Introduction
Notch signaling has been demonstrated to participate
in cell fate determination and in progenitor cell maintenance
during development (1). In mammals,
there are four Notch receptors (Notch1-4), which have distinct
tissue expression patterns and are considered to function in
specific cellular contexts (1).
Notch3, a highly conserved type I transmembrane protein, is
predominantly expressed in vascular smooth muscle cells (VSMCs) has
three domains: An extracellular domain (ECD) with 34 epidermal
growth factor-like repeats, a transmembrane domain and an
intracellular domain (ICD), which serves an important role in VSMC
maturation and differentiation (2).
After binding of a ligand (δ-like protein 1 or protein jagged-1b)
to the ECD, the protein undergoes three proteolytic cleavage steps,
leading to the translocation of the ICD to the nucleus, where it
functions as a nuclear transcription factor (3).
Highly stereotyped mutations of ECD have been
associated with cerebral autosomal-dominant arteriopathy with
subcortical infarcts and leukoencephalopathy (CADASIL), an
inherited small vessel disease that causes stroke and dementia
(4). The typical characteristic of
CADASIL is progressive degeneration of VSMCs in blood vessel walls,
accompanied by the accumulation of Notch3 extracellular region and
the appearance of granular osmiophilic material deposits (5). In CADASIL, >170 different mutations
have been detected (5). Most of
these are missense point mutations complemented by a few small
deletions that lead to gain or loss of a cysteine residue within an
epidermal growth factor-like domain, thus altering the number of
cysteine residues within a given domain from 6 to either 5 or
7(6). This results in an unpaired
cysteine that is predicted to disrupt normal disulfide bridge
formation, causing misfolding of growth factor-like repeats and
increased Notch3 multimerization (7,8).
However, the exact underlying mechanism of pathogenesis in CADASIL
is yet to be elucidated. The role of CADASIL mutants in the
pathogenic mechanism is controversial and several authors have
reported that altered Notch3 function is not the primary
determinant of the disease (9-11).
Based on the uncertain functions of the
CADASIL-Notch3 mutants and the unknown mechanism of CADASIL, the
present study aimed to explore the roles of mutants and wild-type
(WT) Notch3 in the differential regulation of various cellular
phenotypes in several cell lines. The present findings could serve
as a molecular foundation for future studies investigating the
underlying mechanism of Notch3 signaling in CADASIL and may provide
an important insight for the investigation of novel CADASIL
treatments.
Materials and methods
Cells
T/GHA-human aortic VSMCs (CRL-1999), human fetal
lung fibroblasts (IMR-90; CCL-186), human cervical cancer cell
(HeLa; CCL-2), human breast cancer (MCF-7; CRL-3435) and mouse
microglia (BV2 cells;CRL-2468) were obtained from the American Type
Culture Collection. Human breast cancer cells (HCC1937 cells;
TCHu148) were purchased from The Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences. HeLa and MCF-7 cells
were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
whereas the other cell lines were cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin
and 100 µg/ml streptomycin (Beijing Solarbio Science &
Technology Co., Ltd.). Cells were maintained in a humidified
incubator at 37˚C with 5% CO2. HA-VSMCs and IMR-90 cells
were selected to clarify the effect of Notch3 mutants on tunica
media and adventitia, respectively. HeLa, MCF-7 and HCC1937 cells
were selected to assess the role of Notch3 mutants in different
types of cancer, while BV2 cells were used to evaluate the impact
of Notch3 mutants on neuroinflammation.
Western blot analysis
Cells were lysed in a sample buffer containing 2%
SDS, 60 mM Tris-HCl (pH 6.8) and 5% glycerol. Cell lysates were
then boiled for 5 min. Total protein concentration was determined
using a BCA kit (Beyotime Institute of Biotechnology), according to
the manufacturer's protocol, and equal quantities of proteins were
loaded for western blot analysis. In brief, 20 µg proteins were
resolved by 10% SDS-PAGE, transferred to a PVDF membrane (Pall Life
Sciences) and then blocked with 5% milk in TBS with 0.1% Tween-20
for 1 h at room temperature. The membrane was then incubated with
antibodies (1:1,000) against Notch3 (cat. no. SC-5593; Santa Cruz
Biotechnology, Inc.), fibronectin (cat. no. 26836; Cell Signaling
Technology, Inc.), collagen type I (cat. no. SC-59772; Santa Cruz
Biotechnology, Inc.), inducible nitric oxide synthase (iNOS; cat.
no. 13120; Cell Signaling Technology, Inc.), DNA
(cytosine-5)-methyltransferase 1 (DNMT1; cat. no. 5032; Cell
Signaling Technology, Inc.) or β-actin (cat. no. 3700; Cell
Signaling Technology, Inc.) overnight at 4˚C, followed by
incubation with secondary horseradish peroxidase-conjugated
anti-rabbit (1:5,000; cat. no. 7074; Cell Signaling Technology,
Inc.) or anti-mouse (1:5,000; cat. no. 7076; Cell Signaling
Technology, Inc.) antibodies for 1 h at room temperature. Blots
were developed using enhanced chemiluminescence reagents
(LumiGLO® Reagent and Peroxide; Cell Signaling
Technology, Inc.), according to the manufacturer's protocol.
Densitometry was performed using ImageJ software (version 1.8.0;
National Institutes of Health).
Nitric oxide (NO) release assay
Nitrite level in the cell culture medium was
measured as an indicator of NO production. In brief, BV2 cells were
treated with LPS (1 µg/ml, Sigma-Aldrich; Merck KGaA) for 24 h.
Following this, 50 µl supernatant was mixed with an equal volume of
Griess reagent I (Beyotime Institute of Biotechnology), followed by
addition of 50 µl Griess reagent II (Beyotime Institute of
Biotechnology) at room temperature. The absorbance was immediately
measured at 540 nm. The samples were assayed in triplicate and the
concentration of each sample was calculated from a standard curve
generated using sodium nitrite.
Reverse transcription
semi-quantitative PCR analysis
Total RNA was extracted from BV2 cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse transcribed using the PrimeScript™ RT Master Mix (Takara
Biotechnology Co., Ltd.), according to the manufacturer's protocol.
Conditions for reverse transcription were 15 min at 37˚C and 5 sec
at 85˚C. PCR analyses of tumor necrosis factor (TNF)-α, interleukin
(IL)-1β and β-actin mRNA levels were performed using 2X Taq Master
Mix (Vazyme Biotech Co., Ltd.). The forward and reverse primer
pairs used were as follows: TNF-α forward, 5'-ATCCGCGACGTGGAACTG-3'
and reverse, 5'-ACCGCC TGGAGTTCTGGAA-3'; IL-1β forward,
5'-CTTCATCTT-3' and reverse, 5'-TCACACACCAGCAGGTTATCATC-3'; and
β-actin forward, 5'-TGGCACCCAGCACAATGAA-3' and reverse,
5'-CTAAGTCATAGTCCGCCTAGAAGCA-3'. The PCR thermocycling conditions
were as follows: 95˚C for 5 min, followed by 30 cycles of 95˚C for
30 sec, 55˚C for 30 sec and 72˚C for 1 min and 72˚C for 10 min. The
products were separated on a 1.2% agarose gel containing ethidium
bromide and were visualized under a gel imaging system.
