Introduction
The most effective intervention for acute myocardial
infarction is timely revascularization (1-3).
However, re-establishing blood to ischemic areas yields additional
myocardial damage including inflammation, apoptosis and autophagy,
which is known as myocardial ischemia/reperfusion (I/R) injury
(MIRI) (1-3).
The pathophysiological mechanism of MIRI is complex and numerous
studies have confirmed that endoplasmic reticulum (ER) stress
(ERS)-induced apoptosis serves important roles in the progression
of MIRI (4,5). Conventionally, the ER is indispensable
for the biosynthesis, folding, processing, excretion and
transportation of proteins (4,5).
However, the disruption of ER homeostasis caused by MIRI, which is
referred to as ERS, leads to inaccurate synthesis and assembly of
proteins (4,5). Increasing evidence has indicated that
the activation of ERS and the consequent elevation of apoptotic
cascade events act as crucial pathogenic mechanisms in MIRI
(4,5). Targeting ERS-associated apoptosis may
be an important therapeutic approach for MIRI.
Pleckstrin homology-like domain family A member 3
(PHLDA3) serves as a crucial member of the PHLDA family and encodes
a small, 127-amino acid protein containing only a PH domain
(6-8).
PHLDA3 was reported to be highly associated with various
physiological conditions, including pressure overload-induced
cardiac remodeling (9), vascular
development (6) and tumor
development (8). Among these, the
PHLDA3-mediated apoptotic cascade is hypothesized to be a common
pathological mechanism in a wide range of diseases, including
hepatocyte injury and cardiac remodeling (7,10). Han
et al (7) demonstrated that
PHLDA3 was upregulated in liver injury with ERS induction and that
PHLDA3 overexpression was associated with the severity of
hepatocyte injury in a cell model of ERS. However, the roles of
PHLDA3 in myocardial I/R injury associated with ERS and apoptosis
remain unknown.
Stimulation of the p-PI3K/AKT signaling pathway
serves a central role in the protection of MIRI (4,11). The
repression of ERS-induced apoptosis resulting from p-PI3K/AKT
activation was verified as a therapeutic avenue in cardioprotection
during the initiation and development of MIRI (11,12).
Notably, numerous studies reported that PHLDA3 is a negative
modulator in multiple pathological disorders, including vascular
development, cardiac hypertrophy and islets engraftment, mainly by
its inhibition of the p-AKT pathway (6,9,13).
However, whether repression of the p-PI3K/AKT pathway could also be
modulated via PHLDA3 in MIRI remains unknown. The aim of the
current study was to determine whether PHLDA3 served protective
effects on hypoxia/reoxygenation (H/R)-injured cardiomyocytes by
inhibiting ERS-induced apoptosis.
Materials and methods
Chemicals and reagents
DMEM, PBS, trypsin, FBS and collagenase type II were
purchased from Gibco; Thermo Fisher Scientific, Inc. Cell Counting
Kit-8 (CCK-8) and LY294002 (a p-PI3K/AKT inhibitor) were purchased
from Dojindo Molecular Technologies, Inc. and Selleck Chemicals,
respectively. Lactate dehydrogenase (LDH; cat. no. 03010703011) and
creatine kinase (CK; cat. no. 03010702011) were detected by
commercially available ELISA kits (Nanjing Jiancheng Bioengineering
Institute). The following primary antibodies were obtained from
Abcam: PHLDA3 (cat. no. ab22822; 1:500), phosphorylated (p)-PI3K
(cat. no. ab182651; 1:400), PI3K (cat. no. ab180967; 1:300), 78 kDa
glucose-regulated protein (GRP78; cat. no. ab108615; 1:600),
cleaved caspase-12 (cat. no. ab62484; cat. no. 1:600) and GAPDH
(cat. no. ab8245, 1:500). Antibodies against p-AKT (cat. no. 4060;
1:600), AKT (cat. no. 4691; 1:400) and CHOP (cat. no. 5554; 1:600)
were purchased from Cell Signaling Technology, Inc. Horseradish
peroxidase-conjugated goat-anti rabbit secondary antibodies were
obtained from BIOSS. A BCA protein assay kit was purchased from
Pierce, Thermo Fisher Scientific, Inc.
Neonatal rat cardiomyocyte (NRCM)
culture
NRCMs were isolated from the ventricles of 144
1-3-day-old male Sprague-Dawley rats (body weight, 15-20 g) as
previously described (9,14-16).
Animals were purchased from the Animal Experimental Center of
Hainan Medical University (Haikou, China). Animals were bred in a
standard environment with controlled temperature (20-25˚C),
humidity (40-60%) and light conditions (12 h light/dark cycles).
Experiments and animal care were performed in adherence with the
Guide for the Care and Use of Laboratory Animals published by the
US National Institutes of Health (NIH; 8th Edition; 2011) (17) and were approved by the Hainan
Affiliated Hospital of Hainan Medical University.
Cardiomyocyte isolation was performed as previously
described (17). Briefly, rat
hearts were quickly removed following sacrifice by swift
decapitation according to the Guide for the Care and Use of
Laboratory Animals published by the US NIH (17). Large blood vessels were excised
carefully, and the obtained ventricles were rinsed in ice-cold PBS
three times to remove residual blood. Following this, ventricles
were placed in a dish with ice-cold PBS. Heart tissues were
digested with 0.08% collagenase type II and 0.125% trypsin at room
temperature for 5 min. NRCMs were centrifuged at 110 x g for 10 min
at 37˚C and resuspended in DMEM supplemented with 10% FBS and 1%
penicillin/streptomycin (HyClone; Cytiva) in a humidified incubator
at 37˚C with 5% CO2 and 95% O2.
Construction of a
hypoxia/reoxygenation (H/R) injury model and experimental
groups
H/R injury was established as previously described
(14). Briefly, cultured NRCMs were
washed twice with PBS and preserved in serum-free DMEM. Cells were
then incubated at 37˚C in an anaerobic chamber for 4 h. Following
this, cells were transferred to a normal incubator with 95%
O2 and 5% CO2 at 37˚C for an additional 6 h
for reoxygenation.
To determine the possible function of PHLDA3 in
MIRI, primary cardiomyocytes were randomly divided into four
groups: i) Control + adenovirus encoding scrambled short hairpin
(Adsh) RNA group, which consisted of NRCMs cultured in normoxic
condition following AdshRNA transfection; ii) control + adenoviral
vectors encoding PHLDA3 shRNA (AdshPHLDA3) group, which consisted
of NRCMs cultured under normoxic conditions following AdshPHLDA3
transfection; iii) the H/R + AdshRNA group, which consisted of
NRCMs cultured under H/R conditions following AdshRNA transfection;
and iv) H/R + AdshPHLDA3 group, which consisted of NRCMs cultured
in H/R conditions following AdshPHLDA3 transfection. The methods
for experimental design were performed according to a method widely
reported by previous studies (9,18-23).
