Introduction
Loeys-Dietz syndrome (LDS) is a rare hereditary
connective tissue disorder with autosomal dominant inheritance and
is characterized by widespread systemic involvement (1). It was reported that ~25% of patients
diagnosed with LDS have an affected parent and ~75% of probands
have LDS due to a de novo pathogenic variant (2). At present, the pathogenesis of LDS is
not entirely clear, and most researchers believe that it is
associated with mutations in genes of the cytokine family TGF-β
(3). The most typical clinical
triad includes hypertelorism, bifid uvula or cleft palate, and
aortic aneurysm with tortuosity (1). The syndrome shares a number of
clinical characteristics with the well-documented Marfan syndrome
(MFS), but compared with MFS, clinical progression of aortic
aneurysm or dissection in patients with LDS is more rapid,
prognosis of non-surgical treatment is poor, requiring close
monitoring (4). Pre-symptom
diagnosis has clinical significance in the treatment and prognosis
of patients with LDS. LDS has become a focus of researchers
worldwide, leading to improved understanding of the disease
(2); however, LDS remains
insufficiently studied in China. The present report describes a
rare case of a female infant diagnosed with LDS and provides a
review of relevant literature.
Case report
In February 2019, a girl of 2 months and 23 days was
admitted to Nanjing Maternity and Child Health Care Hospital
(Nanjing, China). During a physical health examination of the
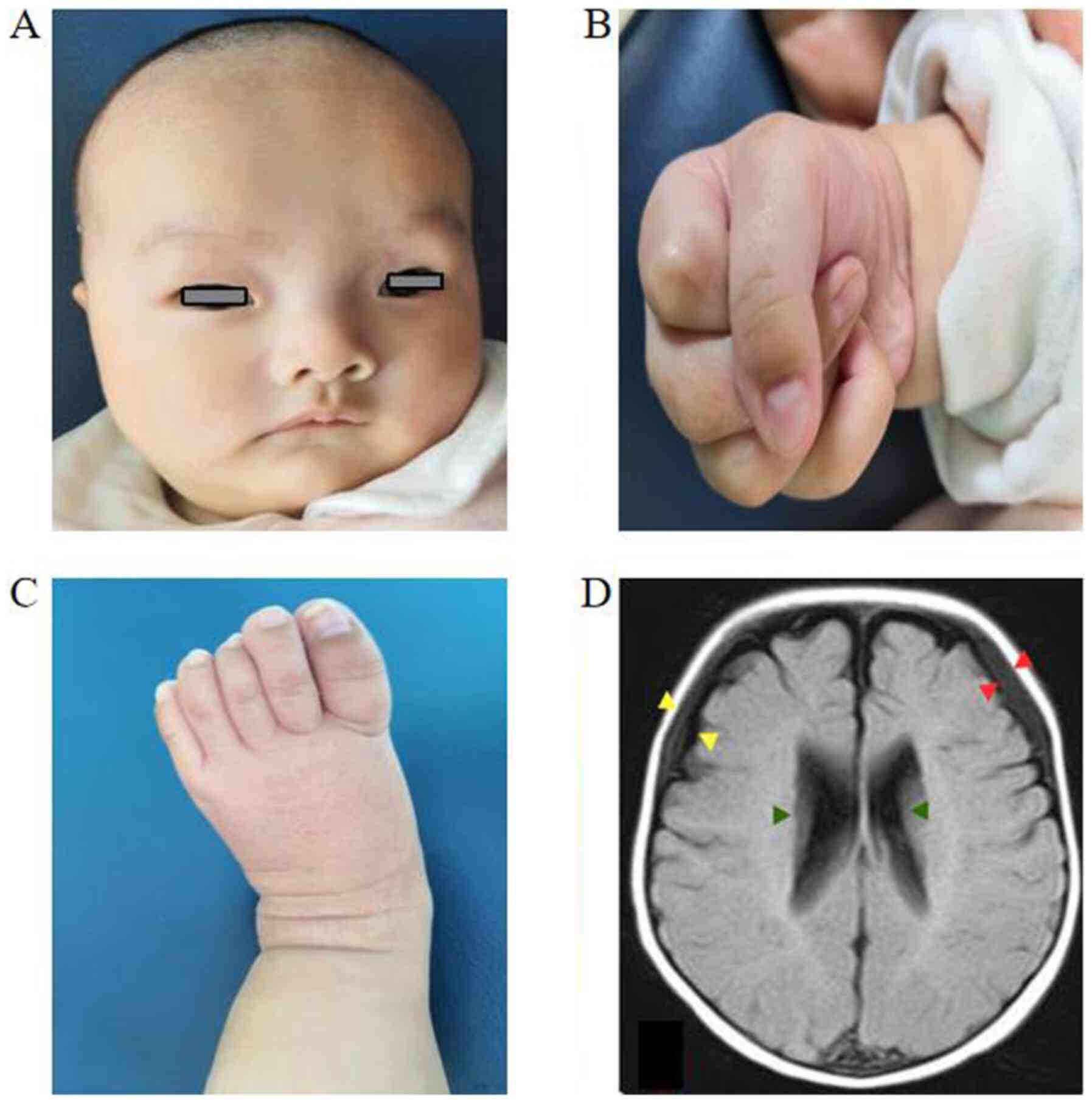
patient at the age of 42 days, facial ocular hypertelorism
(Fig. 1A), ocular exotropia,
