Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is an
aggressive malignant tumor, that originates from T-cell precursors
and has a high degree of genetic, immune phenotypic and clinical
heterogeneity (1,2). It accounts for ~15% childhood ALL and
25% adult ALL worldwide (3).
Administration of glucocorticoids (GC) is an important part of
T-ALL treatment. GCs enter the cell by passive diffusion, where
they bind to the GC receptor (GR; encoded by the NR3C1 gene), which
is a member of the nuclear receptor family of ligand-dependent
transcription factors (4-6).
The activated receptor is then translocated to the nucleus, where
it activates target genes, including NR3C1 itself, BCL-2,
glucocorticoid-induced leucine zipper, Kruppel-like factor-13, NFKB
inhibitor a and period 1, with assistance from chaperone and
transporter proteins, and binds to GR elements (GREs) (7). GR-induced activation or repression of
gene transcription controls apoptosis of normal and malignant
lymphocytes (8). In lymphoid cells,
GR induces the mRNA expression level of BCL2L11, which encodes the
proapoptotic BH3-only factor, BIM, triggering apoptosis (9). In dexamethasone (DEX)-resistant ALL,
the activated GR cannot bind to the BIM intronic region to trigger
apoptosis (10). Therefore,
resistance to GC is one of the most common causes of T-ALL
treatment failure or relapse (11).
Salidroside (SAL) is the main active ingredient of
Rhodiola. It is the glycoside of a phenolic compound. Several
studies have shown that SAL has a potential anti-cancer effect
(12-17).
Therefore, SAL has become potential drug candidate for cancer
treatment. Recently, another study has shown that SAL could improve
the microenvironment of hypoxic tumors and reverse the resistance
to platinum drugs in hepatocellular carcinoma (18). Thus, the human T-ALL GC
DEX-resistant cell line, CEM-C1 and the DEX-sensitive cell line,
CEM-C7 were selected as cell lines to investigate reversal of tumor
resistance caused by SAL.
The proto-oncogene, c-Myc is a transcription factor,
which belongs to the helix-loop helix-leucine zipper protein
family, and functions primarily to maintain cell proliferation,
differentiation, apoptosis and normal cell cycle (19). It has been found that c-Myc was
associated with acute myeloid leukemia drug resistance (20). Mounting evidence also suggests that
downregulation of c-Myc mRNA expression may increase the
sensitivity of tumor cells to chemotherapeutic agents, including
enhancing the sensitivity of breast cancer cells to palbociclib
(21), the sensitivity of human
glioblastoma cells to temozolomide (22), and the sensitivity of malignant
mesothelioma cells to the p21-activated kinase blockage-induced
cytotoxicity (23). In the present
study, it was found that CEM-C1 cells exhibited higher protein
expression levels of c-Myc compared with those in CEM-C7 cells.
Since c-Myc has been associated with drug resistance in various
studies (24-27),
the present study aimed to reveal the anti-leukemic effect and
reversal resistance effect of SAL, and to investigate c-Myc in
T-ALL cells and its association with DEX resistance.
Materials and methods
Reagents
SAL (purity, >99%) was purchased from Chengdu
Ruifensi Biotechnology Co., Ltd. RPMI 1640 culture medium was
purchased from HyClone (GE Healthcare Life Sciences), while fetal
bovine serum was purchased from Zhejiang Tianhang Biotechnology
Co., Ltd., and penicillin-streptomycin was purchased from Beyotime
Institute of Biotechnology. Cell Counting Kit (CCK)-8 assay kit was
purchased from Dojindo Molecular Technologies Inc., while DEX
(Chinese medicine standard, H41020036) was purchased from Shanghai
Shyndec Pharmaceutical Co., Ltd., and the cell cycle detection kit
was purchased from Nanjing KeyGen Biotech Co., Ltd., and the
Annexin V-FITC/PI apoptosis kit was purchased from BD Biosciences.