A total of 20 neonatal rats were used in each group.
Additionally, to detect underlying mechanisms
associated with the p-PI3K/AKT pathway, NRCMs were pretreated with
10 µM LY294002, a p-PI3K/AKT inhibitor, for 30 min prior to
AdshPHLDA3 administration and were subjected to H/R intervention as
previously described (24). PBS was
used as a control. During this experiment, 16 neonatal rats were
used for each group. The protocol for each group was repeated ≥4
times.
Adenoviral construction and
transduction into cardiomyocytes
AdshPHLDA3 or AdshRNA as a control were provided by
Shanghai GeneChem Co., Ltd. and constructed as previously described
(24,25). Briefly, AdshPHLDA3 was synthesized
and generated using an AdMax system (Microbix Biosystems Inc.).
293T cells (SunBio, Inc.) were used to package and amplify acquired
adenoviruses and the final virus concentration was
1x1011 plaque-forming units.
Furthermore, for viral transfection in the in
vitro experiments, NRCMs were seeded at a density of
1x105 cells per 24-well plate were incubated with
AdshPHLDA3 or AdshRNA at 50 multiplicities of infection for 2 h at
37˚C. Following this, the medium was removed and NRCMs were
maintained at 37˚C with complete medium for 48 h.
Cell viability assay
CCK-8 assays were used to measure cell viability in
each group as previously described (24). Optical density values were detected
at a wavelength of 450 nm using a microplate reader (Bio-Rad
Laboratories, Inc.). Cell viability was evaluated as the percentage
relative to the control group.
Measurement of myocardial injury
markers
LDH and CK are commonly used as markers for
myocardial death following MIRI (14,25,26).
Following the indicated treatments, the medium of cultured NRCMs
was collected and commercially available biochemical kits (Nanjing
Jiancheng Bioengineering Institute) were used according to the
manufacturer's protocol to detect levels of LDH and CK. Results are
presented as U/l.
Apoptosis detection
Apoptotic NRCMs were assessed by flow cytometry as
previously described (24).
Following the indicated treatments, NRCMs were maintained in 100 µl
binding buffer containing 5 µl Annexin V-APC and 5 µl
7-minoactinomycin D (7-AAD). Subsequently, stained cells were
observed using a flow cytometer (Beckman Coulter, Inc.). NRCMs
positive for 7-AAD and Annexin V-APC were identified as apoptotic
cells.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was used to detect mRNA levels as previously
described (4,14). Total RNA was extracted from
harvested NRCMs with TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). RT-qPCR was performed using a SYBR-Green
Master Mix kit (Thermo Fisher Scientific, Inc.) on the 7500 ABI
Prism system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The following thermocycling conditions were used for the qPCR: 2
min at 50˚C, 10 min at 95˚C, 40 cycles of 95˚C for 30 sec and 60˚C
for 30 sec. mRNA levels of PHLDA3, CHOP, GRP78 and caspase-12 were
normalized to β-actin. The following primer pairs were used for the
qPCR: PHLDA3 forward, 5'-CATCTACTTCACGCTAGTGACCG-3' and reverse,
5'-TCTGGATGGCCTGTTGATTCT-3'; CHOP forward,
5'-TAGCTTGGCTGACTGAGGAGC-3' and reverse,
5'-CTTCAGCAAGCTGTGCCACT-3'; GRP78 forward,
5'-GATAATCAGCCCACCGTAACAAT-3' and reverse,
5'-GCAAACTTCTCGGCGTCATT-3'; caspase-12 forward,
5'-CATTGCCAATTCCGACAAAC-3' and reverse 5'-CCTTCCTTCTCCATCACTGGA-3'
and β-actin forward, 5'-CGTTGACATCCGTAAAGACCTC-3' and reverse,
5'-TAGGAGCCAGGGCAGTAATCT-3'.
Western blotting
Western blotting was performed to detect the protein
levels of PHLDA3, PI3K, p-PI3K, AKT, p-AKT, CHOP, GRP78, cleaved
caspase-12 and GAPDH in NRCMs after the indicated treatments as
previously described (4,14). Briefly, cardiomyocytes were lysed by
ice-cold RIPA buffer (Beyotime Institute of Biotechnology). Protein
concentrations were detected using a BCA kit (Pierce; Thermo Fisher
Scientific, Inc.). Following this, 50 µg protein/lane was separated
via 10% SDS-PAGE and transferred onto PVDF membranes (EMD
Millipore). PVDF membranes were then incubated with primary
antibodies against PHLDA3, PI3K, p-PI3K, AKT, p-AKT, CHOP, GRP78,
cleaved caspase-12 and GAPDH overnight at 4˚C. Following three
washes with TBS-0.1% Tween-20, membranes were incubated with
horseradish peroxidase-conjugated rabbit anti-rat IgG secondary
antibodies for 2 h at room temperature. Finally, an enhanced
chemiluminescence system (Thermo Fisher Scientific, Inc.) was used
to visualize protein bands. Quantity One 1-D software (version
4.6.9; Bio-Rad Laboratories, Inc.) was used for densitometric
analysis.
Statistical analysis
SPSS software (version 17.0; SPSS, Inc.) was used
for statistical analysis. Data are presented as the mean ± SD from
four independent experiments. Student's t-test or one-way ANOVA
followed by Tukey's post hoc test were used for comparisons between
groups (9,22,27).
P<0.05 was considered to indicate a statistically significant
difference.