micrognathia and high-vaulted palate were evident. The fingers and
toes of the patient were long and thin (Fig. 1B and C). The whole muscle tone was low and joint
relaxation in the distal limbs was observed. Additionally, the
patient presented with contracted finger joints, adduction
deformity of the thumbs, difficulty in extending the knuckles in
both hands and camptodactyly. The heart rhythm was regular with no
murmurs. The outpatient doctor suspected MFS, and recommended
whole-exome sequencing and high-risk infant follow-up at Nanjing
Maternity and Child Health Care Hospital.
The patient was born following a full-term pregnancy
by cesarean delivery. The birth weight was recorded as 3,500 g. The
parents did not exhibit special facial characteristics or a history
of cardiovascular disease. The patient was diagnosed with genu
recurvatum 1 day after birth and immobilized with an orthopedic
cast and fixed with a sling. At 2.5 months of age, the patient
could not hold her head up, her eyes did not track moving objects
and her limbs exhibited low muscle tone. The patient was
subsequently provided physical, cognition and language
rehabilitation training at the hospital. The results of tandem mass
spectrometry showed that the metabolism of organic acids, amino
acids and fatty acids was normal (Non derivatization mass
spectrometry detection kit; PerkinElmer, Inc.) A brain MRI scan
revealed widening of the space between the frontotemporal and
parietal brain, some subdural effusion in the forehead and dilated
bilateral ventricles (Fig. 1D). No
obvious abnormalities were detected by the X-ray examination of