The total RNA extraction kit was purchased from Tiangen Biotech
Co., Ltd., while the reverse transcription and quantitative PCR
(qPCR) kits were purchased from Toyobo Life Science, and the
acridine orange stain was purchased from Biotopped Life Sciences.
The rabbit anti-human c-Myc and GAPDH antibodies were purchased
from ProteinTech Group, Inc., while the rabbit anti-human LC3A/B,
Bax, BCL-2 and cleaved PARP antibodies were purchased from Cell
Signaling Technology, Inc., and the goat anti-rabbit IgG-HRP
antibody was purchased from BIOSS. Lastly, the PCR primers were
synthesized by Shanghai Shenggong Biology Engineering Technology
Service, Ltd.
Cell lines and culture
The CEM-C1 and CEM-C7 cell lines were donated by
Professor Ma Zhigui (Department of Pediatric Hematology and
Oncology, West China Second Hospital of Sichuan University,
Chengdu, China) and were cultured in RPMI 1640 medium supplemented
with 10% fetal bovine serum and 100 µg/ml streptomycin and 100 U/ml
penicillin at 37˚C in a humidified incubator with 5%
CO2. The medium was changed every 2 to 3 days and the
cells were passaged once before the start of the experiments.
Drug dissolution
SAL (1 g) was dissolved in sterile PBS (3 ml), made
into a liquid and frozen in aliquots at -20˚C. The compound was
diluted in RPMI 1640 medium to the required concentration prior to
the experiment.
CCK8 assay
The CEM-C7 and CEM-C1 cells were used in the
logarithmic growth phase and plated in 96-well microplates
(1.5x105 cells/well), then different concentrations of
SAL (5.0, 7.5, 10.0, 12.5 and 15.0 mg/ml) were added. At the same
time, the blank group (containing only culture medium and no cells)
and the control group (containing only cells and culture medium)
were prepared. A total of 4 replicate wells were used for each
group. Following incubation for 20, 44 and 68 h, 10 µl CCK8
solution was added to each well, then the cells were incubated for
another 4 h, after which time the optical density (OD) was measured
using a microplate reader at 450 nm. The experiment was repeated 3
times. The percentage cell inhibition rate (%) was calculated using
the following formula: Cell inhibition=(OD value of control
group-OD value of experimental group)/(OD value of control group-OD
value of blank group) x100%.
The CEM-C7 and CEM-C1 cells were used in the
logarithmic growth phase and plated in 96-well microplates
(1.5x105 cells/well), then they were treated with
different concentrations of DEX. The CEM-C7 cells were treated with
0.25, 0.5, 1.0, 1.5 and 2.0 µg/ml DEX with or without 1.5 mg/ml SAL
(cell inhibition rate <4%), while the CEM-C1 cells were treated
with 25, 50, 100, 150 and 200 µg/ml DEX with or without 1.5 mg/ml
SAL (cell inhibition rate <4%). Following incubation for 44 h,
10 µl CCK8 solution was added to each well, then the cells were
incubated for another 4 h, after which time the OD was measured
using a microplate reader at 450 nm. The experiment was repeated 3
times. The half inhibitory concentration IC50 was
calculated using the GraphPad Prism v8.0.2 software (GraphPad
Software, Inc.). The resistance index (RI) was calculated using the
following equation: RI=IC50 of resistant
cells/IC50 of sensitive cells. The reversal fold (RF)
was calculated as follows: RF=IC50 of resistant
cells/IC50 following addition of the reversal agent.
Observation of cell morphology
The CEM-C1 and CEM-C7 cells, in the logarithmic
growth phase, were treated with 1.5 µg/ml DEX for 48 h, then the
morphological changes in the cells were observed under a light
microscope and images were captured (magnification, x400).