Quantitative PCR (qPCR) analysis
Total RNA extraction and reverse transcription were
carried out as described for semi-quantitative PCR analysis. qPCR
analyses of mRNA expression levels were performed with TB Green
Premix Ex Taq (Takara Biotechnology Co., Ltd.). The forward and
reverse primer pairs were as follows: DNMT1 forward, 5'-GTGGGG
GACTGTGTCTCTGT-3' and reverse, 5'-TGAAAGCTGCA TGTCCTCAC-3'; and
β-actin forward, 5'-ATCGTGCGTGA CATTAAGGAG-3' and reverse,
5'-GAAGGAAGGCTGGAA GAGTG-3'. The qPCR thermocycling conditions were
as follows: 95˚C for 30 sec, followed by 40 cycles of 95˚C for 3
sec, 60˚C for 30 sec. β-actin was used as the reference gene to
normalize the mRNA expression of DNMT1.
Vector construction and
transfection
pCMV-Sport6 Notch3 (Genbank ID: NM_000435.3) was
provided by Dr Michael Wang (University of Michigan, USA). The R90C
and R169C Notch3 CADASIL mutants were generated by PCR mutagenesis
of the pCMV-Sport6 Notch3 vector using the following primers: R90C
forward, 5'-CCC TGTGCTGGCTGTGGTGTCTGCC-3' and reverse,
5'-GGCAGACACCACAGCCAGCCAGGG-3'; and R169C forward,
5'-GTGAGCCCTGCTGCCATGGTG GCA-3' and reverse,
5'-TGCCACCATGGCAGCAGGGCTC ACC-3'. The PCR reaction was performed
using Takara PrimeSTAR Max DNA Polymerase (Takara Biotechnology
Co., Ltd.), according to the manufacturer's protocol. The PCR
thermocycling conditions were as follows: 98˚C for 3 min, followed
by 30 cycles of 98˚C for 30 sec, 60˚C for 30 sec, 72˚C for 5 min
and 72˚C for 10 min. The authenticity of the cloned fragments was
confirmed by sequencing. A total of 2 µg vectors were transfected
into 5x105 cells using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. After 48 h of transfection, cells were
collected for further experiments.
Proliferation assay
A total of 3x105 cells were seeded in
12-well plates and allowed to grow for 24 h prior to plasmid
transfection. At 24 h post-transfection, the cells were trypsinized
at 0, 12, 24 and 48 h. Following this, 10 µl aliquots of cells were
incubated with 10 µl 0.4% trypan blue solution for 5 min at room
temperature. Cells that exhibited trypan blue (viable cells) were
counted using a light microscope (magnification, x100; ECLIPSE Ti;
Nikon Corporation) with a hemocytometer (12).
Wound healing assay
Wound healing assays were performed as described by
Song et al (13). Briefly,
3x105 cells were seeded in 12-well plates overnight and
transfected with the vectors. Cells were grown to confluence for 36
h after transfection and wounded by scratching the monolayer with a
200-µl pipette tip. Following this, cells were washed using
pre-warmed PBS to remove cellular debris and allowed to migrate for
24 h in medium containing 1% FBS. Images of cell migration were
captured at 0 and 24 h after wounding using a light microscope
(magnification, x200; ECLIPSE Ti; Nikon Corporation). The relative
distance between the leading edges was determined using
NIS-Elements Documentation software (version no. 4.10; Nikon
Corporation) and expressed as a migration index (the distance
migrated in 24 h relative to the initial gap).
Statistical analysis
Data were analyzed using one-way ANOVA with Tukey's
post hoc test using the statistical package of SPSS software
(version 18; SPSS, Inc.) for Windows. Data are expressed as the
mean ± standard error of the mean from ≥3 independent experiments.
P<0.05 was considered to indicate a statistically significant
difference.
Results
CADASIL mutants differentially
regulate the expression of extracellular matrix proteins in VSMCs
and fibroblasts
A pathological study demonstrated that marked
fibrotic thickening of arteriolar walls occurs in patients with
CADASIL (14). Based on these data,
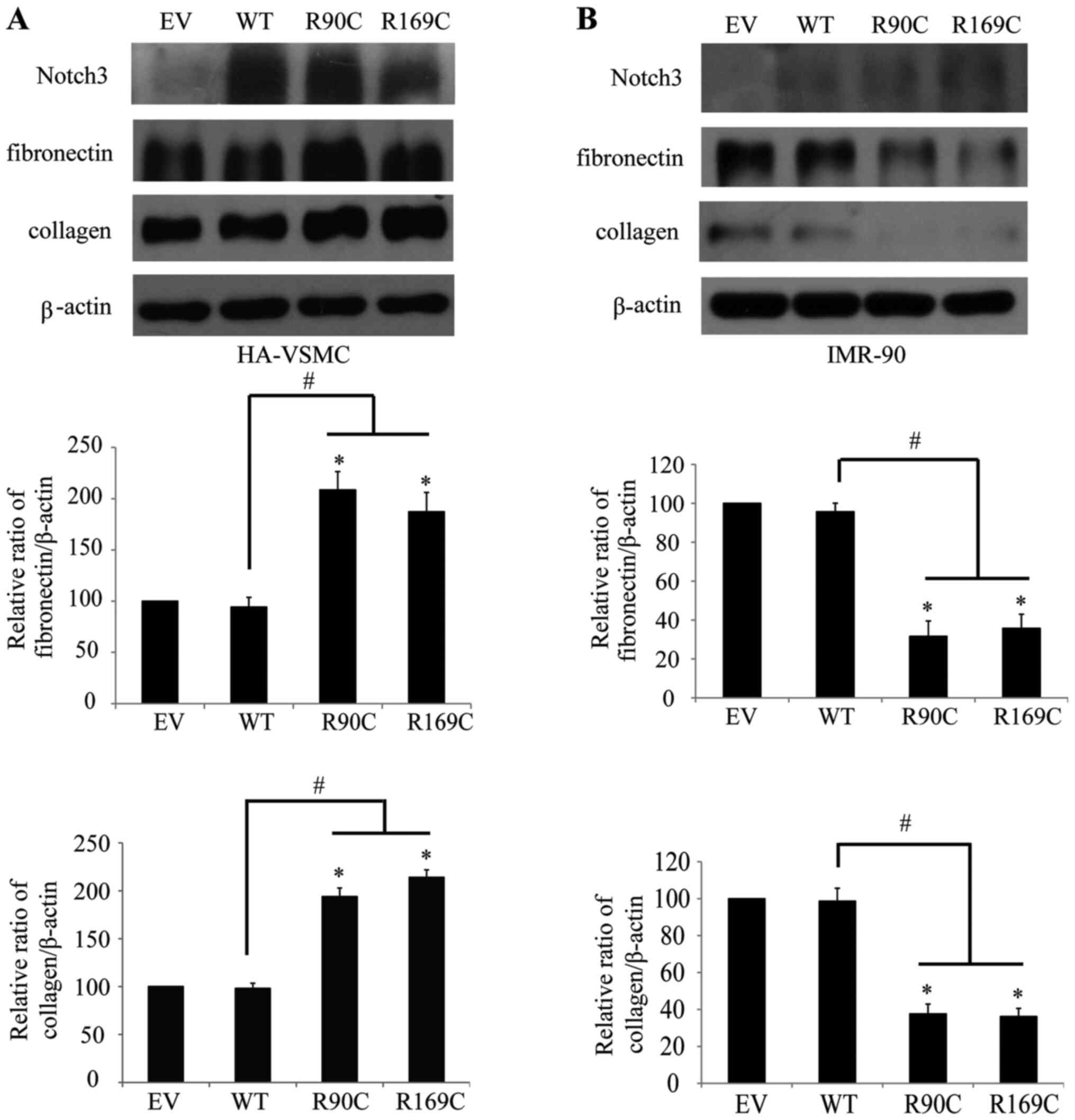
CADASIL mutants R90C and R169C were transfected into HA-VSMCs and
IMR-90 cells to elucidate the molecular mechanism of fibrosis. The
results from the western blot analysis revealed that the expression
level of extracellular matrix collagen and fibronectin were
significantly increased after the transfection of mutants (however,
this did not occur in WT) in HA-VSMCs (Fig. 1A), which is consistent with CADASIL
pathology (15). By contrast, the
results showed a different trend in IMR-90 cells, where collagen
and fibronectin were significantly decreased after transfection of
mutants; however, this did not occur in WT (Fig. 1B). These results indicated that
there was differential regulation of the extracellular matrix
between CADASIL mutants and Notch3 WT in VSMCs and fibroblasts.
