, respectively. (D)
Luciferase activity between miR-17-5p and FUT1 was assessed using
dual-luciferase reporter assay. FUT1 (E) mRNA and (F) protein
levels in OA cartilage tissues and controls were analyzed by
RT-qPCR and western blot analysis, respectively. (G) Correlation
between miR-17-5p and FUT1 in OA cartilage tissues was determined
via Spearman's correlation analysis. FUT1 (H) mRNA and (I) protein
levels in IL-1β-stimulated chondrocytes and controls were detected
by RT-qPCR and western blotting, respectively. Chondrocytes were
untransfected or transfected with miR-NC, miR-17-5p, anti-miR-NC or
anti-miR-17-5p, after which FUT1 (J) mRNA and (K) protein
expression was examined via RT-qPCR and western blotting,
respectively. *P<0.05 vs. si-NC,
*P<0.05 vs. miR-NC, *P<0.05 vs.
Control. miR, microRNA; FUT1, fucosyltransferase 1; siRNA, small
interfering RNA; si-NC, small interfering RNA negative control;
si-FUT1, small interfering RNA targeting FUT1; RT-qPCR, reverse
transcription quantitative-PCR; OA, osteoarthritis; IL,
interleukin; miR-NC, microRNA negative control; anti-miR-NC,
anti-microRNA negative control; anti-miR-17-5p, mir-17-5p
inhibitor. |
FUT1 mRNA and protein levels of FUT1 were increased
in IL-1β-induced chondrocytes compared with controls (Fig. 5H and I). Furthermore, miR-17-5p inhibited FUT1
mRNA and protein expression and miR-17-5p knockdown had the
opposite effect (Fig. 5J and
K). These results revealed that
miR-17-5p directly targeted FUT1 and inhibited FUT1 expression in
chondrocytes.
miR-17-5p knockdown reverses the
effects of IL-1β on cell viability, apoptosis and ECM degradation
mediated by FUT1 knockdown in IL-1β-stimulated chondrocytes
Since miR-17-5p directly regulated FUT1 expression
in chondrocytes, the effect of miR-17-5p on chondrocyte viability,
apoptosis and ECM degradation was investigated. Chondrocytes were
untransfected or transfected with IL-1β, IL-1β+si-NC, IL-1β +
si-FUT1, IL-1β + si-FUT1 + anti-miR-NC or IL-1β + si-FUT1 +
anti-miR-17-5p, after which FUT1 mRNA and protein levels were
analyzed. IL-1β treatment caused a significant increase in FUT1
mRNA and protein levels. FUT1 knockdown significantly reduced these
levels, which were partially rescued by miR-17-5p inhibition
(Fig. 6A and B). The CCK-8 assay revealed that FUT1
knockdown restored IL-1β-mediated suppression in cells. This effect
was reversed by miR-17-5p inhibition (Fig. 6C). IL-1β treatment also mediated
apoptosis. FUT1 knockdown inhibited this apoptosis; however, this
effect was significantly reversed following anti-miR-17-5p
transfection (Fig. 6D). The results
of western blotting revealed that increased levels of MMP13 and
collagen II, and decreased levels of aggrecan were rescued
following si-FUT1 transfection. However, this effect was reversed
following anti-miR-17-5p transfection (Fig. 6E). The results demonstrated that
miR-17-5p downregulation reversed the effects of FUT1 depletion on
cell viability, apoptosis and ECM degradation in IL-1β-induced
chondrocytes.