Reverse transcription-qPCR (RT-qPCR)
analysis
The CEM-C1 and CEM-C7 cells, in the logarithmic
growth phase, were seeded in a 6-well culture plate
(5x106 cells/well). The following experimental groups
were used: Control group (0 mg/ml SAL) and the experimental groups
(5.0, 7.5 and 10.0 mg/ml SAL). The cells were cultured for 48 h,
then RNA was extracted using TRIzol®, according to the
manufacturer's instructions (Invitrogen; Thermo Fisher Scientific,
Inc.). cDNA was generated using RT and the TOYOBO reverse
transcriptase kit. The mRNA expression levels of c-Myc and the
autophagy-related gene, LC3, were detected using
SYBR®-Green I Supermix (Toyobo Life Science), according
to the manufacturer's instructions. The primer sequences are shown
in Table I. The thermocycling
conditions were as follows: Initial denaturation at 95˚C for 30
sec, followed by 40 cycles of 95˚C for 5 sec, 60˚C for 10 sec and
72˚C for 30 sec. Using GAPDH as the internal reference gene, the
relative expression levels of the target genes were expressed using
the 2-∆∆Cq method (28).
The experiment was repeated 3 times.
 | Table ISequences of the primers for
quantitative PCR. |
Table I
Sequences of the primers for
quantitative PCR.
| Primer name | Primer
sequence |
|---|
| c-Myc | F:
5'-CTACCCTCTCAACGACAGCA-3' |
| | R:
5'-AGAGCAGAGAATCCGAGGAC-3' |
| LC3 | F:
5'-CAGCGTCTCCACACCAATCT-3' |
| | R:
5'-TCTCCTGGGAGGCATAGACC-3' |
| GAPDH | F:
5'-CAATGACCCCTTCATTGACC-3' |
| | R:
5'-GACAAGCTTCCCGTTCTCAG-3’ |
Cell cycle analysis using flow
cytometry
The CEM-C1 and CEM-C7 cells, in the logarithmic
growth phase, were seeded in a 6-well culture plate
(3x105 cells/well), cultured for 48 h, then the cells
were collected and washed with PBS solution. The supernatant was
discarded and 500 µl 70% cold ethanol was added. The cells were
fixed overnight at 4˚C. Prior to staining, the ethanol was removed
and the cells were washed with PBS and centrifuged at 300 x g at
4˚C for 5 min. A total of 500 µl PI/RNase A staining working
solution was added to each well. The samples were protected from
light and incubated at room temperature for 30 min. The red
fluorescence was examined at an excitation wavelength of 488 nm.
The experimental groups were the same as those in the
aforementioned RT-qPCR subheading.
Detection of cell apoptosis using flow
cytometry
The CEM-C1 and CEM-C7 cells, in the logarithmic
growth phase, were seeded in a 6-well culture plate
(3x105 cells/well), cultured for 48 h, then the cells
were collected, washed twice with cold PBS and finally resuspended
with 1X binding buffer, to adjust the cell density to
1x106 cells/ml. A total of 100 µl cell suspension was
used in a 5 ml flow cytometer tube and 5 µl PI was mixed with 5 µl
Annexin V-FITC and added to the cells. The samples were shaken and
placed at room temperature for 25 min in the dark. Subsequently,
200 µl 1X binding buffer was added to the cells, and measured using
flow cytometry within 1 h. The experiment was repeated 3 times. The
experimental groups were the same as those in the aforementioned
RT-qPCR subheading. Additionally, according to whether SAL was
combined with DEX, CEM-C1 cells were divided into control group,
SAL group (1.5 mg/ml), DEX group (100 µg/ml) and combination group
(DEX 100 µg/ml + SAL 1.5 mg/ml).
Western blot analysis
The CEM-C1 and CEM-C7 cells, in the logarithmic
growth phase, were seeded into a 6-well culture plate
(5x106 cells/well) and the total protein from each group
was extracted 48 h later using RIPA lysis buffer (Beyotime
Institute of Biotechnology) for 30 min and the total protein
concentration was determined using the BCA method. A total of 25 µg
total protein was extracted and analyzed using SDS-PAGE,
transferred to a PVDF membrane, blocked with 7% skimmed milk at
room temperature for 1 h and incubated with the following primary
antibodies anti-GAPDH (1:15,000 dilution; cat. no. 10494-1-AP),
anti-Bax (1:1,000 dilution; cat. no. 5023), anti-BCL-2 (1:1,000
dilution; cat. no. 4223), anti-cleaved-PARP (1:1,000 dilution; cat.