both hip joints. Color Doppler echocardiography revealed that the
foramen ovale was not fused (3 mm), and the diameters of the
atrioventricular and great vessels were normal. Ophthalmic
examination revealed exotropia with no abnormalities in the ocular
fundus. Peripheral vein blood was collected from the patient and
the parents sent to Chigene Translational Medical Research Center
Co., Ltd. (Beijing, China) for whole exome sequencing. Gene
analyses included: i) screening for mutations using high-throughput
sequencing technology; ii) data analysis was performed using OMIM
(https://omim.org/), Clinvar (https://www.clinicalgenome.org/data-sharing/clinvar/),
HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), dbSNP
(http://www.bioinfo.org.cn/relative/dbSNP%20Home%20Page.htm)
and gnomeAD (Genome Aggregation Database) databases; and iii)
verification of suspected pathogenic mutations using Sanger
sequencing. The exon capture chip of the xGen Exome Research Panel
(version 1.0; Integrated DNA Technologies) was applied for whole
exon group sequencing, which was completed using the Illumina
NovaSeq system 6000 (Illumina, Inc.) series sequencer. Coverage of
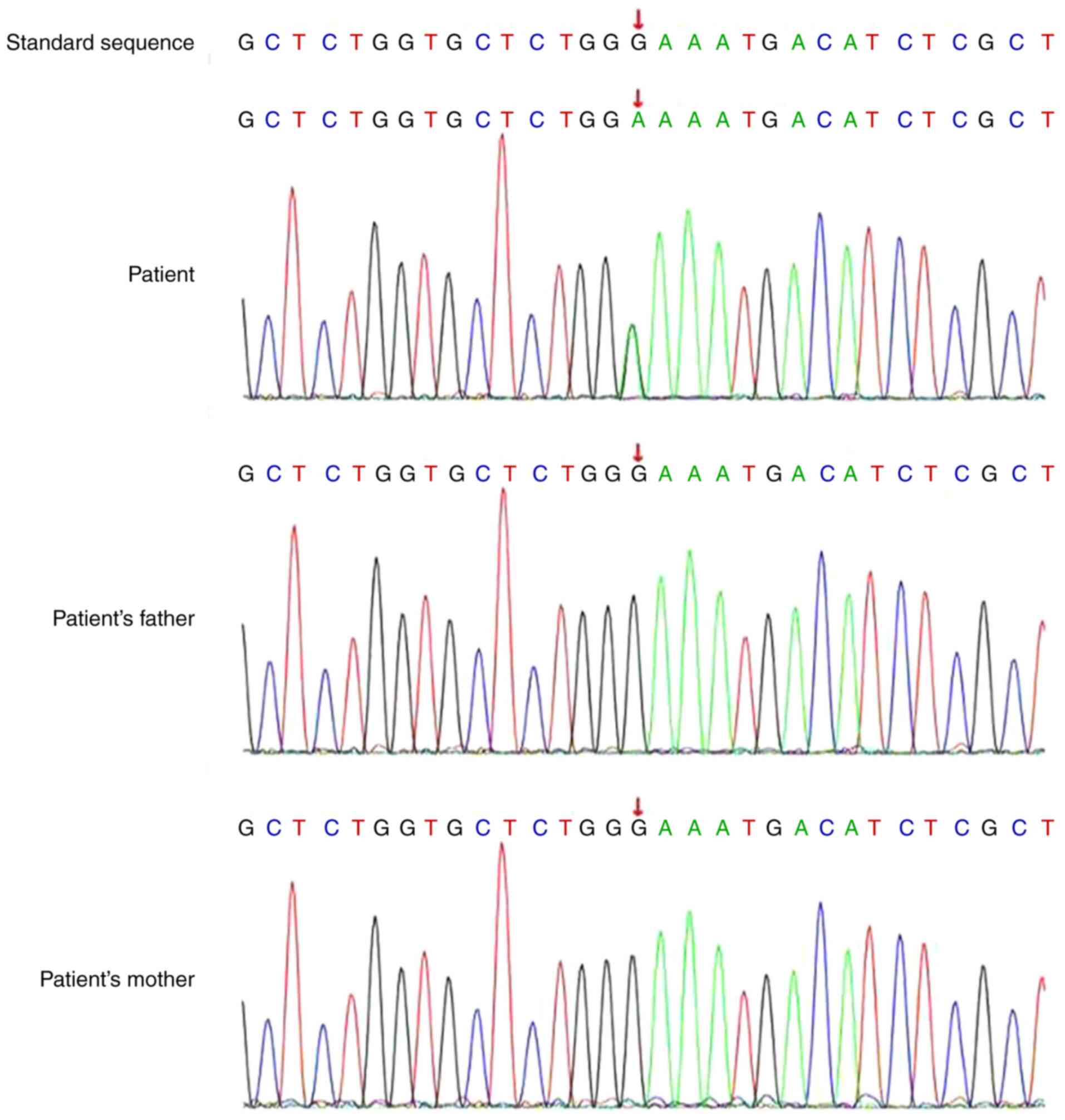
the target sequence was ≥99%. A heterozygous variation, c.1441(exon
6)G>A [p.E481k(p.Glu481Lys)(NM_001024847)], in the transforming
growth factor β receptor 2 (TGFBR2) gene was identified in
the patient. However, neither of the parents carried this mutation
(Fig. 2). The diagnosis of LDS was
confirmed based on the collective findings. The overall development
of the patient continued to improve with systematic rehabilitation.