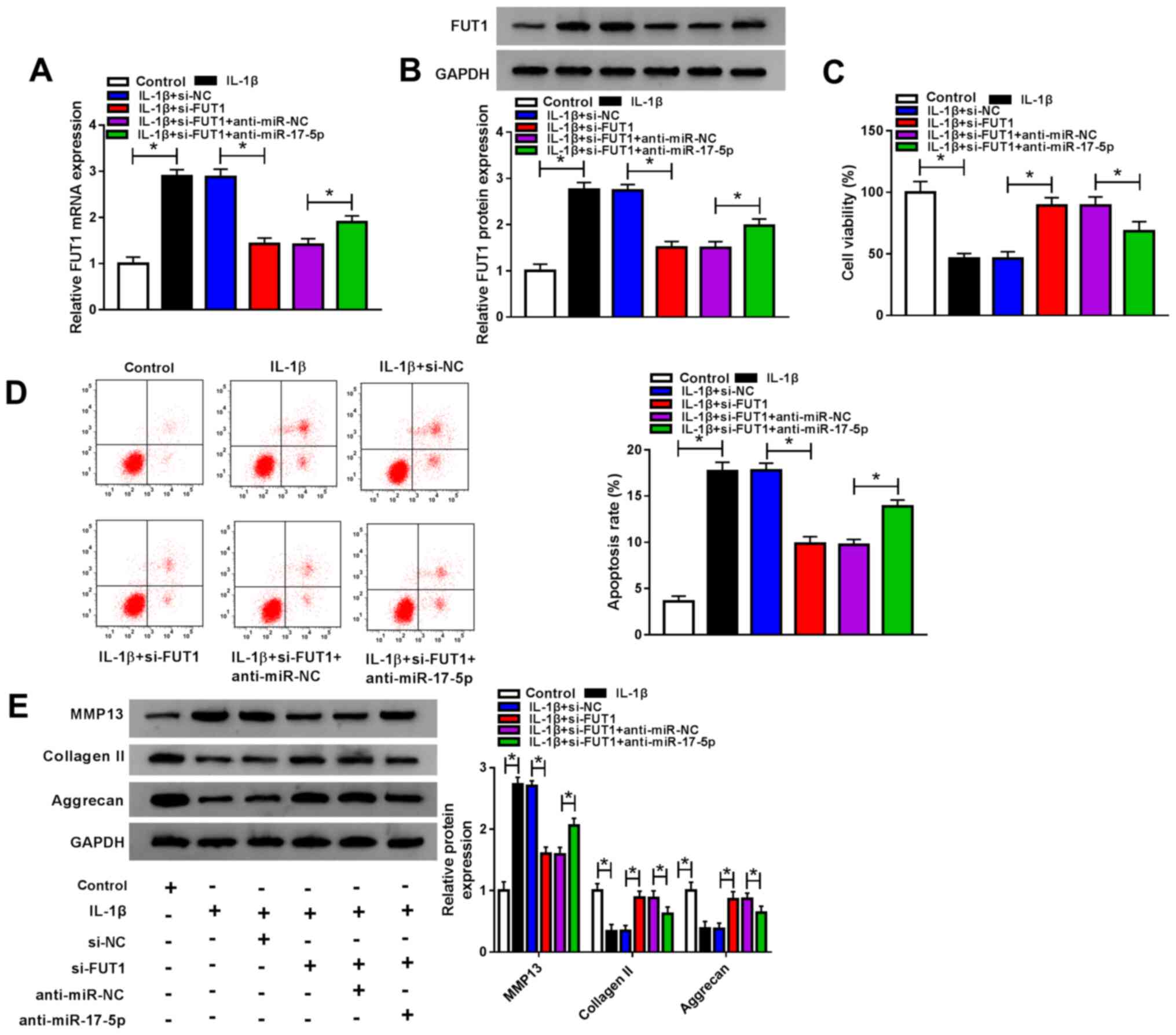 | Figure 6miR-17-5p regulated cell viability,
apoptosis and extracellular matrix degradation by targeting FUT1 in
IL-1β-induced chondrocytes. Chondrocytes were untransfected or
transfected with IL-1β, IL-1β + si-NC, IL-1β + si-FUT1, IL-1β +
si-FUT1 + anti-miR-NC or IL-1β + si-FUT1 + anti-miR-17-5p. (A) FUT1
mRNA expression was measured via reverse transcription quantitative
PCR. (B) FUT 1 protein expression was examined using western
blotting. (C) Chondrocyte viability was assessed via a Cell
Counting kit-8 assay. (D) Chondrocyte apoptosis was evaluated
through flow cytometry analysis. (E) MMP13, collagen II and
aggrecan protein levels were analyzed using western blotting.
