Introduction
Chronic kidney disease (CKD) is a major health
problem faced by individuals worldwide (1). Atherosclerosis and vascular
calcification are highly prevalent in patients with CKD and are
known to be major risk factors for all-cause and cardiovascular
mortalities in these patients (2).
Multiple mechanisms are thought to be associated with vascular
calcification, including hyperphosphatemia, oxidative stress,
inflammation and apoptosis; however, none of these mechanisms is
fully understood (3). Clinical and
epidemiological studies have shown that hyperphosphatemia is the
most important risk factor associated with the initiation of
vascular calcification in patients with CKD, leading to the
hypothesis that maintaining serum phosphorus at appropriate levels
may significantly alleviate vascular calcification and improve
clinical outcomes (4). Moreover,
osteochondrocytic differentiation of vascular smooth muscle cells
(VSMCs) under high phosphate conditions is considered to be a
pivotal pathological process in the initiation of vascular
calcification (5).
Studies have indicated that autophagy plays an
important role in the function of VSMCs and in the development of
vascular diseases, suggesting that it may be a potential target in
the prevention of vascular calcification (6-8).
Autophagy induced by high phosphate conditions has been
demonstrated to counteract the phosphate-induced calcification of
VSMCs (9). High phosphorus promotes
calcification of VSMCs and activates autophagy, which maintains
VSMC stability and allows VSMCs to adapt to unfavorable conditions
(9). First-line drugs used for
treating hyperglycemia, such as metformin, also exhibit protective
effects on the cardiovascular system; however, the underlying
mechanisms are not fully understood. It has previously been
reported that metformin can activate 5' adenosine
monophosphate-activated protein kinase (AMPK) (10) and the AMPK/mammalian target of
rapamycin (mTOR) signaling pathway has been shown to regulate
autophagy, both directly and indirectly (11). AMPK could initiate autophagy either
by directly phosphorylating serine/threonine-protein kinase ULK1
protein (12), or indirectly by
deactivating mTORC1(13). The aim
of the present study was to investigate the function of metformin
in the alleviation of β-glycerophosphate-induced calcification of
VSMCs and the role of AMPK/mTOR-activated autophagy in this
pathological condition.
Materials and methods
Cell culture
The rat thoracic aorta VSMC line A7r5 was purchased
from The Cell Bank of Type Culture Collection of the Chinese
Academy of Sciences the Shanghai Institutes for Biological
Sciences. The cells were inoculated in 35-mm dishes at a density of
2x104 cells and were cultured in high-glucose Dulbecco's
modified Eagle's medium (HDMEM) containing 10% fetal bovine serum
(FBS) (both from Hyclone; Cytiva) unless otherwise indicated. The
cell cultures were incubated at 37˚C in a humidified thermostatic
incubator supplied with 5% CO2. Medium was refreshed
every 2-3 days. To induce calcification, the VSMCs were cultured in
1% FBS-HDMEM containing 10 mmol/l β-glycerophosphate (β-GP;
Sigma-Aldrich; Merck KGaA) for 14 days (9,14). To
determine the effect of metformin on the calcification of VSMCs and
the underlying mechanisms involved, VSMCs were incubated in 1%
FBS-HDMEM containing 10 mmol/l β-GP and 500 µmol/metformin
(Sigma-Aldrich; Merck KGaA), as described previously (15). To investigate the effect of
autophagy on the calcification of VSMCs, the cells were pre-treated
with the autophagy inhibitor, 3-methyladenine (3-MA; Sigma-Aldrich;
Merck KGaA) at 5 mmol/l for 30 min (9). To investigate the involvement of the
AMPK/mTOR signaling pathway in the induction of autophagy by
metformin, the VSMCs were pre-treated with the AMPK inhibitor
compound C (Sigma-Aldrich; Merck KGaA) at 10 µmol/l for 30 min
(16). VSMCs cultured in 1%
FBS-HDMEM were used as the control.
Alizarin Red S staining and
quantification of cell calcium content
After treatment with β-GP for 14 days, VSMCs were
fixed in 95% ethanol for 1 h at room temperature. The cells were
stained with 1% Alizarin Red S solution (Sigma-Aldrich; Merck KGaA)
for 30 min at 37˚C. Cells were then washed three times with PBS to
remove non-specific staining. The positively-stained mineralized
matrix exhibited a reddish/purple color upon direct visualization.
Mineralized nodules were observed at x40 magnification under an
inverted microscope (Nikon TMS; Nikon Corporation) with three
fields for each group. In order to quantify their calcium content,
VSMCs were incubated in triplicate for 14 days before subjecting
them to decalcification with 0.6 mol/l hydrochloric acid (HCl) for
24 h at 37˚C. The calcium content in the HCl supernatants was
determined for each sample using the O-cresolphthalein complexone
method (17). Total protein from
the treated cells was extracted using 1 M NaOH/0.1% SDS and was
quantified using the BCA method. The calcium content was normalized
to the protein content and was expressed as µg calcium per mg
protein.
Transmission electron microscopy
(TEM)
TEM was performed to assess the formation of
autophagosomes in the VSMCs. Briefly, after culture for 72 h, VSMCs
were pre-fixed with 2.5% glutaraldehyde for 2 h at room temperature
and post-fixed with 1% osmium tetroxide for 2 h at 4˚C. After
performing the dehydration process in an ethanol gradient (50% for
15 min, 70% overnight, followed by 80 and 90% each for 15 min, and
100% twice, each for 20 min), the cells were incubated with
propanone and embedded in SPI-Pon-812 resin (Structure Probes,
Inc.). Ultrathin sections were prepared using an ultramicrotome EM
UC7 (Leica Microsystems GmbH), and were then stained with uranyl
acetate and lead nitrate. The stained sections were examined under
the FEI Tecnai G2 Spirit transmission electron microscope (Thermo
Fisher Scientific, Inc.). Three fields of each section were
examined and photographed.