Presently, the patient can walk with assistance, get up on her feet
unaided and, in terms of optical line of sight, move her eyes left
and right, up and down, and track moving objects. However,
responses to different facial expressions are not obvious, eye
contact is poor and exotropia still exists. The patient is
currently 14 months old and is undergoing regular ophthalmic
follow-up. The parents have been advised that a heart B ultrasound
should be performed as soon as possible. Regular follow-ups with a
cardiovascular specialist have been continued.
Discussion
LDS was first reported in 2005 and the most typical
presentation features alterations in cardiovascular, craniofacial,
neurocognitive and skeletal development (5). At present, a total of 619 cases have
been documented in the LDS Mutation Database (http://143.169.238.105/LOVD/diseases).
While the pathogenesis of LDS remains unclear, mutations in the
transforming growth factor β receptor 1 (TGFBR1) and
TGFBR2 genes identified in 2005 are the first known
causative factors (5).
Subsequently, other genes in the TGF-β signaling pathway, including
SMAD3, transforming growth factor β (TGFB)2,
SMAD2 and TGFB3, have been reported to be associated
with LDS (6). Furthermore, LDS is
subdivided into five clinical subtypes based on the pathogenic
genes involved (Table I).
 | Table IFive types of LDS listed in OMIM. |
Table I
Five types of LDS listed in OMIM.
| Subtype of LDS | Affected gene | OMIM ID | No. of patients |
|---|
| LDS1 | TGFBR1 | 609192 | 135 |
| LDS2 | TGFBR2 | 610168 | 251 |
| LDS3 | SMAD3 | 613795 | 95 |
| LDS4 | TGFB2 | 614816 | 73 |
| LDS5 | TGFB3 | 615582 | 53 |
Clinical presentations of LDS, particularly skeletal
features, overlap with those of MFS. Joint relaxation of the distal
limbs in the patient was evident and the fingers and toes were long
and thin, which could easily be misdiagnosed as MFS (7). Abnormal skeletal features in all types
of LDS include pectus deformity, scoliosis and flat feet. Although
evidence of skeletal overgrowth predominantly includes
arachnodactyly and pectus deformities, height and the proportion of
arm to leg length to height are usually within the normal range
(8). Furthermore, camptodactyly,
talipes equinovarus, contractures of joints and extremity
contractures in conjunction with joint hyperextension are common
features of LDS (8). Other common
manifestations include decreased stability of the cervical spine
and anterior displacement of the spine (8).
LDS is commonly associated with various congenital
diseases, including bicuspid aortic valve, atrial septal defect,
patent ductus arteriosus and mitral valve prolapse (1). Aortic root dilatation is the most
frequent and serious clinical manifestation (9,10).
Patients with LDS present with routine involvement of vascular
segments distant from the aortic root, which is a more aggressive
vascular course than that observed in MFS (4). The mean age of death of individuals
with aggressive arterial aneurysms is 26 years (11). Vascular dilatation occurs mainly in
the sinus node, which is further prone to aortic dissection or even
rupture. A study reported early dissection during a young age in
patients with TGFBR1, TGFBR2 or SMAD3
mutations (6).
No specific treatments are currently available for
LDS and the primary aim is to prevent vascular accidents and to
improve prognosis (8). All patients
with LDS require echocardiography at regular intervals (at least
once per year or more frequently) to monitor the status of the
aortic root, ascending aorta and heart valves (8). At follow-up, the decision to undergo
aortic surgery is based on the absolute dimension of the aorta,
rate of progression, valve function, severity of non-cardiac
features, genotype and family history (8). The skeletal system, and craniofacial
and ocular symptoms should be regularly monitored by a
multidisciplinary team.