Results
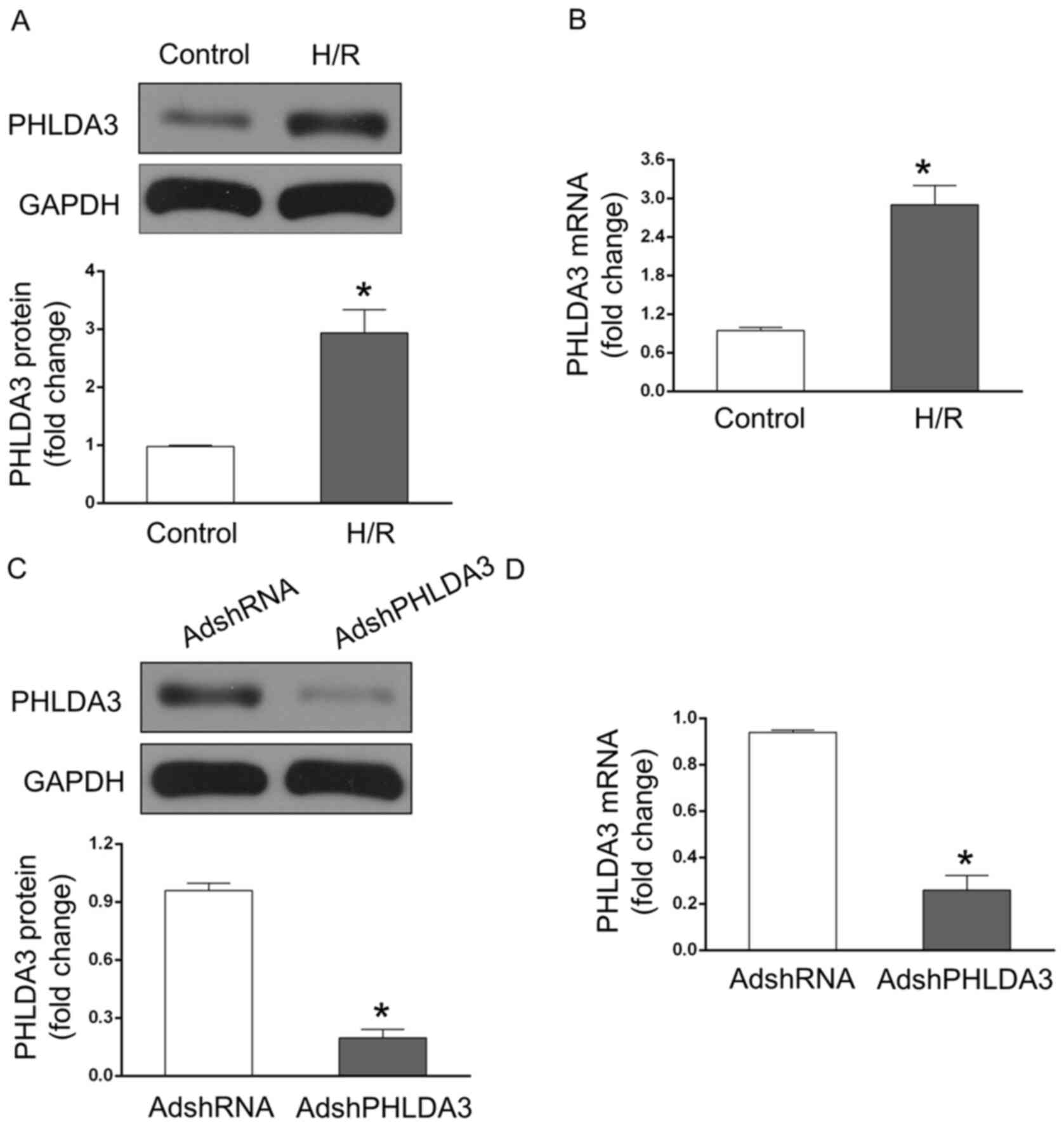
PHLDA3 expression is upregulated
following MIRI
To explore the possible involvement of PHLDA3 during
MIRI, the protein and mRNA levels of PHLDA3 in NRCMs following
exposure to H/R injury were measured. The results indicated that
PHLDA3 protein expression was significantly increased in the H/R
group compared with the control group (Fig. 1A). Similarly, the mRNA levels of
PHLDA3 in H/R-treated NRCMs was significantly higher compared with
the control group (Fig. 1B). These
data suggested that PHLDA3 may play a role in MIRI. Furthermore, to
detect the effects of PHLDA3 in response to MIRI, AdshPHLDA3 was
transfected into NRCMs to inhibit PHLDA3 expression. The results
demonstrated that the protein (Fig.
1C) and mRNA (Fig. 1D) levels
of PHLDA3 in NRCMs significantly decreased following AdshPHLDA3
transfection compared with the AdshRNA group. Additionally, the
mRNA levels of PHLDA3 in H/R-treated NRCMs were measured by
RT-qPCR. The results demonstrated that AdshPHLDA3-transfected NRCMs
decreased the mRNA levels of PHLDA3 following H/R compared with the
H/R + AdshRNA group (data not shown). However, there was no
significant difference between the groups (data not shown).
PHLDA3 inhibition represses
ERS-induced apoptosis in myocardial H/R injury
To investigate whether PHLDA3 inhibition attenuated
H/R-induced myocardial injury, cell viability and LDH/CK release
were detected by CCK-8 assays and ELISA, respectively. The results
of the CCK-8 assays (Fig. 2A)
demonstrated that cell viability was higher in the Control group
relative to the H/R groups. PHLDA3 inhibition following AdshPHLDA3
transfection partially restored the reduced cell viability in
response to H/R injury compared with the AdshRNA group.
Additionally, H/R-induced LDH (Fig.
2B) and CK (Fig. 2C) release
were significantly repressed by PHLDA3 inhibition (AdshPHLDA3 + H/R
group vs. AdshRNA + H/R group.