*P<0.05 as indicated. miR, microRNA; FUT1,
fucosyltransferase 1; IL, interleukin; siRNA, small interfering
RNA; si-NC, small interfering RNA negative control; si-FUT1, small
interfering RNA targeting FUT1; anti-miR-NC, anti-microRNA negative
control; anti-miR-17-5p, miR-17-5p inhibitor; MMP13, matrix
metalloprotein 13. |
TUG1 regulates FUT1 expression by
targeting miR-17-5p in chondrocytes
si-NC, si-TUG1, si-TUG1 + anti-miR-NC or si-TUG1 +
anti-miR-17-5p were transfected into chondrocytes with
untransfected cells as controls to investigate the association
between TUG1, miR-17-5p and FUT1. FUT1 mRNA and protein levels were
significantly decreased following si-TUG1 transfection compared
with si-NC. This was reversed by anti-miR-17-5p transfection
(Fig. 7A and B). Furthermore, Spearman's correlation
analysis revealed a positive correlation between FUT1 and TUG1 mRNA
expression in OA cartilage tissues (Fig. 7C). These data indicated that TUG1
positively modulated FUT1 expression by targeting miR-17-5p in
chondrocytes.
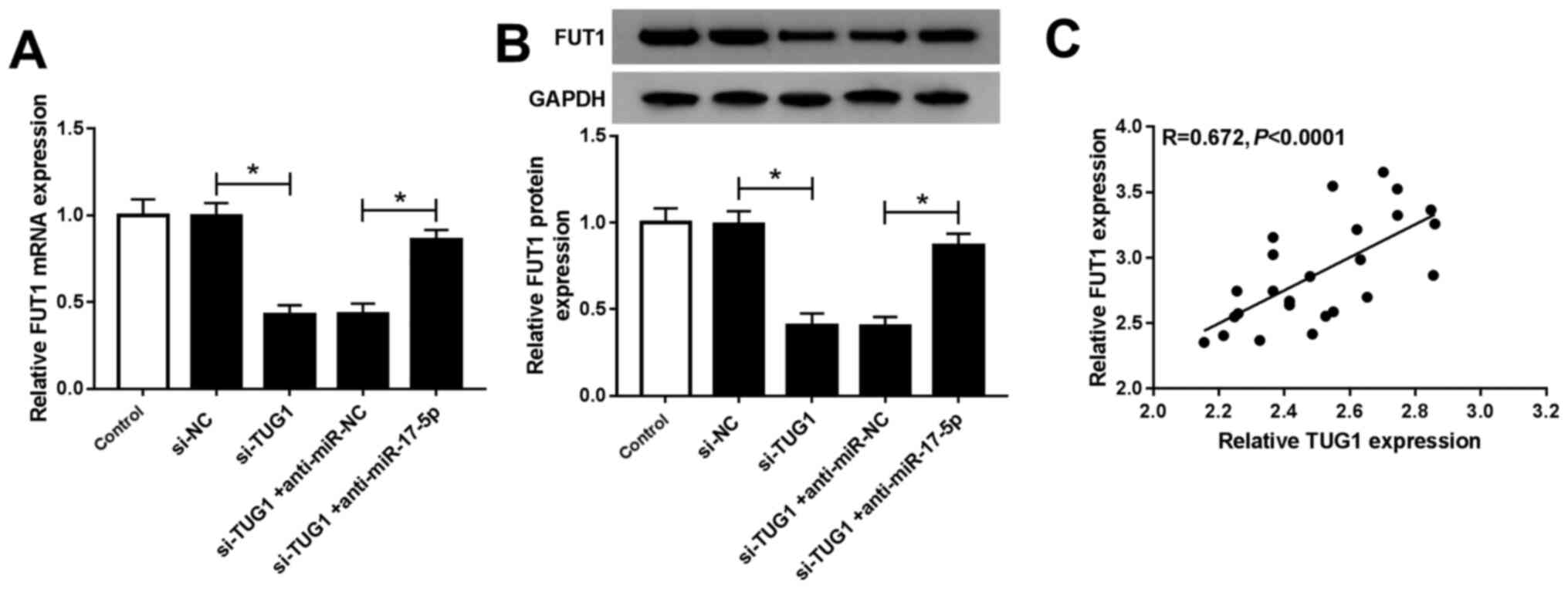 | Figure 7TUG1 upregulated FUT1 expression by
targeting miR-17-5p in chondrocytes. FUT1 (A) mRNA and (B) protein
levels in chondrocytes untransfected or transfected with si-NC,
si-TUG1, si-TUG1 + anti-miR-NC or si-TUG1 + anti-miR-17-5p were
determined using reverse transcription quantitative PCR and western
blotting, respectively. (C) Spearman's correlation analysis was
used to determine the association between FUT1 mRNA and TUG1.