Immunofluorescence
Immunofluorescence analysis was performed to examine
the expression of microtubule-associated protein 1 light chain 3
(LC3). Briefly, the cells were inoculated in 24-well plates at a
density of 3x104 cells/well. After culture for 72 h,
VSMCs were fixed with 4% paraformaldehyde for 20 min at room
temperature and were permeablized by incubation with 0.5% Triton
X-100 for 5 min at room temperature. Thereafter, the cells were
blocked in 100% goat serum blocking solution (Beyotime Institute of
Biotechnology) for 2 h at room temperature, followed by incubation
with anti-LC3 mAb (1:200; cat. no. 3868; Cell Signaling Technology,
Inc.) overnight at 4˚C. The cells were then incubated with
FITC-conjugated secondary antibody (1:150; cat. no. WLA032;
Wanleibio Co., Ltd.) in the dark for 2 h at room temperature.
Nuclei of the cells were stained with DAPI for 5 min at room
temperature. The cells were visualized under a fluorescent
microscope at x200 magnification. Three fields of each sample were
examined and photographed.
Western blot analysis
Total protein from the VSMCs was extracted using
RIPA lysis buffer containing protease and phosphatase inhibitors
(all from Beyotime Institute of Biotechnology). The protein content
in the extracts was quantified using the BCA method. Equal amounts
of protein (40 µg) were subjected to SDS-PAGE [6% for LC3, 12% for
mTOR and phosphorylated (p)-mTOR, and 10% for all other proteins],
followed by the transfer of proteins onto a PVDF membrane. The
membranes were incubated successively with 5% non-fat milk for 1 h
at room temperature, with appropriate primary antibodies overnight
at 4˚C, and with horseradish peroxidase-conjugated secondary
antibody (1:2,000; cat. no. WLA023a; Wanleibio Co., Ltd.) for 1 h
at room temperature. The following primary antibodies were used:
Anti-LC3 (1:500; cat. no. 3868; Cell Signaling Technology, Inc.),
anti-beclin 1 (1:500; cat. no. 3495; Cell Signaling Technology,
Inc.), anti-α-SMA (1:1,000; cat. no. 19245; Cell Signaling
Technology, Inc.), anti-RUNX2 (1:1,000; cat. no. 12556; Cell
Signaling Technology, Inc.), anti-AMPK (1:500; cat. no. 2532; Cell
Signaling Technology, Inc.), anti-Thr172p-AMPK (1:500; cat. no.
2535; Cell Signaling Technology, Inc.), anti-mTOR (1:1,000; cat.
no. ab32028; Abcam), anti-ser2448p-mTOR (1:1,000; cat. no.
ab109268; Abcam), anti-GAPDH (1:1,000; cat. no. ab181602; Abcam)
and anti-β-actin (1:1,000; cat. no. ab179467; Abcam). The membranes
were developed using the ECL (Wanleibio Co., Ltd.) detection
system. All the bands obtained were analyzed using the ImageJ
software (National Institutes of Health). Normalization of the
protein bands was performed using GAPDH or β-actin levels. Data are
presented as relative density ratio of the β-GP group compared with
control group in initial experiments After finding that β-GP
influenced protein expression levels compared to the control group,
further experiments were performed to compare the effect of
treatment with a combination of β-GP metformin, 3-MA and Compound C
in comparison with β-GP alone.
Statistical analysis
Each experiment was performed in triplicate.
Comparisons between two groups were performed using unpaired
t-test. Comparisons between the values of three or more groups were
performed using one-way ANOVA, followed by the Tukey's multiple
comparison test. All the analyses in this study were performed
using GraphPad Prism software (version 7.04; GraphPad Software,
Inc.). The results of the experiments are presented as the mean ±
SEM and P<0.05 was considered statistically significant.
Results
Metformin alleviates
β-glycerophosphate-induced VSMC calcification
VSMCs were treated with β-GP (10 mmol/l) in order to
induce calcification. Alizarin Red S staining and O-cresolphthalein
complexone assay (Fig. 1A) revealed
that β-GP enhanced calcium deposition in VSMCs when compared to
that in the untreated control cells. In addition, the protein
expression level of the smooth muscle cell marker α-SMA was found
to be reduced in β-GP treated cells compared with the untreated
control, whereas the protein expression level of the osteogenic
gene RUNX2 was increased, as shown in the western blotting analysis
(Fig. 1B). To determine whether
metformin could reduce calcification in the VSMCs induced by β-GP,
the cells were first cultured in 1% FBS-HDMEM containing β-GP (10
mmol/l), and were then treated with metformin (500 µmol/l). When
compared with the cells that were treated solely with β-GP, the
calcium deposition was found to be reduced in the metformin-treated
cells (Fig. 1C). Western blot
analysis also revealed that the protein expression level of α-SMA
was increased, whereas the protein expression level of RUNX2 was
decreased in metformin treated cells compared with the β-GP only
treated cells (Fig. 1D). These
results suggest that metformin could potentially alleviate
β-GP-induced calcification of VSMCs.