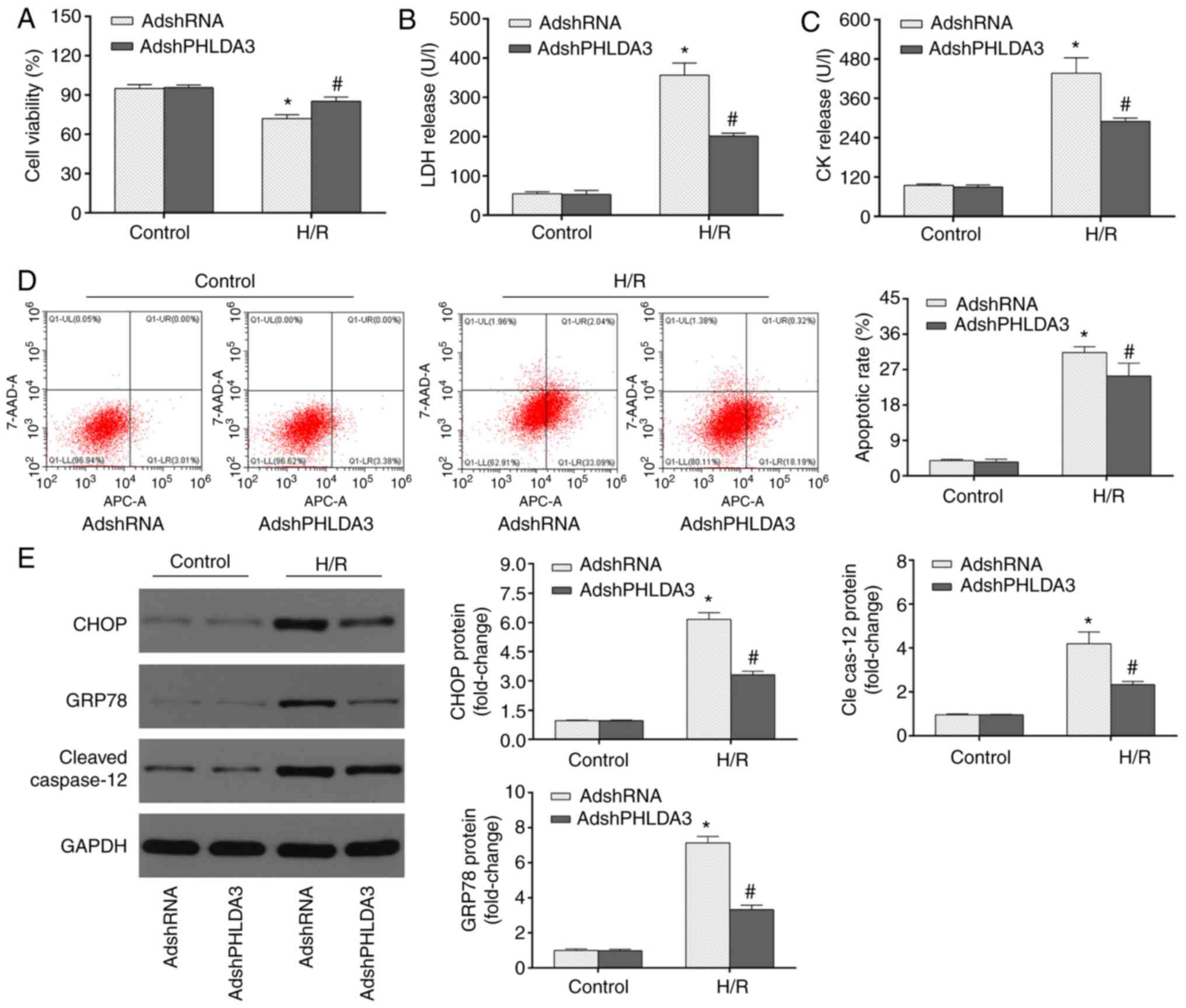 | Figure 2PHLDA3 inhibition alleviates
ERS-induced apoptosis during H/R injury. (A) Cell viability was
assessed using Cell Counting Kit-8 assays following H/R injury. The
release of (B) LDH and (C) CK following H/R was detected by ELISA.
(D) Apoptotic cells were measured by flow cytometry. (E) Expression
of ERS-associated proteins, including GRP78, CHOP and cleaved
caspase-12 was detected using western blotting. The left panel
shows representative blots and the right panel shows the
quantitative results. Data are presented as the mean ± standard
deviation. n=4/group. *P<0.05 vs. AdshRNA or
AdshPHLDA3 + control. #P<0.05 vs. AdshRNA + H/R
group. PHLDA3, pleckstrin homology-like domain family A member 3;
ERS, endoplasmic reticulum stress; H/R, hypoxia/reoxygenation; LDH,
lactate dehydrogenase; GRP78, 78 kDa glucose-regulated protein;
AdshRNA, adenovirus encoding scrambled short hairpin RNA;
AdshPHLDA3, adenoviral vectors encoding PHLDA3 shRNA; 7-AAD,
7-aminoactinomycin D; Cle cas-12, cleaved caspase-12; CK, creatine
kinase. |
To further investigate the pathological mechanisms
of cardioprotective properties following PHLDA3 inhibition and I/R
injury, ERS-induced apoptosis was assessed. MIRI is associated with
the activation of ERS-induced apoptosis (4,12) and
PHLDA3 has been demonstrated to be involved in ERS progression
(7). To determine whether PHLDA3
impacted myocardial H/R injury in an ERS-associated manner,
myocardial apoptotic and ERS-associated proteins, including GRP78,
CHOP and cleaved caspase-12, in NRCMs were assessed. As shown in
Fig. 2D, low apoptotic rates were
observed in the control + AdshRNA and control + AdshPHLDA3 groups;
while H/R significantly increased cardiomyocyte apoptosis compared
with the control group. AdshPHLDA3 transfection substantially
decreased the apoptotic rate following H/R compared with the
AdshRNA-treated cells following H/R insult. Additionally, the
expression levels of myocardial apoptotic and ERS-associated
proteins, including CHOP, GRP78 and cleaved caspase-12, were
detected by western blotting. As shown in Fig. 2E, H/R significantly increased the
protein expression of GRP78, CHOP and cleaved caspase-12 compared
with the control group Furthermore, PHLDA3 inhibition significantly
decreased the levels of these proteins following H/R injury
compared with the H/R + AdshRNA group. These results indicated that
PHLDA3 inhibition mitigated H/R-induced ERS and subsequent
apoptosis in NRCMs.