*P<0.05 as indicated. TUG1, taurine upregulated gene
1; miR, microRNA; siRNA, small interfering RNA; si-NC, small
interfering RNA negative control; si-TUG1, small interfering RNA
targeting TUG1; anti-miR-NC, anti-microRNA negative control;
anti-miR-17-5p, mir-17-5p inhibitor; FUT1, fucosyltransferase
1. |
Discussion
Accumulating evidence has demonstrated that lncRNA
dysregulation serves a vital role in the pathogenesis of OA
(23,24). In the current study, the function
and mechanism of TUG1 in OA was analyzed. The results revealed that
TUG1 and FUT1 were significantly expressed and that miR-17-5p was
significantly inhibited. Additionally, TUG1 knockdown improved cell
viability and inhibited cell apoptosis and ECM degradation via the
miR-17-5p/FUT1 pathway in OA.
IL-1β impairs cartilage formation in the ECM,
induces apoptosis and represses chondrocyte growth (25-27).
In the present study, chondrocytes were exposed to IL-1β to mimic
the OA microenvironment. The results revealed that cell viability
was suppressed and cell apoptosis was induced in IL-1β-stimulated
chondrocytes. As cartilage ECM is mainly composed of collagen II
and aggrecan, and MMP13 is a typical cartilage-degrading enzyme
that serves a vital role in ECM degradation (28,29),
their levels were also evaluated. The results demonstrated
increased MMP13 and decreased collagen II and aggrecan expression,
indicating that ECM degradation was promoted.
The current study reported a significant elevation
in TUG1 expression in OA cartilage tissues and IL-1β-stimulated
chondrocytes. Liang and Ren (30) revealed that emodin increased cell
viability and inhibited cell apoptosis and inflammation via TUG1
elevation in LPS-stimulated ATDC5 cells. Tang et al
(31) demonstrated that TUG1 was
highly expressed in OA cartilage tissues and IL-1β or
TNF-α-stimulated chondrocytes, and that TUG1 promoted the
degradation of chondrocyte ECM. In the present study, TUG1
knockdown promoted IL-1β-induced chondrocyte viability and
inhibited chondrocyte apoptosis and ECM damage.
TUG1 acted as a target for miR-17-5p in
chondrocytes. miR-17-5p has been reported to be a key regulator in
various human conditions, such as triple-negative breast cancer
(32), hepatocellular carcinoma
(33) and colorectal cancer
(34). Furthermore, Hu et al
(10) demonstrated that miR-17-5p
was reduced in OA and its upregulation resulted in chondrocyte
growth and the inhibition of chondrocyte apoptosis and ECM
degradation in IL-1β-induced OA (10). In the current study, miR-17-5p was
weakly expressed in OA and its upregulation increased cell
viability and decreased cell apoptosis and ECM degradation in
IL-1β-stimulated chondrocytes. However, the effect of miR-17-5p on
OA progression was abolished by TUG1 overexpression.
Lai et al (35) demonstrated that FUT1 knockdown
repressed cell growth and metastasis in breast cancer. Kawai et
al (36) reported that FUT1
downregulation inhibited cell growth and promoted cell apoptosis in
NCL-N87 cells. These results indicated that FUT1 was involved in
the development of human disease. In the present study the function
of FUT1 in OA was investigated. FUT1 served as a target for
miR-17-5p in chondrocytes and there was an inverse correlation
between FUT1 and miR-17-5p. Furthermore, the effect of FUT1
knockdown on cell viability, apoptosis and ECM degradation were
partly reversed by the inhibition of miR-17-5p in IL-1β-activated
chondrocytes.
In conclusion, TUG1 knockdown decelerated OA
progression by promoting chondrocyte viability and repressing
chondrocyte apoptosis and cartilage ECM damage. Furthermore, TUG1
modulated OA progression via the miR-17-5p/FUT1 pathway. These
results may further elucidate OA pathogenesis and may provide a
method of effective treatment for patients with OA. However, the
current study had limitations, such as an insufficient sample size.
In addition, the IL-1β-induced chondrocytes used to simulate OA
condition may have limitations, we did not exclude additional
mechanisms such as how TUG1 might affect IL-1β. The role of the
TUG/miR-17-5p/FUT1 pathway in other processes involved in OA
development requires further investigation.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL and JY designed the current study. ZL drafted the
manuscript. JW revised the manuscript for important intellectual
content and performed statistical analysis. All authors read and
approved the final manuscript and agreed to be held accountable for
all aspects of the current work in ensuring that questions related
to the accuracy or integrity of any part of the current work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The current study was approved by the Ethics
Committee of Hanyang Hospital affiliated to Wuhan University of
Science and Technology and patients provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arden N and Nevitt MC: Osteoarthritis:
Epidemiology. Best Pract Res Clin Rheumatol. 20:3–25.