CADASIL mutants downregulate the
levels of NO and iNOS in microglia
It has been reported that the activated Notch
signaling pathway regulate the proliferation and differentiation of
neural precursor cells, mediate the release of inflammatory
mediators and promote angiogenesis (16). Additionally, it serves an important
role in nerve damage repair, inflammatory response and angiogenesis
in ischemic areas (17). Abnormal
regulation of glial cells was observed in patients with CADASIL
(18). Thus, it was hypothesized
that inflammation may be involved in the pathogenesis of CADASIL.
According to the aforementioned findings, R90C and R169C Noctch3
mutants, as well as Notch3 WT, were transfected into BV2 cells to
study their roles in the regulation of the inflammatory response
following LPS administration. The transfection efficiency of R90C,
R169C and WT Notch3 vectors was confirmed by western blotting
(Fig. 2A). mRNA analysis showed
that the expression levels of TNF-α and IL-1β were not altered by
transfection with the mutants compared with those transfected with
WT Notch3 with or without LPS treatment (Fig. 2B). Next, the level of NO in the
culture medium was detected. The results showed that the NO level
of the cells transfected with mutants was significantly lower
compared with the cells transfected with WT with or without LPS
treatment (Fig. 2C). Furthermore,
the protein level of iNOS was in accordance with the change in NO
(Fig. 2D). This indicated that
downregulation of NO, which is a vasodilator factor, may be
involved in the pathogenesis of CADASIL.
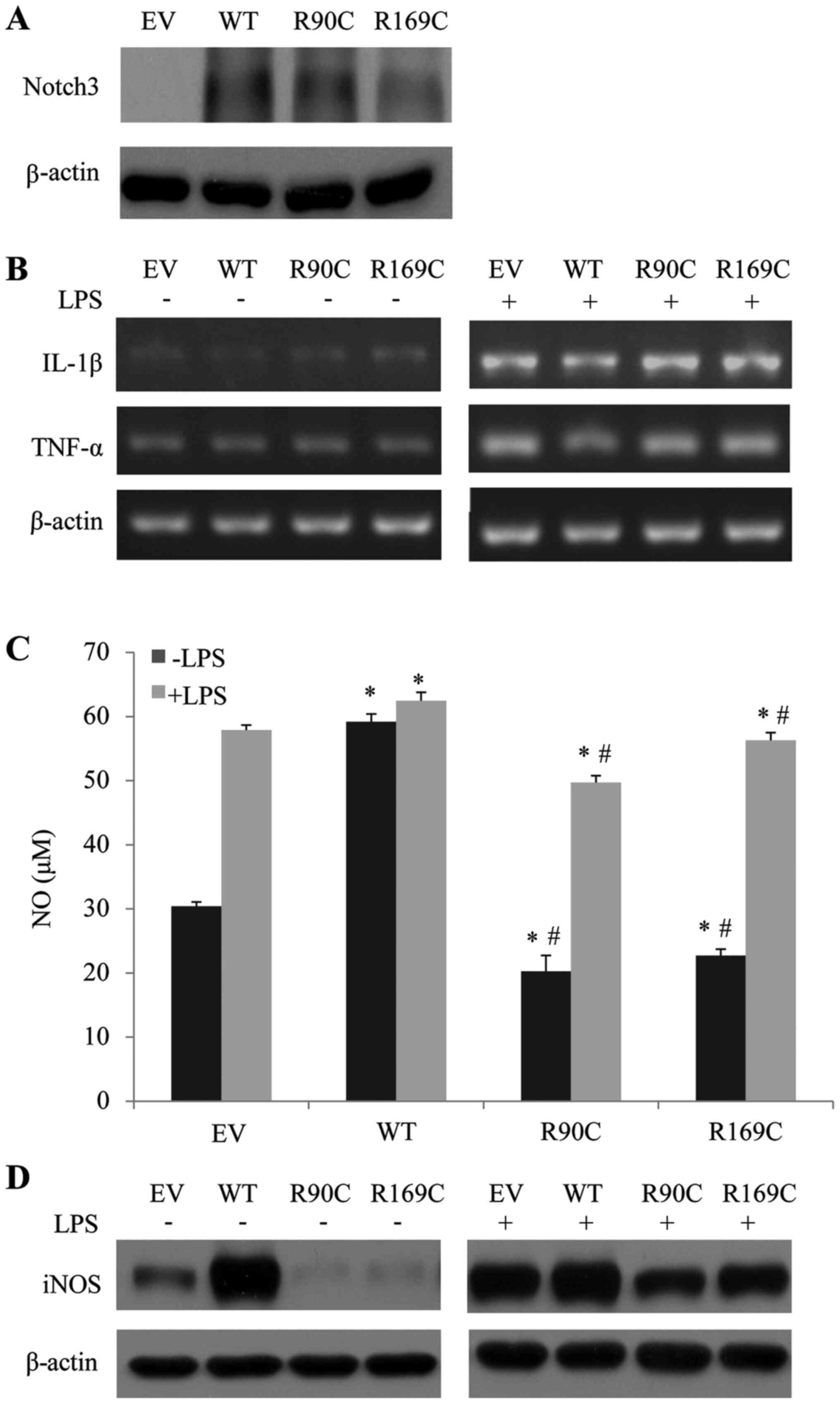 | Figure 2CADASIL mutants downregulate the
level of NO and iNOS in BV2 cells. (A) EV, WT, R90C and R169C were
transfected into BV2 cells and Notch3 protein expression was
detected by western blotting. (B) R90C and R169C CADASIL-Notch3
mutants had no effect on the mRNA expression of TNF-α or IL-1β.
R90C and R169C CADASIL-Notch3 mutants decreased (C) NO and (D) iNOS
levels. The NO level was determined by the Griess method, while the
iNOS level was detected by western blotting 2 days after
transfection. Protein densitometry quantification after
normalization with β-actin levels is shown. Data are presented as
mean ± standard error of the mean (n=3). *P<0.05 vs.