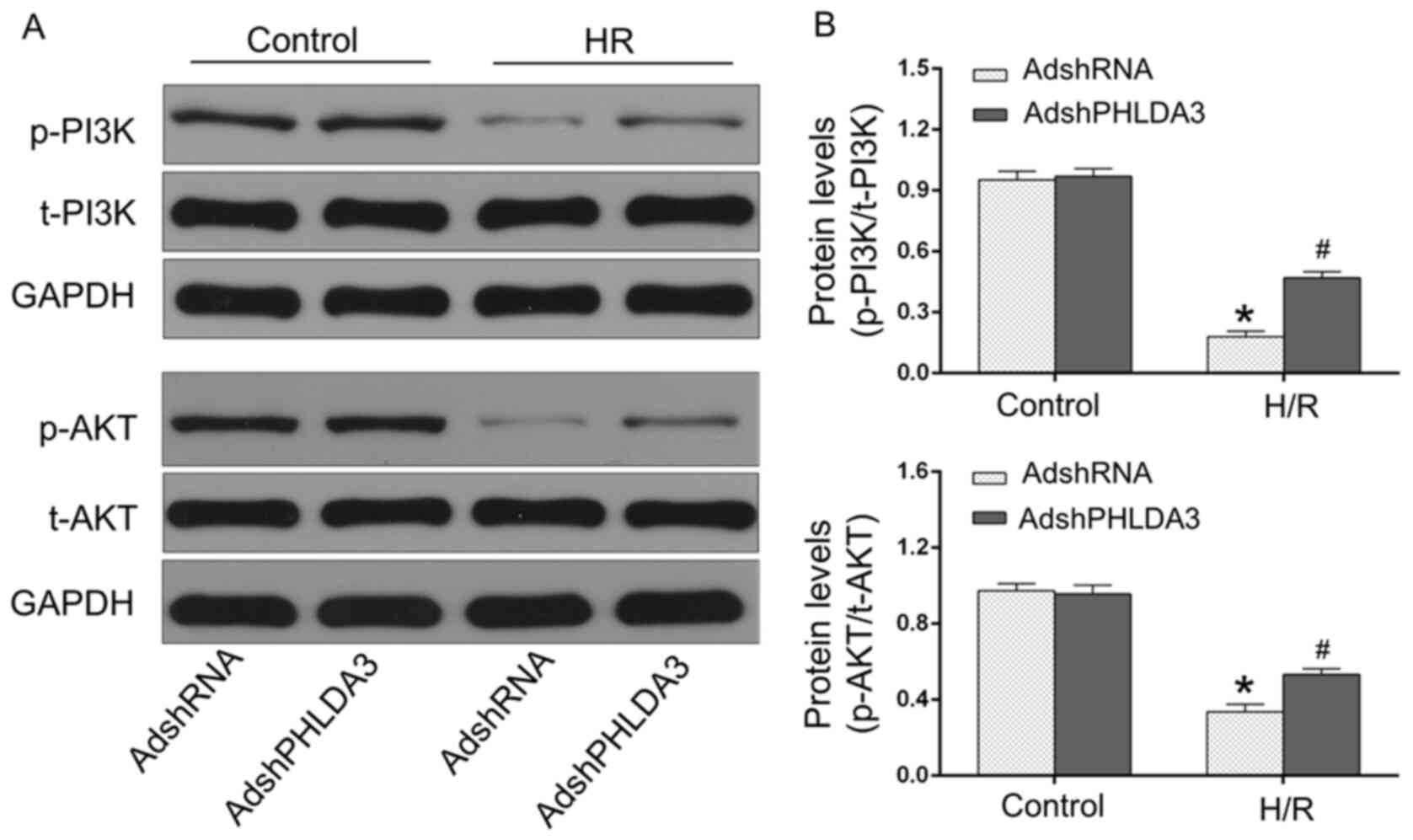
PHLDA3 inhibition promotes the
p-PI3K/AKT signaling pathway following H/R injury
The activation of the p-PI3K/AKT pathway is
considered as a pro-survival mediator in MIRI (4,11,28).
Previous studies demonstrated that PHLDA3-driven alternations of
the p-PI3K/AKT pathway participate in multiple pathological
processes, including cardiac remodeling and liver injury (9,13).
Thus, whether the p-PI3K/AKT pathway was responsible for the
anti-MIRI effects of PHLDA3 inhibition was examined. As shown in
Fig. 3, the levels of p-PI3K and
p-AKT were significantly reduced following H/R injury compared with
normoxic control cells. However, AdshPHLDA3-transfected H/R-treated
NRCMs demonstrated significantly increased p-PI3K and p-AKT
compared with the AdshRNA-transfected H/R group. Additionally,
there were no significant differences in the levels of t-PI3K and
t-AKT between the four groups (data not shown). Therefore, these
results demonstrated the role of the p-PI3K/AKT pathway in the
cardioprotection of PHLDA3 in H/R injury.
Blockage of the p-PI3K/AKT pathway
inhibits the protective effects of PHLDA3 inhibition following H/R
injury
Based on the aforementioned results, a rescue study
was performed. Adenovirus-transfected and H/R-treated cells were
treated with the p-PI3K inhibitor LY294002. Cell viability
(Fig. 4A), LDH release (Fig. 4B), apoptosis (Fig. 4C) and the mRNA expression of
ERS-associated mediators (Fig.
4D-F) were significantly different between the AdshRNA and
AdshPHLDA3 groups under H/R + PBS conditions. Treatment with
LY294002 inhibited the protective effects of PHLDA3 inhibition on
AdshPHLDA3-transfected cell survival compared with the H/R + PBS +
AdshPHLDA3-treated NRCMs (Fig. 4A
and B). Similar results were
observed for apoptosis rates (Fig.
4C) and expression ERS-associated molecules (Fig. 4D-F) following LY294002 treatment.
LY294002 administration abolished the protective effects of PHLDA3
inhibition on apoptosis and ERS-associated mediators compared with
H/R + PBS + AdshPHLDA3-treated NRCMs. Overall, these results
indicated that the cardioprotective contributions of PHLDA3
inhibition following H/R may be dependent on the p-PI3K/AKT pathway
activation and subsequent decrease in expression of ERS-associated
apoptosis.