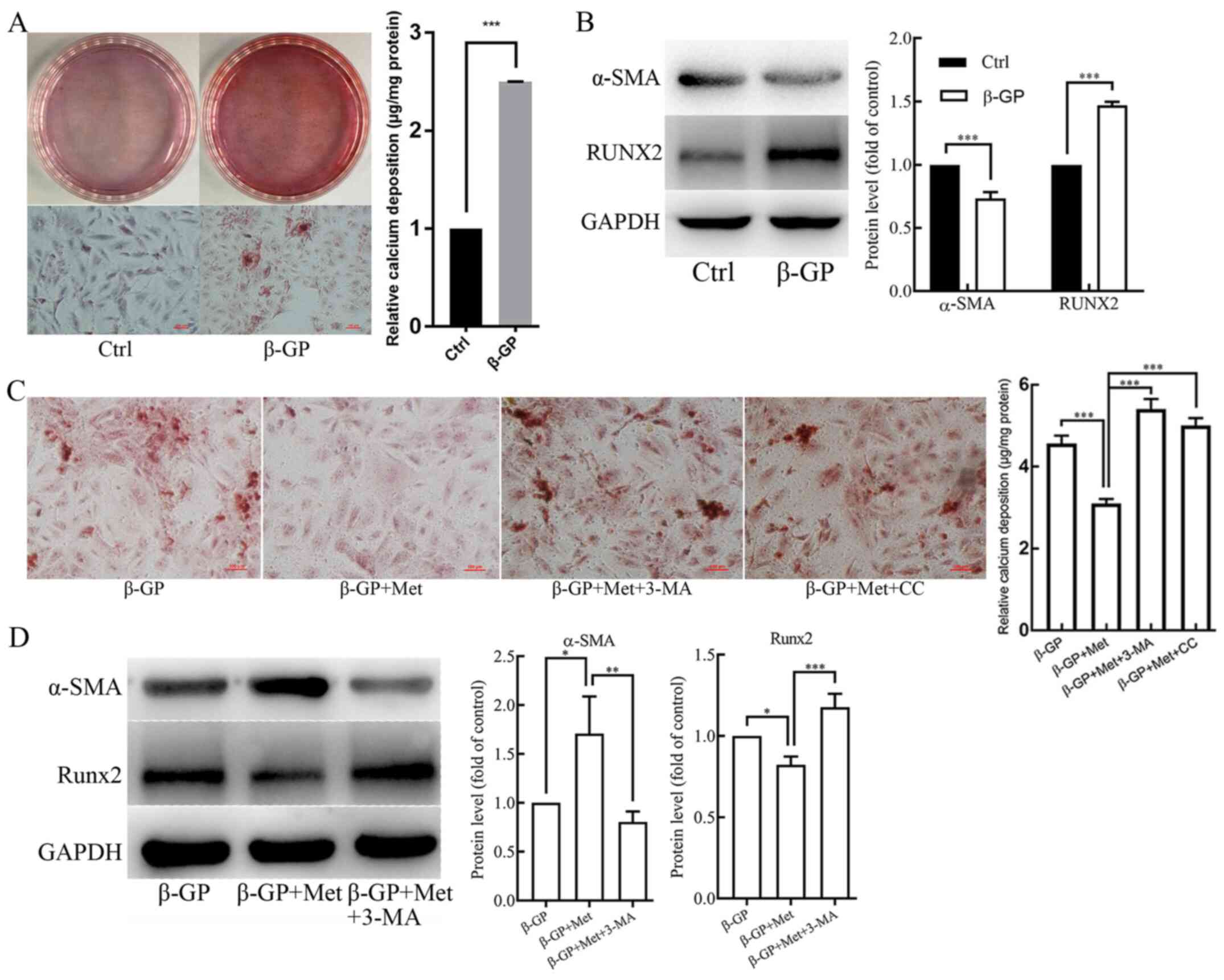 | Figure 1The effects of β-GP, Met, 3-MA and CC
on VSMC calcification. VSMCs were treated with β-GP (10 mmol/l) to
induce calcification and with metformin (500 µmol/l) to examine its
effect on calcification. 3-MA (5 mmol/l) and CC (10 µmol/l) were
added to the cells to investigate the effects of autophagy and the
AMPK signaling pathway, respectively. (A) Alizarin red S staining
and O-cresolphthalein complexone method calcium quantitation
showing calcium deposition in VSMCs treated with vehicle or β-GP.
(B) Western blots showing protein levels of α-SMA and Runx2 in
VSMCs treated with vehicle or β-GP. (C) Alizarin red S staining and
O-cresolphthalein complexone method calcium quantitation showing
calcium deposition in VSMCs treated with β-GP, MET, 3-MA, or CC.
(D) Western blots showing protein levels of α-SMA and Runx2 in
VSMCs treated with β-GP, Met, or 3-MA. Western bands of interest
were normalized against GAPDH, and data are provided as relative
density ratios compared with control group or β-GP group. Scale
bars in Alizarin red S staining represent 100 µm. Representative
images are shown. Data are presented as mean ± SEM, n=3.
*P<0.05, **P<0.01,
***P<0.001. β-GP, β-glycerophosphate; Met, metformin;
3-MA, 3-methyladenine; CC, compound C; VSMC, vascular smooth muscle
cell; α-SMA, α-smooth muscle actin; RUNX2, runt-related
transcription factor 2. |
Metformin promotes
β-glycerophosphate-induced autophagy in VSMCs
To elucidate the potential effect of metformin on
autophagy in VSMCs, the cells were subjected to TEM to examine
their autophagic activity. TEM results revealed an increase in the
accumulation of autophagosomes in the cells that were treated with
β-GP compared with untreated control cells, which was found to be
further increased in cells that were treated with both β-GP and
metformin (Fig. 2A). Fluorescence
microscopy results showed that the formation of the green
fluorescent LC3 puncta in VSMCs increased after treatment with β-GP
when compared to the cells that were treated only with the vehicle,
and the green fluorescent LC3 puncta were found to be further
increased after treatment with both β-GP and metformin (Fig. 2B). Additionally, the protein
expression levels of LC3II/I and beclin 1 were examined in the
VSMCs using western blot analysis. As shown in Fig. 2C, the expression levels of LC3II/I
and beclin 1 were significantly increased in VSMCs that were
treated with β-GP as compared with cells treated with vehicle.
After finding that β-GP increased the protein expression levels of
LC3II/I and beclin 1 compared to the untreated control group,
further experiments compared cells treated with multiple drugs with
β-GP alone. β-GP and metformin treatment further increased the
expression levels of LC3II/I and beclin 1 in comparison to β-GP
alone (Fig. 2D). These results
suggest that β-GP may potentially stimulate autophagy in VSMCs and
that metformin may further promote autophagic activity.