EV-transfected cells. #P<0.05 vs. WT-transfected
cells. EV, empty vector; WT, wild-type; NO, nitric oxide; iNOS,
inducible nitric oxide synthase; CADASIL, cerebral
autosomal-dominant arteriopathy with subcortical infarcts and
leukoencephalopathy; TNF-α, tumor necrosis factor-α; IL-1β,
interleukin-1β. |
CADASIL mutants promote the
proliferation and migration of HeLa cells; however, they inhibit
those of breast cancer cells
It has been reported that Notch3 protein, which is
present in numerous types of tumors, serves an important role in
promoting tumor progression (19).
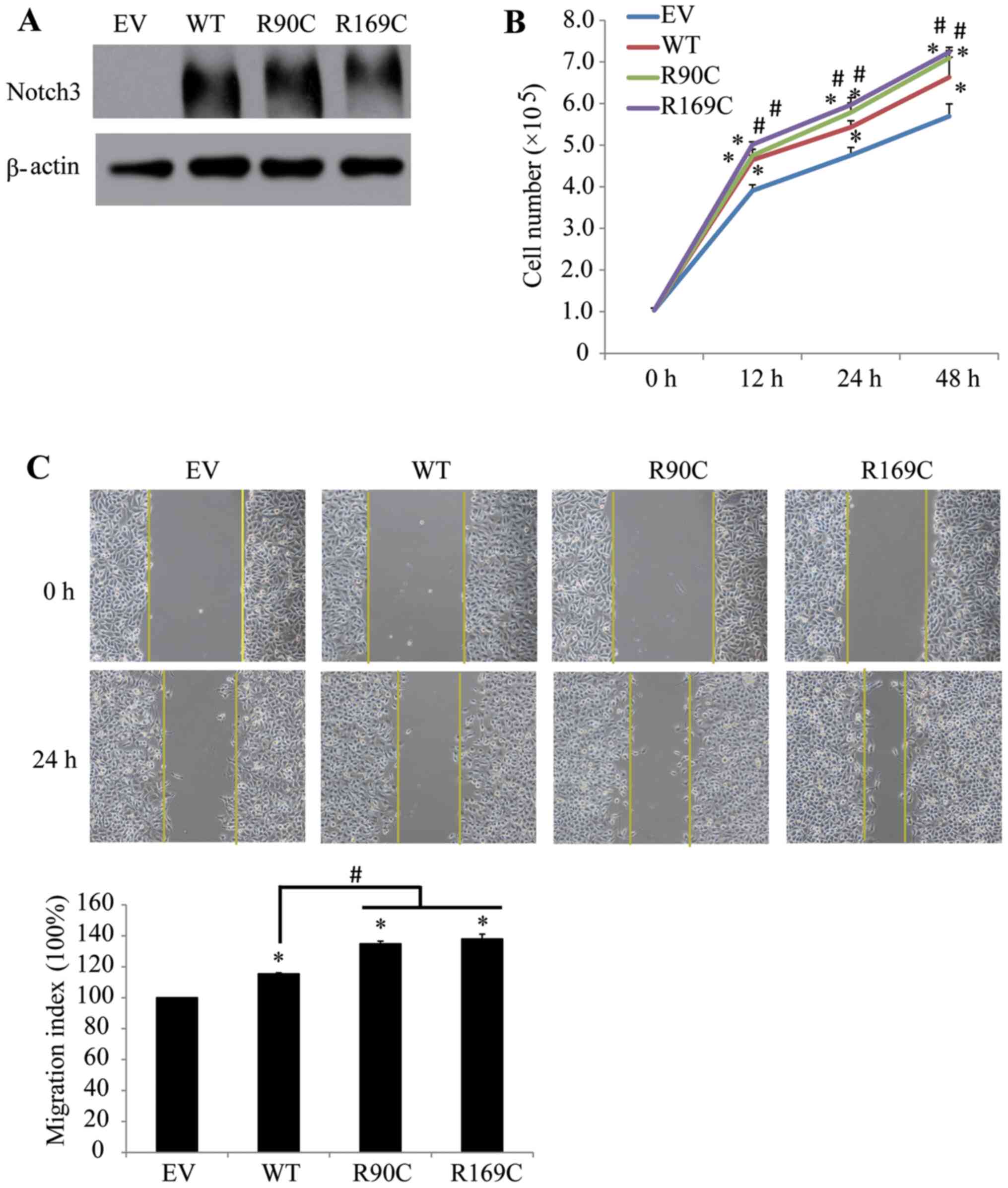
In the present study, HeLa cells were used to assess the effects of
the mutants on cell proliferation and migration, in which no
apparent difference was observed among Notch3-transfected and
EV-transfected cells. All Notch3 vectors effectively enhanced
Notch3 expression in HeLa cells (Fig.
3A). Exogenous expression of WT Notch3 led to a significant
increase in cell proliferation (Fig.
3B) and migration (Fig. 3C);
however, R90C and R169C exhibited a stronger promoter effect on
HeLa cells compared with WT. The aforementioned results showed that
both WT and CADASIL mutants were involved in the regulation of the
phenotype in HeLa cells; however, the regulatory effects of CADASIL
mutants were more apparent compared with WT. Additional cancer
cells were employed to explore whether the malignancy-promoting
effect of Notch3 is cell-specific or general. Notch3 proteins were
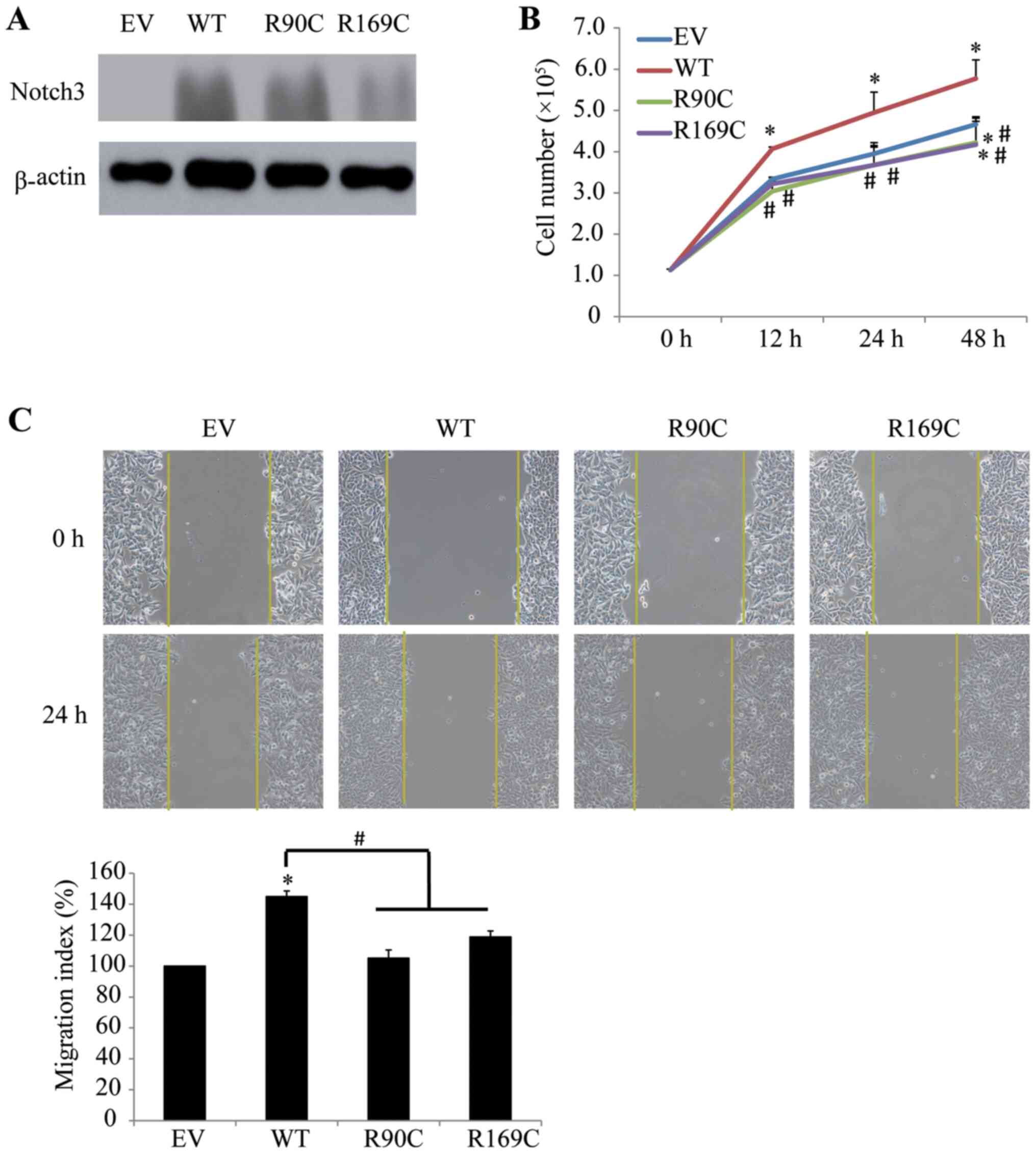
remarkably increased in MCF-7 cells by Notch3 mutants and WT
vectors transfection (Fig. 4A).