In the current case, the patient presented with
clinical manifestations and multiple system abnormalities,
including long and thin fingers and toes, contraction of finger
joints, deformation of the thumbs and general recurvatum skeletal
system changes. Furthermore, the symptoms, including ocular
hypertelorism, ocular exotropia, micrognathia and high-vaulted
arch, were consistent with the classic symptoms of LDS (1,5).
Due to overlapping clinical phenotypes, no clear
standard diagnosis for LDS is available and gene detection is used
as an effective diagnostic tool (12). The patient in the present report
exhibited a heterozygous c.1441(exon 6)G>A
[p.E481k(p.Glu481Lys)(NM_001024847)] mutation in the TGFBR2
gene located at chr3:30715708. This pathogenic abnormality has been
documented in the LDS Mutation Database. Based on the clinical
manifestations and gene testing results, the patient was diagnosed
with LDS. To the best of our knowledge, the patient in the present
report is the youngest reported case of LDS diagnosis in China.
However, no aorta-related changes were evident in the current case,
despite rapidly progressive aortic aneurysmal disease being a
distinct feature of LDS (1). Aortic
dissection in 3-month-old infants and cerebral hemorrhage in
children as young as 3 years has been reported (13,14).
The John Hopkins group reported that LDS1 and LDS2 are the more
aggressive subtypes and recommend root surgery at a threshold of
4.0 cm (15,16). Therefore, the present report
recommended that parents should ensure continued cardiovascular
follow-up for the patient.
Future studies will include a preliminary in
vitro experimental study on the mutation site to investigate
the impact of the mutation on coding proteins and downstream
signaling pathways. It is of great significance to diagnose LDS in
the early stage of life, as treatment strategies can be optimized,
precise treatments can be designed, and quality of life will be
improved. When pediatric patients diagnosed with LDS reach school
age, doctors or genetics professionals should provide necessary
documentation regarding their diagnosis, physical education
restrictions, impact of skeletal and joint features, and the
psychological impact of LDS (8).
All this information can aid the patient and educational
institutions may develop personalized education programs.
In conclusion, LDS is a rare autosomal dominant
connective tissue disease involving multiple systems, particularly
the cardiovascular system. The progress of LDS is rapid and
presents a serious threat to the life of pediatric patients. To
further the understanding of the clinical manifestations of LDS,
early detection and diagnosis, close monitoring of cardiovascular
changes and enhanced follow-up are necessary for suspected
cases.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81671359),
the Subproject of Key Research and Development Program of China
(grant no. 2016YFC1000204-6), the Jiangsu Provincial Medical
Innovation Team (grant no. CXTDA2017001), The ‘Six Talent Peak’
High-level Talents Training Project of Jiangsu Province (grant no.
WSN-165), the Key Project Supported by Medical Science and
Technology Development Foundation Nanjing Department of Health
(grant no. zkx18044) and the 333 High Level Talents Training
Project of Jiangsu Province.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CZ collected data, drafted the manuscript and
revised the manuscript. MT monitored the data collection and
revised the manuscript. XC designed the present study and was
responsible for the integrity of the present study. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the independent
Ethical Committee Review Board of Women's Hospital of Nanjing
Medical University, Nanjing Maternity and Child Health Care
Hospital (Nanjing, China). Written consent for participation was
obtained from the patient's parents.