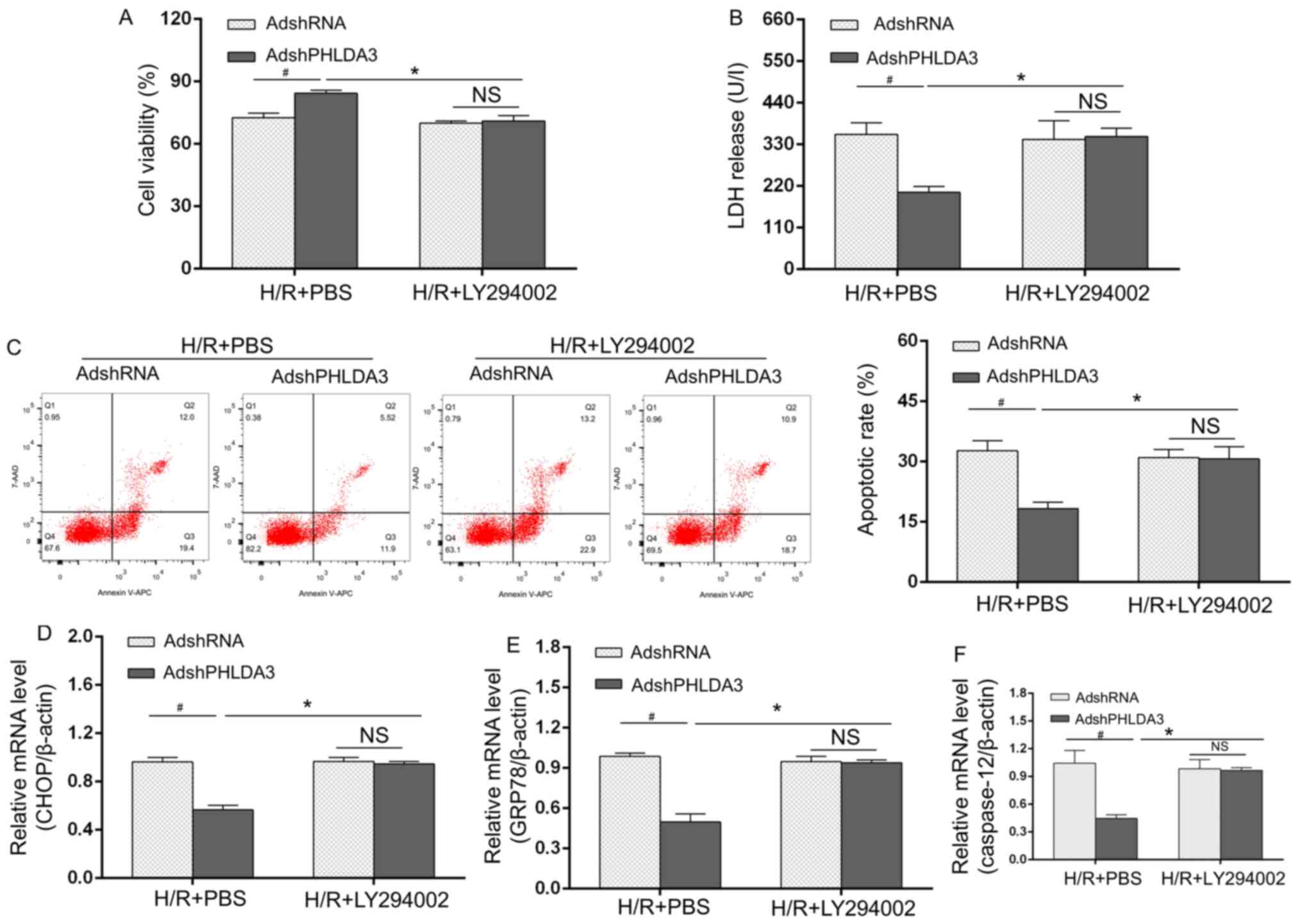 | Figure 4Blockage of the p-PI3K/AKT pathway
inhibits the protective effects of PHLDA3 inhibition following H/R
injury. (A) Cell Counting Kit-8 assays were used to evaluate cell
viability. (B) LDH release was detected by ELISA. (C) Apoptotic
rate in each group. mRNA levels of ERS markers, including (D) CHOP,
(E) GRP78 and (F) caspase-12, were measured by reverse
transcription-quantitative PCR. Data are presented as the mean ±
standard deviation. n=4/group. *P<0.05 vs. H/R +
AdshPHLDA3 + PBS. #P<0.05 vs. H/R + AdshRNA + PBS.
PHLDA3, pleckstrin homology-like domain family A member 3; H/R,
hypoxia/reoxygenation; LDF, lactate dehydrogenase; GRP78, 78 kDa
glucose-regulated protein; AdshRNA, adenovirus encoding scrambled
short hairpin RNA; AdshPHLDA3, adenoviral vectors encoding PHLDA3
shRNA; NS, not significant. |
Discussion
Prior studies have demonstrated that PHLDA3
alleviated pressure overload-induced cardiac remodeling (9) and impaired hemangioblast specification
and vascular development (6).
However, the role of PHLDA3 and possible molecular mechanisms in
MIRI have not been elucidated. The current study demonstrated that
PHLDA3 expression was upregulated in cardiomyocytes following H/R
injury. Adenovirus-mediated PHLDA3 inhibition, however, reduced
myocardial H/R injury, which was characterized by the increase of
cell activity and the decreased release of LDH/CK. Additionally,
PHLDA3 inhibition ameliorated cardiomyocyte apoptosis and inhibited
the protein expression of ERS-associated molecules, including
GRP78, CHOP and cleaved caspase-12. The mechanistic study observed
that PHLDA3 inhibition promoted the p-PI3K/AKT signaling pathway,
while LY294002, a p-PI3K/AKT inhibitor, reversed its protective
roles against H/R-induced damage and ERS-associated apoptosis.
These results indicated that PHLDA3 inhibition exhibited a
significant role in ameliorating H/R injury and ERS-associated
apoptosis mainly via the p-PI3K/AKT-dependent pathway.
Extensive studies have indicated that PHLDA3
exhibits pleiotropic effects in a variety of pathological
conditions, including liver injury (7), renal tubular cell death (29) and tumor initiation (8). The underlying pathogeneses are closely
associated with apoptosis-regulatory activity (7,13).
Notably, recent studies have revealed that PHLDA3 serves important
roles in cardiovascular diseases (6,9). For
example, in in vivo and in vitro models of pressure
overload-induced cardiac remodeling, Liu et al (9) demonstrated that PHLDA3 overexpression
acted as a potent therapeutic agent for the protection against
hypertrophy and fibrosis. Wang et al (6) indicated that PHLDA3 upregulation
affected the specification of hemangioblasts and vascular
development. However, whether PHLDA3 participates in the
development of MIRI remains unclear and the potential pathological
mechanisms are unknown. Notably, a previous study demonstrated that
PHLDA3 was a crucial mediator for the progression of ERS and
apoptotic response (7). Han et
al demonstrated that PHLDA3 overexpression enhanced the levels
of ERS markers and facilitated ERS progression, by which it acted
as a possible therapeutic agent in limiting ERS-associated
hepatocyte apoptosis and cell death (7). In terms of the possible involvement of
PHLDA3 in ERS-associated apoptosis, it was hypothesized that PHLDA3
may exert protective effects on cardiomyocytes and serve an
important role in ERS-associated apoptosis following H/R insult.
The results of the current study provided evidence that PHLDA3
inhibition had an advantageous role in H/R-injured NRCMs and that
it reduced H/R-induced and ERS-associated apoptosis.
The PHLDA family of genes consists of three members:
PHLDA1, PHLDA2 and PHLDA3 (13,30).
The present study and another previous investigation (9) have revealed that PHLDA3 served
important roles in cardiovascular diseases (13). Notably, the contributions of other
members of PHLDA family have also been demonstrated in
cardiovascular insult. For instance, Guo et al (30) demonstrated that PHLDA1 knockdown
improved and PHLDA1 overexpression enhanced oxidative
stress-induced cardiac injury in MIRI. However, further studies are
required to explore whether PHLDA2 participates in cardiovascular
diseases, particularly in MIRI.
Timely and effective reperfusion of the ischemic
heart has been confirmed to be the most efficacious treatment for
acute myocardial infarction (4,14).