Notably, opposite outcomes to those observed in HeLa cells were
observed in MCF-7 cells transfected with mutants, since the
expression of exogenous Notch3 mutants significantly inhibited the
proliferation of MCF-7 cells at 48 h compared with the results upon
transfection with EV at 12, 24 and 48 h, and those transfected with
WT (Fig. 4B). The results of the
wound healing assay showed that overexpression of Notch3 mutants
significantly inhibited the migration of MCF-7 cells compared with
cells transfected with WT (Fig.
4C). There was no significant difference between transfected
mutants and EV on the migration of MCF-7 cells. In view of the
aforementioned results, it was concluded that this differential
regulation may be associated with the type of cancer cells. To
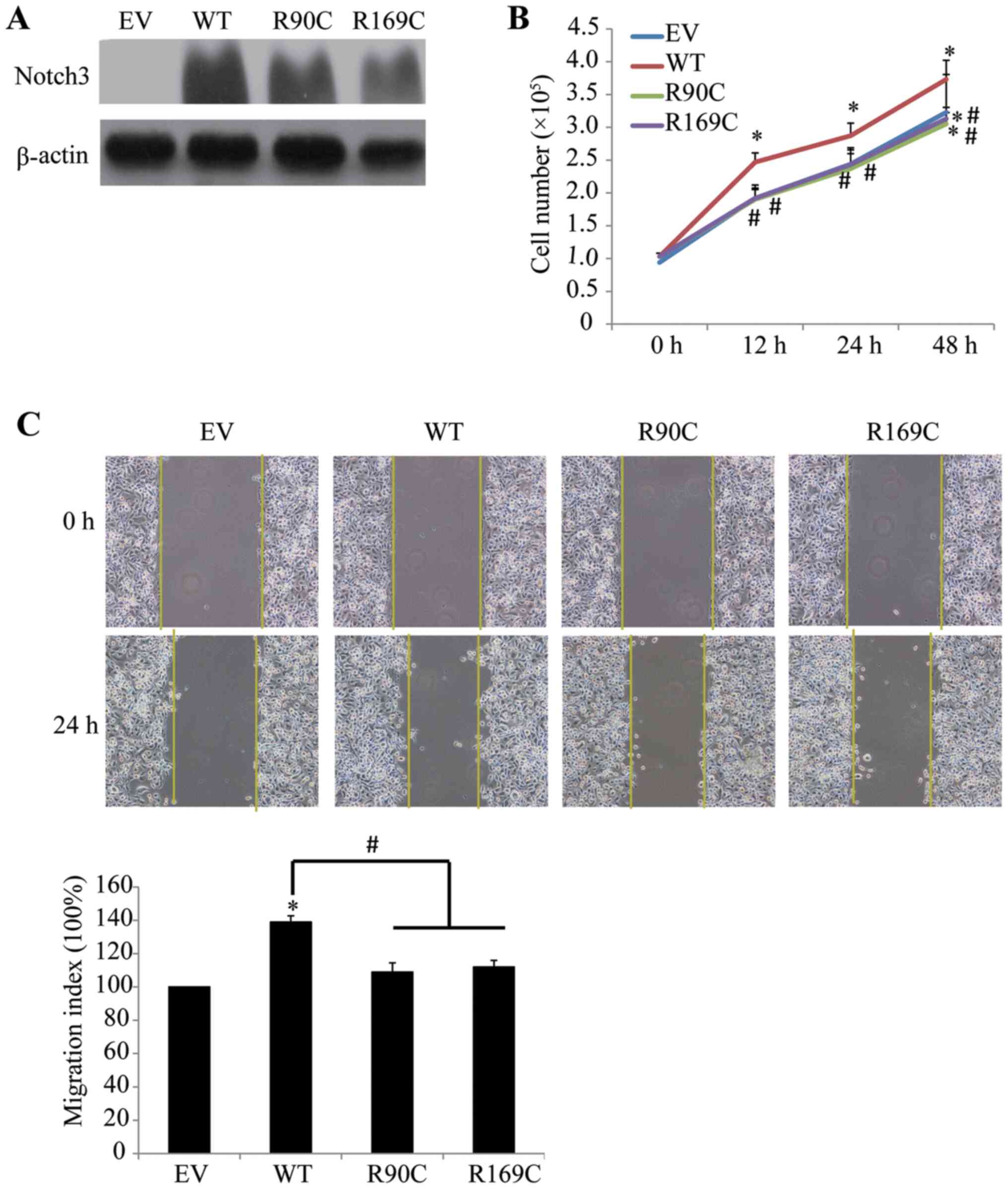
further verify this conclusion, HCC1937 cells, which are also
breast cancer cells, were selected for further investigation.
HCC1937 cells and MCF-7 cells exhibited similar changes in
proliferation and migration following transfection of CADASIL
mutants (Fig. 5A-C). These results
suggested that the differential regulation of phenotype between
CADASIL mutants R90C and R169C and WT was associated with cell
type.