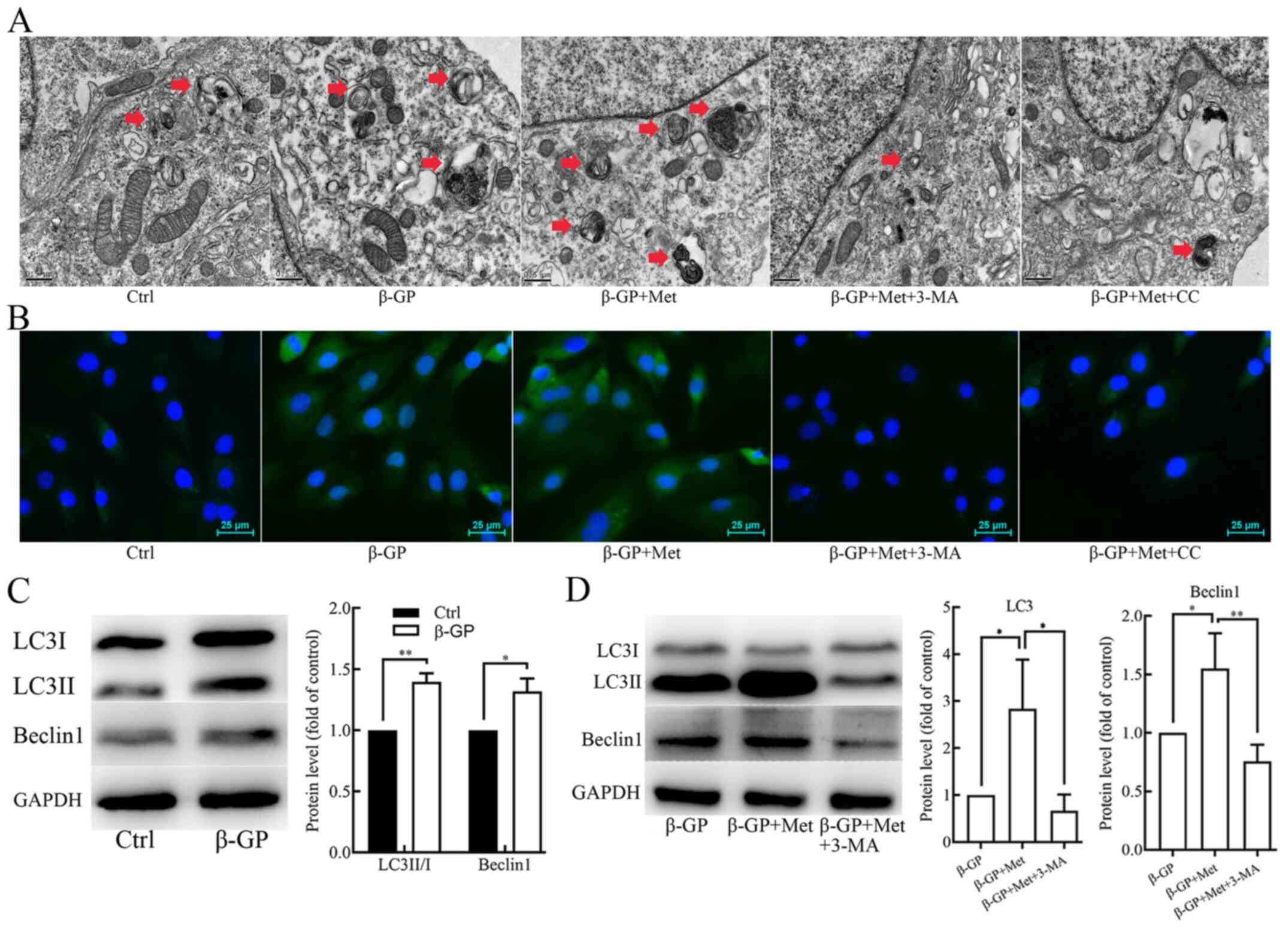 | Figure 2The effects of β-GP, Met, 3-MA and CC
on autophagic activity in VSMCs. (A) TEM showing autophagosomes
(red arrows) in VSMCs treated with β-GP, Met, 3-MA or CC. Scale
bars represent 0.5 µm. (B) Immunofluorescence staining showing the
expression of LC3 in VSMCs treated with β-GP, Met, 3-MA, or CC.
Scale bars represent 25 µm. Cells were treated with 1% FBS-HDMEM
containing β-GP, β-GP + Met, β-GP + Met + 3-MA, β-GP + Met + CC, or
solely 1% FBS-HDMEM for 72 h, before being subjected to TEM or
fluorescence microscopy (magnification, x200). (C) Western blots
showing protein levels of LC3 and beclin 1 in VSMCs treated with
vehicle or β-GP. (D) Western blots showing protein levels of LC3
and beclin 1 in VSMCs treated with β-GP, metformin, or 3-MA.
Western bands of interest were normalized against GAPDH, and data
are presented as relative density ratios compared with control
group or β-GP group. Representative images are shown. Data are
presented as mean ± SEM, n=3. *P<0.05,
**P<0.01. β-GP, β-glycerophosphate; Met, metformin;
3-MA, 3-methyladenine; CC, compound C; VSMC, vascular smooth muscle
cell; TEM, transmission electron microscopy; LC3,
microtubule-associated protein 1 light chain 3; HDMEM, high-glucose
Dulbecco's modified Eagle's medium. |
Metformin alleviates
β-glycerophosphate-induced VSMC calcification by enhancing
autophagy
To further determine whether metformin alleviated
the β-GP-induced VSMC calcification by promoting autophagy in
VSMCs, cells were pre-treated with the autophagy inhibitor, 3-MA (5
mmol/l) for 30 min before treatment with β-GP and metformin. The
results of Alizarin Red S staining and an O-cresolphthalein
complexone assay (Fig. 1C) showed
an evident increase in calcium deposition in VSMCs that were
pre-treated with 3-MA when compared to the cells that were not
subjected to 3-MA pre-treatment. However, the number of
autophagosomes observed under TEM (Fig.
2A) and green fluorescent LC3 puncta observed under the
fluorescence microscope (Fig. 2B)
were significantly decreased. Western blot analyses revealed
significantly lower levels of LC3II/I and Beclin 1 in VSMCs that
were pre-treated with 3-MA when compared to the cells that were not
subjected to 3-MA pre-treatment (Fig.
2D). In addition, the protein expression level of α-SMA was
found to be reduced, whereas the protein expression level Runx2 was
found to be increased when compared to the cells that were not
subjected to 3-MA pre-treatment (Fig.