no. 9185), anti-LC3A/B (1:1,000 dilution; cat. no. 12741) and
anti-c-Myc (1:2,000 dilution; cat. no. 10828-1-AP) overnight at
4˚C. The membrane was washed 3 times with PBS with 0.07% Tween-20
(PBST), then the secondary antibody (HRP-labeled goat anti-rabbit
antibody; 1:2,000; cat. no. bs-0295G-HRP) was added and the
membrane was incubated for 1 h at room temperature. The membrane
was washed with PBST three times and developed using an enhanced
chemiluminescence kit (EMD Millipore). The protein expression level
was measured using densitometry of the bands with ImageJ v1.4.3.67
(National Institute of Health). The protein expression levels were
normalized to GAPDH. The experiments were repeated three times. The
experimental groups were the same as those in the aforementioned
RT-qPCR subheading. Additionally, according to whether SAL was
combined with DEX, CEM-C1 cells were divided into control group,
SAL group (1.5 mg/ml), DEX group (100 µg/ml) and combination group
(DEX 100 µg/ml + SAL 1.5 mg/ml).
Acridine orange staining
The CEM-C1 and CEM-C7 cells, in the logarithmic
growth phase, were seeded in a 6-well culture plate
(3x105 cells/well), cultured for 48 h, then washed with
PBS, and stained with acridine orange staining solution (10 µg/ml)
for 30 min in the dark at room temperature. The cells were observed
and images were captured using a fluorescence microscope
(magnification, x400). The experimental groups were the same as
those in the aforementioned RT-qPCR subheading.
Statistical analysis
The SPSS v23.0 software (IBM Corp.) was used for
data analysis. The quantitative data are presented as the mean ±
SD. Comparisons between two groups was performed using an
independent Student's t-test, while one-way ANOVA was used for the
comparison of multiple groups. Tukey's post hoc test was used when
the homogeneity of variance was equal, while the Tamhane's T2 test