2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Michael JW, Schlüter-Brust KU and Eysel P:
The epidemiology, etiology, diagnosis, and treatment of
osteoarthritis of the knee. Dtsch Arztebl Int. 107:152–162.
2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rahmati M, Nalesso G, Mobasheri A and
Mozafari M: Aging and osteoarthritis: Central role of the
extracellular matrix. Ageing Res Rev. 40:20–30. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Van der Kraan P, Buma P, Van Kuppevelt T
and Van Den Berg W: Interaction of chondrocytes, extracellular
matrix and growth factors: Relevance for articular cartilage tissue
engineering. Osteoarthritis Cartilage. 10:631–637. 2002.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cross M, Smith E, Hoy D, Nolte S, Ackerman
I, Fransen M, Bridgett L, Williams S, Guillemin F, Hill CL, et al:
The global burden of hip and knee osteoarthritis: Estimates from
the global burden of disease 2010 study. Ann Rheum Dis.
73:1323–1330. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Johnsson P, Lipovich L, Grandér D and
Morris KV: Evolutionary conservation of long non-coding RNAs;
sequence, structure, function. Biochim Biophys Acta.
1840:1063–1071. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Niu Y, Ma F, Huang W, Fang S, Li M, Wei T
and Guo L: Long non-coding RNA TUG1 is involved in cell growth and
chemoresistance of small cell lung cancer by regulating LIMK2b via
EZH2. Mol Cancer. 16(5)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Huang MD, Chen WM, Qi FZ, Sun M, Xu TP, Ma
P and Shu YQ: Long non-coding RNA TUG1 is up-regulated in
hepatocellular carcinoma and promotes cell growth and apoptosis by
epigenetically silencing of KLF2. Mol Cancer.
14(165)2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hu J, Wang Z, Shan Y, Pan Y, Ma J and Jia
L: Long non-coding RNA HOTAIR promotes osteoarthritis progression
via miR-17-5p/FUT2/β-catenin axis. Cell Death Dis.
9(711)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chen K, Zhu H, Zheng M-Q and Dong QR:
lncRNA MEG3 inhibits the degradation of the extracellular matrix of
chondrocytes in osteoarthritis via targeting miR-93/TGFBR2 Axis.
Cartilage: Jun 28, 2019 (Epub ahead of print). doi:
10.1177/1947603519855759.
|
|
12
|
Li L, Lv G, Wang B and Kuang L: The role
of lncRNA XIST/miR-211 axis in modulating the proliferation and
apoptosis of osteoarthritis chondrocytes through CXCR4 and MAPK
signaling. Biochem Biophys Res Commun. 503:2555–2562.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531.
2004.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Huang J, Zhao L, Fan Y, Liao L, Ma PX,
Xiao G and Chen D: The microRNAs miR-204 and miR-211 maintain joint
homeostasis and protect against osteoarthritis progression. Nat
Commun. 10(2876)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Meng F, Li Z, Zhang Z, Yang Z, Kang Y,
Zhao X, Long D, Hu S, Gu M, He S, et al: MicroRNA-193b-3p regulates
chondrogenesis and chondrocyte metabolism by targeting HDAC3.
Theranostics. 8:2862–2883. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cai C, Min S, Yan B, Liu W, Yang X, Li L,
Wang T and Jin A: miR-27a promotes the autophagy and apoptosis of
IL-1β treated-articular chondrocytes in osteoarthritis through
PI3K/AKT/mTOR signaling. Aging (Albany NY). 11:6371–6384.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yang B, Kang X, Xing Y, Dou C, Kang F, Li
J, Quan Y and Dong S: Effect of microRNA-145 on IL-1β-induced
cartilage degradation in human chondrocytes. FEBS Lett.
588:2344–2352. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Pilyugin M and Irminger-Finger I: Long
non-coding RNA and microRNAs might act in regulating the expression
of BARD1 mRNAs. Int J Biochem Cell Biol. 54:356–367.