1D). Therefore, these results indicate that metformin
alleviates β-GP-induced VSMC calcification by promoting autophagic
activity.
Metformin promotes autophagy by
activating the AMPK/mTOR signaling pathway
In order to understand the role of the AMPK/mTOR
signaling pathway in the induction of autophagy by metformin, VSMCs
were pre-treated with AMPK inhibitor compound C (10 µmol/l) for 30
min before treatment with β-GP and metformin. Western blot analyses
revealed that p-AMPK levels were increased and p-mTOR levels were
reduced in VSMCs that were treated with both, β-GP and metformin
when compared to the cells that were treated only with β-GP
(Fig. 3A). In addition, the protein
expression levels of LC3II/I and Beclin 1 were shown to be
significantly increased (Fig. 3B),
and also the protein expression level of α-SMA was increased, but
the protein expression levels of Runx2 was found to be decreased
(Fig. 3B) in the cells that were
treated with both β-GP and metformin in comparison to the cells
that were treated only with β-GP. When the VSMCs were pre-treated
with the Compound C, a significant reduction in p-AMPK and increase
in p-mTOR levels was observed (Fig.
3A). In addition, the protein expression levels of LC3II/I and
Beclin 1 were found to be reduced (Fig.
3B) along with the expression level of α-SMA, but the protein
expression level of Runx2 was found to be increased (Fig. 3B). The number of autophagosomes
observed under TEM (Fig. 2A) and
the green fluorescent LC3 puncta observed under the fluorescence
microscope (Fig. 2B) were evidently
reduced in the VSMCs that were pre-treated with compound C.
However, the results of Alizarin Red S staining and and an
O-cresolphthalein complexone assay (Fig. 1C) showed a significant increase in
the levels of calcium deposition. These results suggested that
metformin could activate the AMPK/mTOR signaling pathway in VSMCs
that were treated with β-GP, leading to an enhancement in cellular
autophagic activity, in turn alleviating the β-GP-induced
calcification of the VSMCs.
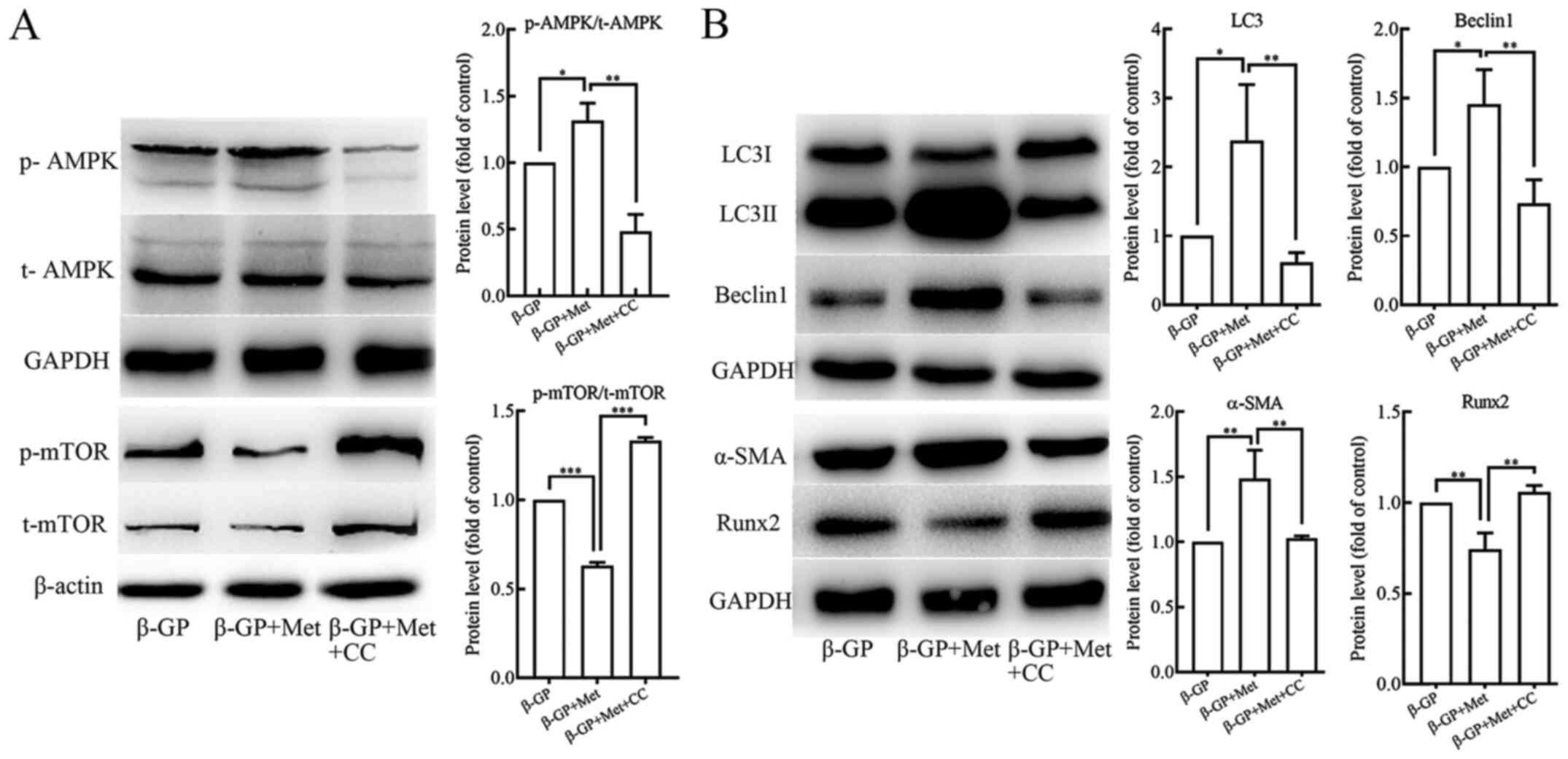 | Figure 3The effects of β-GP, Met and CC on
the AMPK signaling pathway and on cell calcification and autophagy
activity in VSMCs. (A) Western blots showing protein levels of p-
and t-AMPK and mTOR in VSMCs treated with β-GP, Met or CC. (B)
Western blots showing protein levels of LC3, beclin 1, α-SMA and
Runx2 in VSMCs treated with β-GP, MET, or CC. Western bands of
interest were normalized against GAPDH or β-actin, and data are
presented as relative density ratios compared with β-GP group.