Nevertheless, the accompanying I/R injury can strongly offset this
beneficial effect (4,14). The ERS-elicited apoptotic response
is considered to be one of the most critical mechanisms of MIRI
(5,11). Previous studies have reported that
MIRI results in severe ERS due to markedly enhanced GRP78 levels
following MIRI, which subsequently leads to the activation of
caspase-12 and CHOP, eliciting myocardial apoptosis (5,11). The
present study demonstrated that MIRI markedly induced myocardial
death, apoptosis and ERS. However, PHLDA3 inhibition reduced the
levels of ERS-associated proteins including GRP78, CHOP and
caspase-12, and ameliorated the parameters associated with the
severity of H/R injury. Therefore, PHLDA3 inhibition protected
cardiomyocytes against H/R damage, possibly by mitigating
ERS-associated apoptosis.
Based on the aforementioned findings, the potential
molecular mechanisms by which PHLDA3 inhibition exerts protective
effects against H/R injury and ERS-associated apoptosis was
investigated. It is well known that p-PI3K and its downstream
target serine/threonine kinase p-AKT serve critical roles in
protecting against MIRI and that the activation of this pathway
inhibits ERS-associated myocardial apoptosis (4,11).
Importantly, previous studies have demonstrated the close
association between PHLDA3 and the p-PI3K/AKT signaling pathway
(6,13). For instance, Wang et al
(6) demonstrated that PHLDA3
overexpression inhibited the activation of p-AKT in vascular
development. P-AKT was elevated in PHLDA3-deficient islets
following early islet transplantation (13). Moreover, PHLDA3 was reported to
repress p-AKT and p-AKT-regulated biological processes in
pancreatic endocrine tissue (8).
Consistently, the current study confirmed that PHLDA3 inhibition
ameliorated H/R-induced apoptosis and increased p-PI3K and p-AKT
levels. To further verify the association between the p-PI3K/AKT
pathway and PHLDA3, a specific inhibitor of p-PI3K/AKT, LY294002,
was added before H/R. Notably, the cardioprotective efficacy and
inhibitory effects on ERS-related apoptosis following PHLDA3
inhibition were abolished in the presence of LY294002. Therefore,
these results indicated that PHLDA3 inhibition decreased
ERS-associated apoptosis and H/R-induced cell damage in a
p-PI3K/AKT-dependent manner. To the best of our knowledge, other
molecular mechanisms resulting from PHLDA3 in MIRI have not been
reported. A previous study investigated the effect of PHLDA3 on the
regulation of the inositol requiring enzyme/X-box binding protein 1
pathway and demonstrated PHLDA3 be involved in liver injury
(7). Further studies are required
to detect other molecular mechanisms of PHLDA3 apart from the
p-PI3K/AKT pathway in MIRI.
In summary, the present study demonstrated that the
downregulation of PHLDA3 alleviated MIRI by attenuating ERS-induced
apoptosis in a p-PI3K/AKT-dependent manner. Moreover, numerous
other signaling pathways are involved in the pathophysiological
processes of MIRI (31-33).
Further studies are necessary to explore whether other signaling
pathways are involved in the effect of PHLDA3 in MIRI. Moreover,
the present study reported the effects of PHLDA3 in H/R-injured
NRCMs in an in vitro study. Further investigations will
study the effects of PHLDA3 on myocardial histological morphology
in I/R-treated rat hearts in vivo. In conclusion, the
present results indicated that PHLDA3 may be a prospective
therapeutic option for MIRI.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KL, YC and FA conceived and designed the
experiments. YQL, KZ and WTZ made substantial contributions to the
acquisition of data and manuscript revision, and perfomed the
statistical analysis. All authors contributed to the preparation
and revision of the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Experiments and animal care were performed in
adherence with the Guide for the Care and Use of Laboratory Animals
published by the US National Institutes of Health (NIH; 8th
Edition; 2011) (17) and were
approved by the Hainan Affiliated Hospital of Hainan Medical
University (Haikou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dai SH, Wu QC, Zhu RR, Wan XM and Zhou XL:
Notch1 protects against myocardial Ischaemia-reperfusion injury via
regulating mitochondrial fusion and function. J Cell Mol Med.
24:3183–3191. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Satomi S, Morio A, Miyoshi H, Nakamura R,
Tsutsumi R, Sakaue H, Yasuda T, Saeki N and Tsutsumi YM:
Branched-chain amino acids-induced cardiac protection against
ischemia/reperfusion injury. Life Sci. 245(117368)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kohler D, Granja T, Volz J, Koeppen M,
Langer HF, Hansmann G, Legchenko E, Geisler T, Bakchoul T, Eggstein
C, et al: Red blood cell-derived semaphorin 7A promotes
thrombo-inflammation in myocardial ischemia-reperfusion injury
through platelet GPIb. Nat Commun. 11(1315)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhang BF, Jiang H, Chen J, Guo X, Li Y, Hu
Q and Yang S: Nobiletin ameliorates myocardial ischemia and
reperfusion injury by attenuating endoplasmic reticulum
stress-associated apoptosis through regulation of the PI3K/AKT
signal pathway. Int Immunopharmacol. 73:98–107. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gao J, Guo Y, Liu Y, Yan J, Zhou J, An X
and Su P: Protective effect of FBXL10 in myocardial ischemia
reperfusion injury via inhibiting endoplasmic reticulum stress.
Respir Med. 161(105852)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wang X, Li J, Yang Z, Wang L, Li L, Deng
W, Zhou J, Wang L, Xu C, Chen Q and Wang QK: Phlda3 overexpression
impairs specification of hemangioblasts and vascular development.
Febs J. 285:4071–4081. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Han CY, Lim SW, Koo JH, Kim W and Kim SG:
PHLDA3 overexpression in hepatocytes by endoplasmic reticulum
stress via IRE1-Xbp1s pathway expedites liver injury. Gut.
65:1377–1388. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ohki R, Saito K, Chen Y, Kawase T, Hiraoka
N, Saigawa R, Minegishi M, Aita Y, Yanai G, Shimizu H, et al:
PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine
tumors. Proc Natl Acad Sci USA. 111:E2404–E2413. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Liu J, Liu X, Hui X, Cai L, Li X, Yang Y,
Shu S, Wang F, Xia H and Li S: Novel role for pleckstrin
homology-like domain family a, member 3 in the regulation of
pathological cardiac hypertrophy. J Am Heart Assoc.