2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mizuochi T, Taniguchi T, Shimizu A and
Kobata A: Structural and numerical variations of the carbohydrate
moiety of immunoglobulin G. J Immunol. 129:2016–2020.
1982.PubMed/NCBI
|
|
20
|
Hu J, Wang Z, Pan Y, Ma J, Miao X, Qi X,
Zhou H and Jia L: miR-26a and miR-26b mediate osteoarthritis
progression by targeting FUT4 via NF-κB signaling pathway. Int J
Biochem Cell Biol. 94:79–88. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ma B, Simala-Grant JL and Taylor DE:
Fucosylation in prokaryotes and eukaryotes. Glycobiology.
16:158R–184R. 2006.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lu J, Deng ZH, Li YS and Lei GH: Long
noncoding RNAs in osteoarthritis. Joint Bone Spine. 84:553–556.
2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xing D, Liang Jq, Li Y, Lu J, Jia Hb, Xu
Ly and Ma Xl: Identification of long noncoding RNA associated with
osteoarthritis in humans. Orthop Surg. 6:288–293. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
Aida Y, Maeno M, Suzuki N, Shiratsuchi H,
Motohashi M and Matsumura H: The effect of IL-1beta on the
expression of matrix metalloproteinases and tissue inhibitors of
matrix metalloproteinases in human chondrocytes. Life Sci.
77:3210–3221. 2005.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Daheshia M and Yao JQ: The interleukin
1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol.
35:2306–2312. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lopez-Armada MJ, Carames B, Lires-Dean M,
Cillero-Pastor B, Ruiz-Romero C, Galdo F and Blanco FJ: Cytokines,
tumor necrosis factor-alpha and interleukin-1beta, differentially
regulate apoptosis in osteoarthritis cultured human chondrocytes.
Osteoarthritis Cartilage. 14:660–669. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Takahashi A, de Andrés MC, Hashimoto K,
Itoi E, Otero M, Goldring MB and Oreffo ROC: DNA methylation of the
RUNX2 P1 promoter mediates MMP13 transcription in chondrocytes. Sci
Rep. 7(7771)2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Larkin J, Lohr TA, Elefante L, Shearin J,
Matico R, Su JL, Xue Y, Liu F, Genell C, Miller RE, et al:
Translational development of an ADAMTS-5 antibody for
osteoarthritis disease modification. Osteoarthritis Cartilage.
23:1254–1266. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liang Z and Ren C: Emodin attenuates
apoptosis and inflammation induced by LPS through up-regulating
lncRNA TUG1 in murine chondrogenic ATDC5 cells. Biomed
Pharmacothery. 103:897–902. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tang LP, Ding JB, Liu ZH and Zhou GJ:
lncRNA TUG1 promotes osteoarthritis-induced degradation of
chondrocyte extracellular matrix via miR-195/MMP-13 axis. Euro Rev
Med Pharmacol Sci. 22:8574–8581. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li J, Lai Y, Ma J, Liu Y, Bi J, Zhang L,
Chen L, Yao C, Lv W, Chang G, et al: miR-17-5p suppresses cell
proliferation and invasion by targeting ETV1 in triple-negative
breast cancer. BMC Cancer. 17(745)2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yang F, Yin Y, Wang F, Wang Y, Zhang L,
Tang Y and Sun S: miR-17-5p Promotes migration of human
hepatocellular carcinoma cells through the p38 mitogen-activated
protein kinase-heat shock protein 27 pathway. Hepatology.
51:1614–1623. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ma Y, Zhang P, Wang F, Zhang H, Yang Y,
Shi C, Xia Y, Peng J, Liu W, Yang Z and Qin H: Elevated oncofoetal
miR-17-5p expression regulates colorectal cancer progression by
repressing its target gene P130. Nat Commun. 3(1291)2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lai TY, Chen IJ, Lin RJ, Liao GS, Yeo HL,
Ho CL, Wu JC, Chang NC, Lee AC and Alice LY: Fucosyltransferase 1
and 2 play pivotal roles in breast cancer cells. Cell Death Discov.
5(74)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kawai S, Kato S, Imai H, Okada Y and
Ishioka C: Suppression of FUT1 attenuates cell proliferation in the
HER2-overexpressing cancer cell line NCI-N87. Oncol Rep. 29:13–20.
2013.PubMed/NCBI View Article : Google Scholar
|