Representative images are shown. Data are presented as mean ± SEM,
n=3. *P<0.05, **P<0.01,
***P<0.001. β-GP, β-glycerophosphate; Met, metformin;
3-MA, 3-methyladenine; CC, compound C; VSMC, vascular smooth muscle
cell; p, phosphorylated; t, total; LC3, microtubule-associated
protein 1 light chain 3; α-SMA, α-smooth muscle actin; RUNX2,
runt-related transcription factor 2; AMPK, 5' adenosine
monophosphate-activated protein kinase; mTOR, mammalian target of
rapamycin. |
Discussion
The occurrence of vascular calcification in patients
with CKD is thought to be due to the transformation of vascular
smooth muscle cells from a contractile to an osteogenic phenotype,
which may be induced by hyperphosphatemia (18). However, the mechanisms underlying
this transformation remain to be elucidated. In the present study,
using an in vitro cellular model of VSMC calcification, it
was demonstrated that metformin inhibited the VSMC contractile to
osteogenic phenotypic transformation induced by β-GP and thus
reduced their calcification. In addition, the results suggested
that metformin alleviated the calcification of VSMCs by promoting
their autophagic activity, which was indicated by an increased
number of autophagosomes, green fluorescent LC3 puncta and
expression levels of LC3II/I and Beclin 1 in metformin treated
cells when compared with controls. These metformin-induced
increases were later reversed by treatment with the autophagy
inhibitor 3-MA. Metformin appeared to promote the autophagic
activity of VSMCs by activating the AMPK/mTOR signaling pathway, as
indicated by the elevated levels of p-AMPK and reduced levels of
p-mTOR that were thought to be induced by metformin when compared
with a control and could be reversed by treatment with compound
C.
Metformin is widely used in the treatment of type 2
diabetes in clinical practice, typically at an oral dose of
500-1,000 mg in the form of standard immediate-tablet formulation
(19). Metformin has also been
shown to exhibit protective effects on the cardiovascular system.
Previous clinical studies demonstrated that metformin could
markedly improve below-the-knee arterial calcification in patients
with type 2 diabetes (20) and
coronary atherosclerosis in a diabetes prevention study (21). Additionally, in an in vivo
study in rats, it was demonstrated that metformin could potentially
alleviate vitamin D3- and nicotine-induced vascular calcification
via the AMPK pathway (22). In
cellular biology experiments, metformin has also been shown to
inhibit nuclear factor-κB (23) and
activate the AMPK-phosphatase and tensin homolog pathways (24) to alleviate the inflammatory response
in human and rat VSMCs, respectively. Furthermore, a study
indicated that metformin inhibited human aortic VSMC proliferation
and migration through activation of the AMPK signaling pathway and
up-regulation of expression levels of p53 and interferon
γ-inducible protein 16(25). In the
present study, it was demonstrated that metformin increased the
expression level of α-SMA and down-regulated the expression level
of Runx2 when compared with a control treatment. Additionally, it
was demonstrated that metformin could significantly alleviate
calcium deposition in VSMCs in in vitro experiments. Taken
together these results suggested that metformin potentially
repressed β-GP-induced contractile-to-osteogenic phenotypic
switching in VSMCs and alleviated their calcification.
Autophagy is involved in the degradation of damaged
and senescent organelles, as well as the long lived proteins, which
is critical for survival, development and differentiation of cells,
and also for the maintenance of homeostasis (26). It is a conserved cellular process
that takes place in all the eukaryotic cells and plays a pivotal
role in multiple human diseases (27). It has previously been demonstrated
that autophagy dysregulation is involved in a wide array of
vascular diseases, and also in the arterial medial calcification of
the tunica media in chronic kidney diseases (8). In a study that was performed using a
diabetes mouse model, metformin was found to prevent cardiomyopathy
by up-regulating the autophagic activity in the myocardial cells
(28). The present study indicated
that β-GP increased LC3II/I expression in comparison to the control
group, suggesting that cellular autophagic activity was activated
in a high phosphate environment. This is supported by the findings
of previous publications that demonstrated that β-GP could promote
cellular autophagic activity (9,29). In
the present study, metformin could further stimulate the autophagic
activity in the VSMCs treated with β-GP, as shown by the marked
increase in the number of autophagosomes, green fluorescent LC3
puncta and LC3II/I and Beclin 1 protein expression levels in
comparison with controls. Intracellular accumulation of the LC3
puncta observed under fluorescence microscope has been shown to be
associated with autophagic activity in cells (30). In the present study, when the cells
were pre-treated with the frequently used autophagy inhibitor,
3-MA, before treatment with both β-GP and metformin (31) an increase in the number of
autophagosomes and green fluorescent LC3 puncta and a reduction the
expression levels of LC3II/I and Beclin 1 were observed in
comparison to cells that had not been pre-treated. In addition, the
α-SMA expression level was decreased, but the expression level of
Runx2 and calcium deposition was found to be increased after
treatment with 3-MA in comparison with untreated cells. These
results suggest that the effect of metformin on the phenotypic
switching and subsequent calcification of the VSMCs may be due to
stimulation of the autophagic activity in the VSMCs.