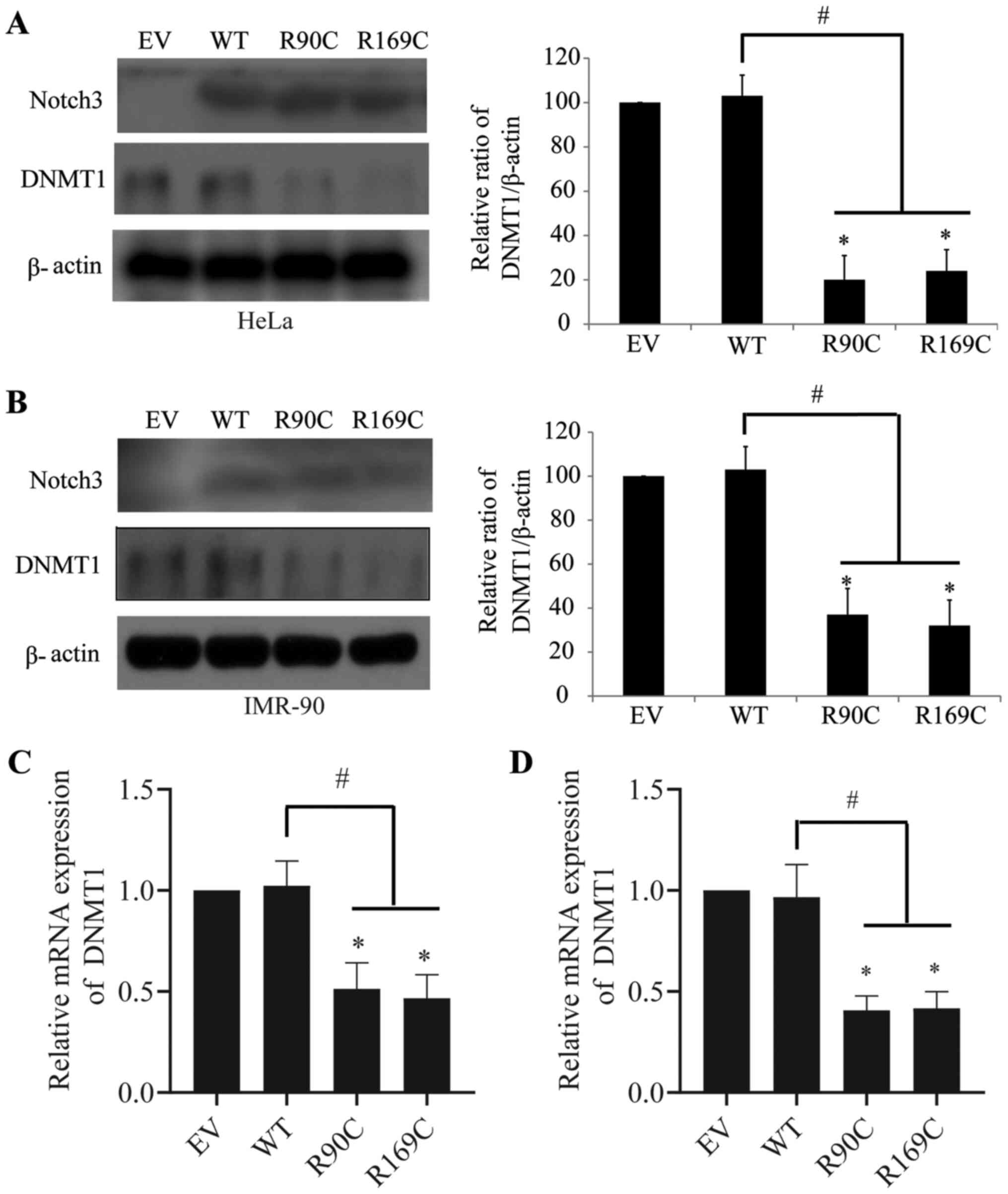
CADASIL mutants downregulate DNMT1 in
HeLa and IMR-90 cells
DNMT1 serves a critical role in the maintenance of
the genetic stability of methylation sites during replication
(20). Abnormal expression of DNMT1
can lead to unusual DNA methylation and further affect gene
expression and disease progression (20). The present study assessed the
regulation of CADASIL mutants and Notch3 WT on DNMT1 as a
preliminary exploration of the underlying mechanism. The results
showed that CADASIL mutants significantly downregulated the protein
expression of DNMT1 in HeLa (Fig.
6A) and IMR-90 (Fig. 6B) cells,
whereas WT had no effect. To further explore the mechanism
underlying CADASIL mutants-mediated reduction of DNMT1, mRNA
expression of DNMT1 was analyzed. The data implied that CADASIL
mutants reduced the mRNA expression of DNMT1 in HeLa (Fig. 6C) and IMR-90 (Fig. 6D) cells, indicating that the
regulatory events may occur at the transcriptional level. This
suggested that there may be deregulation of DNA methylation in
CADASIL, which may provide a novel insight for the exploration of
subsequent pathogenesis.
Discussion
Mutations in genes encoding Notch proteins lead to a
range of dysfunctions and diseases (21). Notch1 mutations have been reported
in a wide variety of human diseases, including Adams-Oliver
syndrome (22), left-sided
congenital heart disease (23) and
bicuspid aortic valve disease (24). Notch2 mutations are involved in the
development of Alagille syndrome (25), while mutations in Notch3 can cause
CADASIL. Diseases associated with Notch4 mutations have not been
thoroughly studied (21). The most
common mutations of Notch3 in CADASIL are R133C, R141C, R182C,
R153C, R169C and R90C (26). In the
present study, R90C and R169C CADASIL-Notch3 mutants and Notch3 WT
had different impacts on several cell lines, suggesting that Notch3
mutants may have more biological functions beyond current
knowledge.
Stenosis of cerebral white matter arterioles with
fibrosis and thickening of their walls have been attributed to
accumulation of various extracellular matrix components, which can
lead to decreased cerebral blood flow to such a severity that
lacunar infarcts occur in CADASIL (18). It had been reported that, in
diseased arteries, types I, III and VI collagen spread from an
external location (adventitia) into the vascular media, while type
IV collagen accumulated in an internal pattern (intima and media)
(27), which is consistent with the
present results. No major leakage of plasma fibrinogen or
fibronectin was observed in a previous study (15). In the present study, fibronectin was
observed to increase in HA-VSMCs following transfection with
mutants, whereas in IMR-90 cells, collagen and fibronectin were
decreased following transfection. Oide et al (28) reported that extensive arterial
medial smooth muscle cells and arterial adventitia extracellular
matrix were lost in CADASIL, which led to diffuse
leukoencephalopathy. It is commonly known that the adventitia is
primarily composed of fibroblasts. According to the aforementioned
results, it was suggested that Notch3 mutants affected the
physiological characteristics of arteries by two mechanisms: i)
Upregulation of extracellular matrix in VSMCs, which may induce
fibrotic thickening of arteriolar; and ii) downregulation of
extracellular matrix in fibroblasts, which may deprive the
structural and functional ability of the cerebrovascular system to
regulate the cerebral blood flow, most likely due to the loss of
arterial medial smooth muscle cells and mural extracellular
matrix.