8(e11830)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kawase T, Ohki R, Shibata T, Tsutsumi S,
Kamimura N, Inazawa J, Ohta T, Ichikawa H, Aburatani H, Tashiro F
and Taya Y: PH domain-only protein PHLDA3 is a p53-regulated
repressor of Akt. Cell. 136:535–550. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Shen D, Chen R, Zhang L, Rao Z, Ruan Y, Li
L, Chu M and Zhang Y: Sulodexide attenuates endoplasmic reticulum
stress induced by myocardial ischaemia/reperfusion by activating
the PI3K/Akt pathway. J Cell Mol Med. 23:5063–5075. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yu L, Li B, Zhang M, Jin Z, Duan W, Zhao
G, Yang Y, Liu Z, Chen W, Wang S, et al: Melatonin reduces
PERK-eIF2alpha-ATF4-mediated endoplasmic reticulum stress during
myocardial ischemia-reperfusion injury: Role of RISK and SAFE
pathways interaction. Apoptosis. 21:809–824. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sakata N, Yamaguchi Y, Chen Y, Shimoda M,
Yoshimatsu G, Unno M, Sumi S and Ohki R: Pleckstrin homology-like
domain family a, member 3 (PHLDA3) deficiency improves islets
engraftment through the suppression of hypoxic damage. PLoS One.
12(e187927)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yang G, Zhang X, Weng X, Liang P, Dai X,
Zeng S, Xu H, Huan H, Fang M, Li Y, et al: SUV39H1 mediated SIRT1
trans-repression contributes to cardiac ischemia-reperfusion
injury. Basic Res Cardiol. 112(22)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang L, Li C, Zhu Q, Li N and Zhou H:
Liraglutide, a glucagon-like peptide-1 analog, inhibits high
glucose-induced oxidative stress and apoptosis in neonatal rat
cardiomyocytes. Exp Ther Med. 17:3734–3740. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li L, Sha Z, Wang Y, Yang D, Li J, Duan Z,
Wang H and Li Y: Pre-treatment with a combination of Shenmai and
Danshen injection protects cardiomyocytes against
hypoxia/reoxygenation- and H2O2-induced injury by inhibiting
mitochondrial permeability transition pore opening. Exp Ther Med.
17:4643–4652. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chen L, Huang J, Ji Y, Zhang X, Wang P,
Deng K, Jiang X, Ma G and Li H: Tripartite motif 32 prevents
pathological cardiac hypertrophy. Clin Sci (Lond). 130:813–828.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wu G, Liu Y, Huang H, Tang Y, Liu W, Mei
Y, Wan N, Liu X and Huang C: SH2B1 is critical for the regulation
of cardiac remodelling in response to pressure overload. Cardiovasc
Res. 107:203–215. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ye P, Xiang M, Liao H, Liu J, Luo H, Wang
Y, Huang L, Chen M and Xia J: Dual-Specificity phosphatase 9
protects against nonalcoholic fatty liver disease in mice through
ASK1 suppression. Hepatology. 69:76–93. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Xiang M, Luo H, Wu J, Ren L, Ding X, Wu C,
Chen J, Chen S, Zhang H, Yu L, et al: ADAM23 in cardiomyocyte
inhibits cardiac hypertrophy by targeting FAK-AKT signaling. J Am
Heart Assoc. 7(e8604)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu R, van Berlo JH, York AJ, Vagnozzi RJ,
Maillet M and Molkentin JD: DUSP8 regulates cardiac ventricular
remodeling by altering ERK1/2 signaling. Circ Res. 119:249–260.
2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xin G, Xu-Yong L, Shan H, Gang W, Zhen C,
Ji-Jun L, Ping Y and Man-Hua C: SH2B1 protects cardiomyocytes from
ischemia/reperfusion injury via the activation of the PI3K/AKT
pathway. Int Immunopharmacol. 83(105910)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang BF, Jiang H, Chen J, Hu Q, Yang S,
Liu XP and Liu G: LncRNA H19 ameliorates myocardial
infarction-induced myocardial injury and maladaptive cardiac
remodelling by regulating KDM3A. J Cell Mol Med. 24:1099–1115.
2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sun Y, Liu L, Yuan J, Sun Q, Wang N and
Wang Y: RP105 protects PC12 cells from oxygenglucose
deprivation/reoxygenation injury via activation of the PI3K/AKT
signaling pathway. Int J Mol Med. 41:3081–3089. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Guo X, Jiang H, Yang J, Chen J, Yang J,
Ding JW, Li S, Wu H and Ding HS: Radioprotective 105 kDa protein
attenuates ischemia/reperfusion-induced myocardial apoptosis and
autophagy by inhibiting the activation of the TLR4/NF-NF-κB
signaling pathway in rats. Int J Mol Med. 38:885–893.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hu X, Zhang K, Chen Z, Jiang H and Xu W:
The HMGB1IL17A axis contributes to hypoxia/reoxygenation injury via
regulation of cardiomyocyte apoptosis and autophagy. Mol Med Rep.
17:336–341. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang HB, Duan MX, Xu M, Huang SH, Yang J,
Yang J, Liu LB, Huang R, Wan CX, Ma ZG, et al: Cordycepin
ameliorates cardiac hypertrophy via activating the AMPKα pathway. J
Cell Mol Med. 23:5715–5727. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhou YH, Han QF, Gao L, Sun Y, Tang ZW,
Wang M, Wang W and Yao HC: HMGB1 protects the heart against
ischemia-reperfusion injury via PI3K/AkT pathway-mediated
upregulation of VEGF expression. Front Physiol.
10(1595)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lee CG, Kang YJ, Kim HS, Moon A and Kim
SG: Phlda3, a urine-detectable protein, causes p53 accumulation in
renal tubular cells injured by cisplatin. Cell Biol Toxicol.
31:121–130. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Guo Y, Jia P, Chen Y, Yu H, Xin X, Bao Y,
Yang H, Wu N, Sun Y and Jia D: PHLDA1 is a new therapeutic target
of oxidative stress and ischemia reperfusion-induced myocardial
injury. Life Sci. 245(117347)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yang J, Yang C, Yang J, Ding J, Li X, Yu
Q, Guo X, Fan Z and Wang H: RP105 alleviates myocardial ischemia
reperfusion injury via inhibiting TLR4/TRIF signaling pathways. Int
J Mol Med. 41:3287–3295. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kong T, Liu M, Ji B, Bai B, Cheng B and
Wang C: Role of the extracellular signal-regulated kinase 1/2
signaling pathway in ischemia-reperfusion injury. Front Physiol.
10(1038)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Huang CK, Kafert-Kasting S and Thum T:
Preclinical and clinical development of noncoding RNA therapeutics
for cardiovascular disease. Circ Res. 126:663–678. 2020.PubMed/NCBI View Article : Google Scholar
|