The AMPK/mTOR signaling is an important pathway
involved in the process of cellular autophagy. AMPK directly
phosphorylates unc-51-like autophagy activating kinase 1 (ULK1) and
subsequently activates autophagy (32), however, it may also deactivate the
mTORC1 complex, which prevents the phosphorylation of ULK1 and its
association with AMPK, and thus may indirectly activate the process
of autophagy (12). By
investigating the mechanism by which metformin activated the
autophagic activity in the VSMCs, it was demonstrated that
metformin increased the phosphorylation of AMPK and reduced
phosphorylation of mTOR in VSMCs that were treated with β-GP. In
addition, the protein expression levels of the autophagy-associated
proteins, including LC3II/I and Beclin 1, as well as the number of
autophagosomes and green fluorescent LC3 puncta were found to be
increased. Moreover, the protein expression level of α-SMA was also
increased, but the protein expression level of Runx2 and calcium
content were decreased. However, upon treatment of these cells with
the AMPK inhibitor, Compound C, the expression levels of p-AMPK and
p-mTOR were markedly reversed, and the expression levels of the
autophagy-associated proteins and α-SMA were decreased, but the
expression level of Runx2 and calcium deposition were increased.
Therefore, these results indicate that the effect of metformin on
the autophagic activity of the VSMCs was implemented, at least in
part, by activation of the AMPK/mTOR signaling pathway, which
subsequently inhibited the phenotypic transition towards
osteogenesis in high-phosphorus milieu and thus alleviated its
calcification.
The major causes of lethality in patients with CKD
are the cardiovascular events (33). As vascular calcification is an
independent risk factor contributing to the lethal cardiovascular
events, it is necessary to decelerate the progression of vascular
calcification in these patients and to implement it in the clinical
practices. In a study conducted using a rat model of CKD, which was
induced by an adenine diet, metformin was shown to protect the rats
from developing severe CKD by reducing the inflammation and
fibrosis in the kidney, as well as exhibited a favorable effect on
the associated comorbidities such as vascular calcification
(34).
Several limitations must be contemplated while
considering the results of the present study. In the present study,
VSMCs were treated with β-glycerophosphate, metformin,
3-methyladenine, and/or compound C to induce calcification, to
examine the effect of metformin on calcification and to investigate
the role of autophagy, along with the involvement of the AMPK/mTOR
signaling pathway. However, this is a preliminary study that, was
conducted using a model system and to address the hypothesis.
Therefore, future studies are warranted to further elucidate the
underlying mechanisms. In addition, based on the fact that the
living organisms respond to the pharmaceutical agents in a more
complex manner, further in vivo studies based on animal
models are necessary to before concluding the full biological
effect of metformin on vascular calcification in a living organism.
Moreover, even if it seems to be a very promising and novel
interventional method for the treatment of vascular calcification
in patients with a chronic kidney disease, a number of pre-clinical
and clinical studies should be performed before the direct
implementation of metformin in clinical practices.
In the present study, it was demonstrated that
metformin could promote cellular autophagy and could potentially
exert a beneficial impact on the β-GP-induced calcification of the
VSMCs by activating the AMPK/mTOR signaling pathway. Therefore,
metformin may exhibit a potential role in reducing vascular
calcification independent of its hypoglycemic action. Nevertheless,
as metformin may also induce lactate acidosis in CKD patients, its
use in these cohorts is limited (35). Lalau et al (36) have previously reported that in
moderate-to-severe CKD patients, it is possible to adjust the dose
of metformin according to patient renal function, in order to exert
a safe but a pharmacologically efficacious function. In general,
apart from reducing the blood glucose levels, metformin may exhibit
a dual function in protecting the kidney, as well as in alleviating
the vascular calcification (34).
In addition, in the work conducted by Zhang et al (22), the role of metformin in vitamin D3
and nicotine-induced vascular calcification was investigated. In
this study, a dose of 300 mg/kg metformin was administered to
170-200 g rats, which was clearly an overdose for the CKD patients
with a compromised renal function. In the present study, 500 µmol/l
metformin was used in the treatment of VSMCs, which was based on an
established protocol for in vitro experiments (15). In the future, it will be important
to conduct in vivo experiments in order to further
investigate the role of metformin, potentially by using a rat model
of vascular calcification with compromised renal function. Also,
the dose of metformin should be carefully tested and adjusted in
order to provide a potential therapeutic value to its clinical
usage in CKD patients.
In the present study, it was demonstrated that
metformin, apart from its most common use in reducing blood glucose
levels in diabetic patients, could also exert a beneficial effect
on the β-GP-induced calcification of the VSMCs by promoting the
cellular autophagic activity and activating the AMPK/mTOR signaling
pathway. Therefore, we could provide relevant information regarding
a novel therapeutic agent that could be applied in the treatment of
vascular calcification in CKD patients; however, further studies
should be performed in the future to better understand the
underlying mechanisms and validate the effectiveness of
metformin.
Acknowledgements
The authors would like to thank Dr Zhiyuan Wu
[University of Erlangen-Nuremberg, Germany
(Friedrich-Alexander-Universität Erlangen-Nürnberg)] for assistance
with imaging.
Funding
This work was supported by grants from the National
Natural Science Foundation of China (grant no. 81770766) and the
Provincial Natural Science Foundation of Liaoning (grant no.
20170540999). Professor Li Yao was also funded by the Shenyang
Scientific Innovation Research and Development Project (grant no.
1900192) and Dr Tianhua Xu was also funded by Youth Mainstay
Supporting Project of China Medical University (grant no.
QGZD2018005).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XQ and LY designed the experiments; XQ performed the
majority of experiments; QX performed the immunofluorescence
experiment; TX and PW conducted the TEM; ZS and YH performed all
the statistical analysis; XQ, QX, TX and LY wrote the manuscript.
All the authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Coresh J: Update on the burden of CKD. J
Am Soc Nephrol. 28:1020–1022. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Go AS, Chertow GM, Fan D, McCulloch CE and
Hsu CY: Chronic kidney disease and the risks of death,
cardiovascular events, and hospitalization. N Engl J Med.
351:1296–1305. 2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bardeesi ASA, Gao J, Zhang K, Yu S, Wei M,
Liu P and Huang H: A novel role of cellular interactions in
vascular calcification. J Transl Med. 15(95)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cannata-Andia JB and Martin KJ: The
challenge of controlling phosphorus in chronic kidney disease.