Neuroinflammation mediated by microglia serves an
important role in damage of the central nervous system and
prognosis of the disease (18). In
a previous study, activation of the Notch signaling pathway caused
nerve damage, which contributed to the infiltration of inflammatory
cells via microglia activation (17). Abnormal regulation of glial cells
was observed in patients with CADASIL (29). Thus, a microglial cell model of
CADASIL was established to study the regulation of CADASIL mutants
on the inflammatory response. Both NO and iNOS levels observed in
two mutants groups were much lower compared with the WT group with
or without LPS treatment. Considering that NO is a factor that
contributes to vasodilatation and patients with CADASIL often
suffer ischemic stroke or subcutaneous infarction, limited NO
availability may contribute to an increase in the resting vasomotor
tone and, therefore, may serve an important role in cerebral
ischemia (30). It has been
reported that cerebral vasoreactivity enhanced by L-arginine, which
is the substrate for NOS, may have therapeutic implications for
CADASIL (31). In terms of a
pathogenic role of impaired cerebral hemodynamics and endothelial
dysfunction in CADASIL, it was hypothesized that Notch3 mutants may
also affect the synthesis of NO in endothelial cells. However,
further studies are required to explore this.
Notch3 primarily serves a role in promoting cancer
development and it has been detected in multiple types of tumor,
including ovarian, cervical, breast and colon cancer (19). Compared with the effects of Notch3
WT, CADASIL mutants promoted the proliferation and migration of
HeLa cells and inhibit these of breast cancer cells. A previous
study observed hyperplasia in organs, including the liver, kidney
and prostate in patients with CADASIL (32). The autopsy examination of a Japanese
case revealed that, besides the vascular and neurological lesions
characteristic of CADASIL, multiple neoplastic lesions were also
observed, including carcinoid tumorlets and diffuse idiopathic
pulmonary neuroendocrine cell hyperplasia in the lungs, renal cell
carcinoma, prostatic adenocarcinoma and adenomatoid tumor of the
epididymis (33). Combined with the
present data, this suggested that Notch3 mutants may contribute to
the increased or decreased risks of different cancer types in
CADASIL.
Cerebral small vessel disease (CSVD) is one of the
most common nervous system disease worldwide (1920-2017) and is
considered an important pathological process of subcortical
structures such as lacunar infarcts, white matter lesions and
microbleeds (34). CSVD is an
important cerebral microvascular pathogenesis, since it is the
cause of 20% of strokes worldwide (1920-2017) and the most common
cause of cognitive impairment and dementia, including vascular
dementia (34). It has been
reported that CSVD contributes to the occurrence of Alzheimer's
disease (AD) and CADASIL (35). The
association between epigenetics and CSVD has not yet been
established; however, research on cerebrovascular diseases and DNA
methylation are gradually increasing, particularly on DNA
methylation abnormalities in stroke (36). A previous study that discovered the
mechanism of cerebral small vessel injury aided the present study
(36). Furthermore, another
previous study reported that DNA methylation served an important
role in gene transcription, gene imprinting and embryo development
(20). DNMT1 primarily maintained
the genetic stability of the methylation site during the
replication process and its abnormal expression lead to
dysregulation of DNA methylation, therefore affecting gene
expression and disease progression (20). To the best of our knowledge, the
present study is the first to report that CADASIL mutants
significantly inhibited DNMT1 mRNA and protein expression in both
HeLa and IMR-90 cells, whereas WT Notch3 had no effect.
Multiple studies have revealed that abnormal DNA
methylation was associated with AD (37,38).
Both the mRNA and protein levels of DNMT1 and DNMT3a, as well as
those of methyl CpG binding protein 2, have been demonstrated to be
downregulated in AD (39). However,
the exact underlying mechanism remains to be discovered. It was
reported that a pathogenic presenilin mutation in AD caused a
specific increase in p53 protein (40), which was found to block the
transcription of the DNMT1 gene by binding to its promoter region
in hepatocellular carcinoma, ovarian cancer and pancreatic ductal
adenocarcinoma (41). Whether the
downregulation of DNMT1 in CADASIL and AD was also regulated by
p53-mediated transcriptional inhibition needs to be further
explored. The present data suggested that there may be an abnormal
regulation of DNA methylation in CADASIL, which provides a novel
approach for the exploration of subsequent pathogenesis.
In summary, the present study revealed that Notch3
mutants had differential effects depending on cell type, suggesting
that Notch3 mutants may have more biological functions beyond our
current knowledge. These results provided novel avenues for further
research on the pathogenesis of CADASIL and future studies need to
be carried out to elucidate the specific pathways or underlying
mechanisms of this differential regulation.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Natural Science
Foundation of Zhejiang Province, China (grant no. LQ19H080003).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YuZ, KY and CL conceived and designed the current
study. ZH and RZ conducted the research. YiZ and YS analyzed data
and wrote the manuscript. KY and CL revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray SJ: Notch signalling in context. Nat
Rev Mol Cell Biol. 17:722–735. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Domenga V, Fardoux P, Lacombe P, Monet M,
Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z,
et al: Notch3 is required for arterial identity and maturation of
vascular smooth muscle cells. Genes Dev. 18:2730–2735.
2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang T, Baron M and Trump D: An overview
of Notch3 function in vascular smooth muscle cells. Prog Biophys
Mol Biol. 96:499–509. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Joutel A, Corpechot C, Ducros A, Vahedi K,
Chabriat H, Mouton P, Alamowitch S, Domenga V, Cécillion M,
Marechal E, et al: Notch3 mutations in CADASIL, a hereditary
adult-onset condition causing stroke and dementia. Nature.
383:707–710. 1996.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Hervé D and Chabriat H: Cadasil. J Geriatr
Psychiatry Neurol. 23:269–276. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Joutel A, Vahedi K, Corpechot C, Troesch
A, Chabriat H, Vayssière C, Cruaud C, Maciazek J, Weissenbach J,
Bousser MG, et al: Strong clustering and stereotyped nature of
Notch3 mutations in CADASIL patients. Lancet. 350:1511–1515.
1997.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dichgans M, Ludwig H, Müller-Höcker J,
Messerschmidt A and Gasser T: Small in-frame deletions and missense
mutations in CADASIL: 3D models predict misfolding of Notch3
EGF-like repeat domains. Eur J Hum Genet. 8:280–285.
2000.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Duering M, Karpinska A, Rosner S, Hopfner
F, Zechmeister M, Peters N, Kremmer E, Haffner C, Giese A, Dichgans
M, et al: Co-aggregate formation of CADASIL-mutant NOTCH3: A
single-particle analysis. Hum Mol Genet. 20:3256–3265.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Cognat E, Baron-Menguy C, Domenga-Denier
V, Cleophax S, Fouillade C, Monet-Leprêtre M, Dewerchin M and
Joutel A: Archetypal Arg169Cys mutation in NOTCH3 does not drive
the pathogenesis in cerebral autosomal dominant arteriopathy with
subcortical infarcts and leucoencephalopathy via a loss-of-function
mechanism. Stroke. 45:842–849. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Joutel A: Pathogenesis of CADASIL:
Transgenic and knock-out mice to probe function and dysfunction of
the mutated gene, Notch3, in the cerebrovasculature. BioEssays.
33:73–80. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Rutten JW, Haan J, Terwindt GM, van Duinen
SG, Boon EM and Lesnik Oberstein SA: Interpretation of NOTCH3
mutations in the diagnosis of CADASIL. Expert Rev Mol Diagn.