Patient consent for publication
Written informed consent for publication of data and
images of the patient and their genetic data was retrospectively
obtained from the patient's parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Van Laer L, Dietz H and Loeys B:
Loeys-Dietz syndrome. Adv Exp Med Biol. 802:95–105. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Loeys BL and Dietz HC: Loeys-Dietz
Syndrome. 2008 Feb 28 [Updated 2013 Jul. 11]. In:
GeneReviews™ [Internet]. Pagon RA, Adam MP, Bird TD,
et al (eds) University of Washington, Seattle, WA,
1993-2013. Available from: urihttps://www.ncbi.nlm.nih.gov/books/NBK1133/simplehttps://www.ncbi.nlm.nih.gov/books/NBK1133/.
|
|
3
|
Ritelli M, Chiarelli N, Dordoni C,
Quinzani S, Venturini M, Maroldi R, Calzavara-Pinton P and Colombi
M: Further delineation of Loeys-Dietz syndrome type 4 in a family
with mild vascular involvement and a TGFB2 splicing mutation. BMC
Med Genet. 15(91)2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Maleszewski JJ, Miller DV, Lu J, Dietz HC
and Halushka MK: Histopathologic findings in ascending aortas from
individuals with Loeys-Dietz syndrome (LDS). Am J Surg Pathol.
33:194–201. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Loeys BL, Chen J, Neptune ER, Judge DP,
Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et
al: A syndrome of altered cardiovascular, craniofacial,
neurocognitive and skeletal development caused by mutations in
TGFBR1 or TGFBR2. Nat Genet. 37:275–281. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Yang H, Ma Y, Luo M, Zhu G, Zhang Y, Li B,
Shu C and Zhou Z: Genetic profiling and cardiovascular phenotypic
spectrum in a Chinese cohort of Loeys-Dietz syndrome patients.
Orphanet J Rare Dis. 15(6)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Singh KK, Rommel K, Mishra A, Karck M,
Haverich A, Schmidtke J and Arslan-Kirchner M: TGFBR1 and TGFBR2
mutations in patients with features of Marfan syndrome and
Loeys-Dietz syndrome. Hum Mutat. 27:770–777. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
MacCarrick G, Black JH III, Bowdin S,
El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, Sponseller
PD, Loeys B and Dietz HC III: Loeys-Dietz syndrome: A primer for
diagnosis and management. Genet Med. 16:576–587. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Van Hemelrijk C, Renard M and Loeys B: The
Loeys-Dietz syndrome: An update for the clinician. Curr Opin
Cardiol. 25:546–551. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
van de Laar IM, van der Linde D, Oei EH,
Bos PK, Bessems JH, Bierma-Zeinstra SM, van Meer BL, Pals G,
Oldenburg RA, Bekkers JA, et al: Phenotypic spectrum of the
SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet.
49:47–57. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Loeys BL, Schwarze U, Holm T, Callewaert
BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S,
Manouvrier S, et al: Aneurysm syndromes caused by mutations in the
TGF-beta receptor. N Engl J Med. 355:788–798. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Van Laer L, Proost D and Loeys BL:
Educational paper. Connective tissue disorders with vascular
involvement: From gene to therapy. Eur J Pediatr. 172:997–1005.
2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Malhotra A and Westesson PL: Loeys-Dietz
syndrome. Pediatr Radiol. 39(1015)2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Williams JA, Loeys BL, Nwakanma LU, Dietz
HC, Spevak PJ, Patel ND, François K, DeBacker J, Gott VL, Vricella
LA and Cameron DE: Early surgical experience with Loeys-Dietz: A
new syndrome of aggressive thoracic aortic aneurysm disease. Ann
Thorac Surg. 83:S757–S763, S785-S790. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Patel ND, Crawford T, Magruder JT, Alejo
DE, Hibino N, Black J, Dietz HC, Vricella LA and Cameron DE:
Cardiovascular operations for Loeys-Dietz syndrome:
Intermediate-term results. J Thorac Cardiovasc Surg. 153:406–412.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Krohg-Sørensen K, Lingaas PS, Lundblad R,
Seem E, Paus B and Geiran OR: Cardiovascular surgery in Loeys-Dietz
syndrome types 1-4. Eur J Cardiothorac Surg. 52:1125–1131.
2017.PubMed/NCBI View Article : Google Scholar
|