Nephrol Dial Transplant. 31:541–547. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Durham AL, Speer MY, Scatena M, Giachelli
CM and Shanahan CM: Role of smooth muscle cells in vascular
calcification: Implications in atherosclerosis and arterial
stiffness. Cardiovasc Res. 114:590–600. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tai S, Hu XQ, Peng DQ, Zhou SH and Zheng
XL: The roles of autophagy in vascular smooth muscle cells. Int J
Cardiol. 211:1–6. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Salabei JK and Hill BG: Autophagic
regulation of smooth muscle cell biology. Redox Biol. 4:97–103.
2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nussenzweig SC, Verma S and Finkel T: The
role of autophagy in vascular biology. Circ Res. 116:480–488.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Dai XY, Zhao MM, Cai Y, Guan QC, Zhao Y,
Guan Y, Kong W, Zhu WG, Xu MJ and Wang X: Phosphate-induced
autophagy counteracts vascular calcification by reducing matrix
vesicle release. Kidney Int. 83:1042–1051. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Piwkowska A, Rogacka D, Jankowski M,
Dominiczak MH, Stepinski JK and Angielski S: Metformin induces
suppression of NAD(P)H oxidase activity in podocytes. Biochem
Biophys Res Commun. 393:268–273. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Egan D, Kim J, Shaw RJ and Guan KL: The
autophagy initiating kinase ULK1 is regulated via opposing
phosphorylation by AMPK and mTOR. Autophagy. 7:643–644.
2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang S, Song P and Zou MH: AMP-activated
protein kinase, stress responses and cardiovascular diseases. Clin
Sci (Lond). 122:555–573. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shioi A, Nishizawa Y, Jono S, Koyama H,
Hosoi M and Morii H: Beta-glycerophosphate accelerates
calcification in cultured bovine vascular smooth muscle cells.
Arterioscler Thromb Vasc Biol. 15:2003–2009. 1995.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cao X, Li H, Tao H, Wu N, Yu L, Zhang D,
Lu X, Zhu J, Lu Z and Zhu Q: Metformin inhibits vascular
calcification in female rat aortic smooth muscle cells via the
AMPK-eNOS-NO pathway. Endocrinology. 154:3680–3689. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
She C, Zhu LQ, Zhen YF, Wang XD and Dong
QR: Activation of AMPK protects against hydrogen peroxide-induced
osteoblast apoptosis through autophagy induction and NADPH
maintenance: New implications for osteonecrosis treatment? Cell
Signal. 26:1–8. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhu D, Mackenzie NC, Shanahan CM, Shroff
RC, Farquharson C and MacRae VE: BMP-9 regulates the osteoblastic
differentiation and calcification of vascular smooth muscle cells
through an ALK1 mediated pathway. J Cell Mol Med. 19:165–174.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shroff R, Long DA and Shanahan C:
Mechanistic insights into vascular calcification in CKD. J Am Soc
Nephrol. 24:179–189. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bailey CJ: Metformin: Historical overview.
Diabetologia. 60:1566–1576. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mary A, Hartemann A, Liabeuf S, Aubert CE,
Kemel S, Salem JE, Cluzel P, Lenglet A, Massy ZA, Lalau JD, et al:
Association between metformin use and below-the-knee arterial
calcification score in type 2 diabetic patients. Cardiovasc
Diabetol. 16(24)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Goldberg RB, Aroda VR, Bluemke DA,
Barrett-Connor E, Budoff M, Crandall JP, Dabelea D, Horton ES,
Mather KJ, Orchard TJ, et al: Effect of long-term metformin and
lifestyle in the diabetes prevention program and its outcome study
on coronary artery calcium. Circulation. 136:52–64. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang X, Xiao J, Li R, Qin X, Wang F, Mao
Y, Liang W, Sheng X, Guo M, Song Y and Ji X: Metformin alleviates
vascular calcification induced by vitamin D3 plus nicotine in rats
via the AMPK pathway. Vascular pharmacol. 81:83–90. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Isoda K, Young JL, Zirlik A, MacFarlane
LA, Tsuboi N, Gerdes N, Gerdes N, Schönbeck U and Libby P:
Metformin inhibits proinflammatory responses and nuclear
factor-kappaB in human vascular wall cells. Arterioscler Thromb
Vasc Biol. 26:611–617. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kim SA and Choi HC: Metformin inhibits
inflammatory response via AMPK-PTEN pathway in vascular smooth
muscle cells. Biochem Biophys Res Commun. 425:866–872.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hao B, Xiao Y, Song F, Long X, Huang J,
Tian M, Deng S and Wu Q: Metformin-induced activation of AMPK
inhibits the proliferation and migration of human aortic smooth
muscle cells through upregulation of p53 and IFI16. Int J Mol Med.
41:1365–1376. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:24–41. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662.
2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xie Z, Lau K, Eby B, Lozano P, He C,
Pennington B, Li H, Rathi S, Dong Y, Tian R, et al: Improvement of
cardiac functions by chronic metformin treatment is associated with
enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes.
60:1770–1778. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xu M, Liu L, Song C, Chen W and Gui S:
Ghrelin improves vascular autophagy in rats with vascular
calcification. Life Sci. 179:23–29. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326.
2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Webster AC, Nagler EV, Morton RL and
Masson P: Chronic kidney disease. Lancet. 389:1238–1252.
2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Neven E, Vervaet B, Brand K,
Gottwald-Hostalek U, Opdebeeck B, De Mare A, Verhulst A, Lalau JD,
Kamel S, De Broe ME and D'Haese PC: Metformin prevents the
development of severe chronic kidney disease and its associated
mineral and bone disorder. Kidney Int. 94:102–113. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lipska KJ, Bailey CJ and Inzucchi SE: Use
of metformin in the setting of mild-to-moderate renal
insufficiency. Diabetes Care. 34:1431–1437. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lalau JD, Kajbaf F, Bennis Y,
Hurtel-Lemaire AS, Belpaire F and De Broe ME: Metformin treatment
in patients with type 2 diabetes and chronic kidney disease stages
3A, 3B, or 4. Diabetes Care. 41:547–553. 2018.PubMed/NCBI View Article : Google Scholar
|