14:593–603. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yamaguchi N, Oyama T, Ito E, Satoh H,
Azuma S, Hayashi M, Shimizu K, Honma R, Yanagisawa Y, Nishikawa A,
et al: NOTCH3 signaling pathway plays crucial roles in the
proliferation of ErbB2-negative human breast cancer cells. Cancer
Res. 68:1881–1888. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Song G, Zhang Y and Wang L: MicroRNA-206
targets notch3, activates apoptosis, and inhibits tumor cell
migration and focus formation. J Biol Chem. 284:31921–31927.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Miao Q, Paloneva T, Tuisku S, Roine S,
Poyhonen M, Viitanen M and Kalimo H: Arterioles of the lenticular
nucleus in CADASIL. Stroke. 37:2242–2247. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Dong H, Blaivas M and Wang MM:
Bidirectional encroachment of collagen into the tunica media in
cerebral autosomal dominant arteriopathy with subcortical infarcts
and leukoencephalopathy. Brain Res. 1456:64–71. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Grandbarbe L, Michelucci A, Heurtaux T,
Hemmer K, Morga E and Heuschling P: Notch signaling modulates the
activation of microglial cells. Glia. 55:1519–1530. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yao L, Cao Q, Wu C, Kaur C, Hao A and Ling
EA: Notch signaling in the central nervous system with special
reference to its expression in microglia. CNS Neurol Disord Drug
Targets. 12:807–814. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Dichgans M: Cognition in CADASIL. Stroke.
40 (Suppl):S45–S47. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Aburjania Z, Jang S, Whitt J,
Jaskula-Stzul R, Chen H and Rose JB: The Role of Notch3 in Cancer.
Oncologist. 23:900–911. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Edwards JR, Yarychkivska O, Boulard M and
Bestor TH: DNA methylation and DNA methyltransferases. Epigenetics
Chromatin. 10(23)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Meester JAN, Verstraeten A, Alaerts M,
Schepers D, Van Laer L and Loeys BL: Overlapping but distinct roles
for NOTCH receptors in human cardiovascular disease. Clin Genet.
95:85–94. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Southgate L, Sukalo M, Karountzos ASV,
Taylor EJ, Collinson CS, Ruddy D, Snape KM, Dallapiccola B, Tolmie
JL, Joss S, et al: Haploinsufficiency of the NOTCH1 Receptor as a
Cause of Adams-Oliver Syndrome With Variable Cardiac Anomalies.
Circ Cardiovasc Genet. 8:572–581. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kerstjens-Frederikse WS, van de Laar IM,
Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, Klein
Wassink-Ruiter JS, van der Zwaag PA, du Marchie Sarvaas GJ, Bergman
KA, et al: Cardiovascular malformations caused by NOTCH1 mutations
do not keep left: Data on 428 probands with left-sided CHD and
their families. Genet Med. 18:914–923. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mohamed SA, Aherrahrou Z, Liptau H, Erasmi
AW, Hagemann C, Wrobel S, Borzym K, Schunkert H, Sievers HH and
Erdmann J: Novel missense mutations (p.T596M and p.P1797H) in
NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res
Commun. 345:1460–1465. 2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
McDaniell R, Warthen DM, Sanchez-Lara PA,
Pai A, Krantz ID, Piccoli DA and Spinner NB: NOTCH2 mutations cause
Alagille syndrome, a heterogeneous disorder of the notch signaling
pathway. Am J Hum Genet. 79:169–173. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Haritunians T, Chow T, De Lange RP,
Nichols JT, Ghavimi D, Dorrani N, St Clair DM, Weinmaster G and
Schanen C: Functional analysis of a recurrent missense mutation in
Notch3 in CADASIL. J Neurol Neurosurg Psychiatry. 76:1242–1248.
2005.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Miao Q, Paloneva T, Tuominen S, Pöyhönen
M, Tuisku S, Viitanen M and Kalimo H: Fibrosis and stenosis of the
long penetrating cerebral arteries: The cause of the white matter
pathology in cerebral autosomal dominant arteriopathy with
subcortical infarcts and leukoencephalopathy. Brain Pathol.
14:358–364. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Oide T, Nakayama H, Yanagawa S, Ito N,
Ikeda S and Arima K: Extensive loss of arterial medial smooth
muscle cells and mural extracellular matrix in cerebral autosomal
recessive arteriopathy with subcortical infarcts and
leukoencephalopathy (CARASIL). Neuropathology. 28:132–142.
2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Miao Q, Kalimo H, Bogdanovic N, Kostulas
K, Börjesson-Hanson A and Viitanen M: Cerebral arteriolar pathology
in a 32-year-old patient with CADASIL. Neuropathol Appl Neurobiol.
32:455–458. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
DeMartino AW, Kim-Shapiro DB, Patel RP and
Gladwin MT: Nitrite and nitrate chemical biology and signalling. Br
J Pharmacol. 176:228–245. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Peters N, Freilinger T, Opherk C,
Pfefferkorn T and Dichgans M: Enhanced L-arginine-induced
vasoreactivity suggests endothelial dysfunction in CADASIL. J
Neurol. 255:1203–1208. 2008.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ling Y, De Guio F, Jouvent E, Duering M,
Hervé D, Guichard JP, Godin O, Dichgans M and Chabriat H: Clinical
correlates of longitudinal MRI changes in CADASIL. J Cereb Blood
Flow Metab. 39:1299–1305. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hassan WA, Udaka N, Ueda A, Ando Y and Ito
T: Neoplastic lesions in CADASIL syndrome: Report of an autopsied
Japanese case. Int J Clin Exp Pathol. 8:7533–7539. 2015.PubMed/NCBI
|
|
34
|
Søndergaard CB, Nielsen JE, Hansen CK and
Christensen H: Hereditary cerebral small vessel disease and stroke.
Clin Neurol Neurosurg. 155:45–57. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Müller K, Courtois G, Ursini MV and
Schwaninger M: New Insight Into the Pathogenesis of Cerebral
Small-Vessel Diseases. Stroke. 48:520–527. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Baccarelli A, Wright R, Bollati V,
Litonjua A, Zanobetti A, Tarantini L, Sparrow D, Vokonas P and
Schwartz J: Ischemic heart disease and stroke in relation to blood
DNA methylation. Epidemiology. 21:819–828. 2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Altuna M, Urdánoz-Casado A, Sánchez-Ruiz
de Gordoa J, Zelaya MV, Labarga A, Lepesant JMJ, Roldán M,
Blanco-Luquin I, Perdones Á, Larumbe R, et al: DNA methylation
signature of human hippocampus in Alzheimer's disease is linked to
neurogenesis. Clin Epigenetics. 11(91)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yokoyama AS, Rutledge JC and Medici V: DNA
methylation alterations in Alzheimer's disease. Environ Epigenet.
3(dvx008)2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bihaqi SW and Zawia NH: Alzheimer's
disease biomarkers and epigenetic intermediates following exposure
to Pb in vitro. Curr Alzheimer Res. 9:555–562. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Bialopiotrowicz E, Szybinska A, Kuzniewska
B, Buizza L, Uberti D, Kuznicki J and Wojda U: Highly pathogenic
Alzheimer's disease presenilin 1 P117R mutation causes a specific
increase in p53 and p21 protein levels and cell cycle dysregulation
in human lymphocytes. J Alzheimers Dis. 32:397–415. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Enane FO, Saunthararajah Y and Korc M:
Differentiation therapy and the mechanisms that terminate cancer
cell proliferation without harming normal cells. Cell Death Dis.
9:912–926. 2018.PubMed/NCBI View Article : Google Scholar
|