was used when the variance was unequal. P<0.05 was considered to
indicate a statistically significant difference.
Results
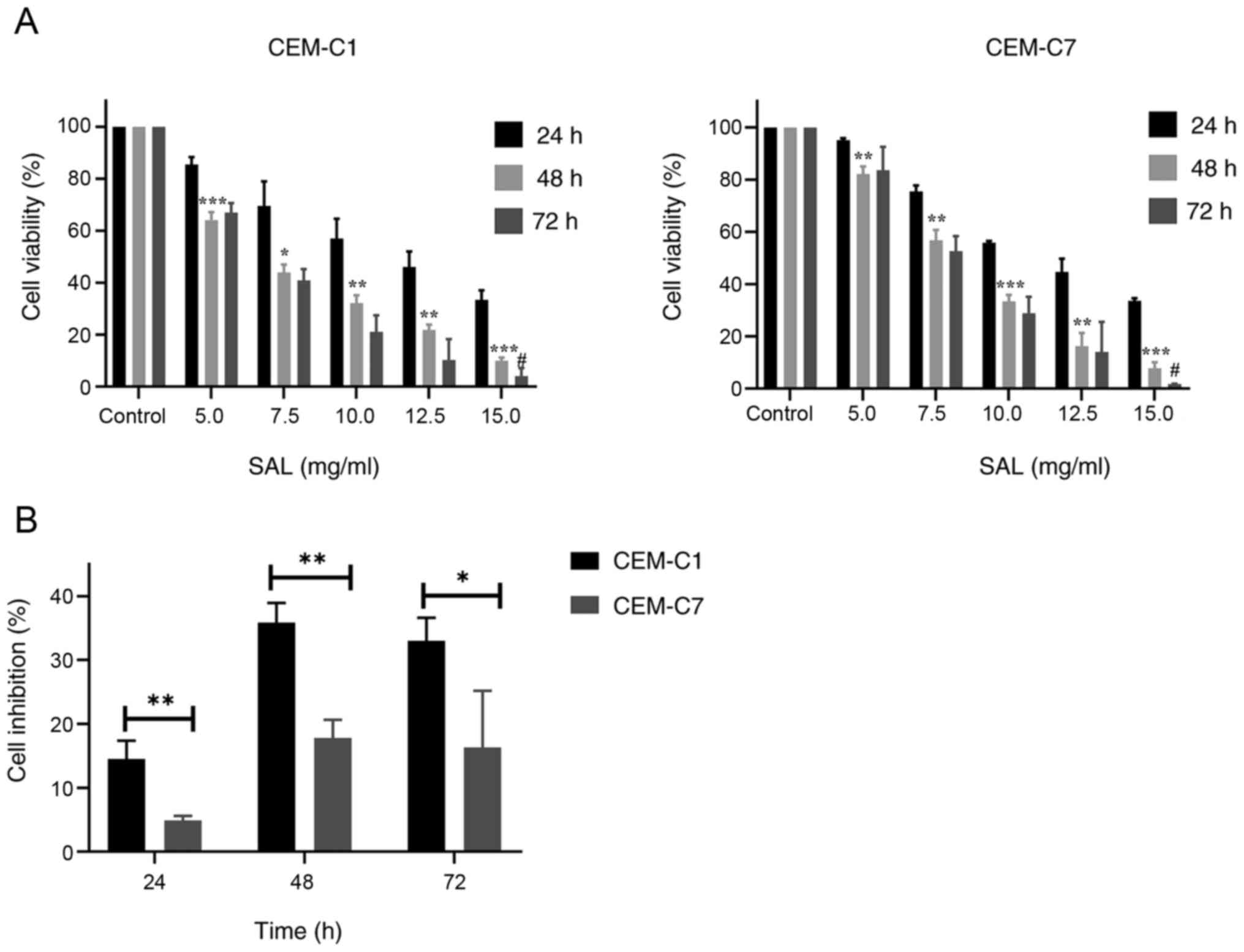
SAL inhibits the proliferation of the
T-ALL cells
To investigate the anti-proliferative activity of
SAL on the T-ALL cells, cell proliferation was determined using a
CCK8 assay. As depicted in Fig. 1A,
SAL effectively inhibited the proliferation of the CEM-C1 and
CEM-C7 cells in a dose-and time-dependent manner. The
IC50 of the CEM-C1 cells at 24, 48 and 72 h was 11.26,
6.69 and 6.45 mg/ml, respectively, while the IC50 of the
CEM-C7 cells at 24, 48 and 72 h was 11.42, 8.03 and 7.73 mg/ml,
respectively (data not shown). No significant difference was found
in the IC50 values between the 48 and 72 h time points
(P>0.05). Based on this finding, 48 h was selected as the
intervention time point. In subsequent experiments, different
concentrations of SAL (5.0, 7.5 and 10.0 mg/ml) to treat the cells
were selected to detect the effect on cell cycle, apoptosis, and
autophagy. The results also showed that SAL was more effective at
inhibiting CEM-C1 cell viability compared with that in the CEM-C7
cells, which indicated that the DEX-resistant cells were more
sensitive to SAL, as shown in Fig.
1B.
Effect of DEX on the morphology of the
CEM-C1 and CEM-C7 cells
The CEM-C1 and CEM-C7 cells were treated with 1.5
µg/ml DEX for 48 h and cellular morphology was assessed using a
light microscope. As shown in Fig.
2, the morphology of the DEX-resistant, CEM-C1 cells changed
from slender and irregular shapes to round shapes and no notable
reduction in cell viability was noted using microscopy compared
with that in the control group. However, the DEX-sensitive CEM-C7
cells showed a large number of cell fragments and increased cell
death compared with that in the control cells. It was suggested
that CEM-C1 cells exhibited strong resistance to DEX.
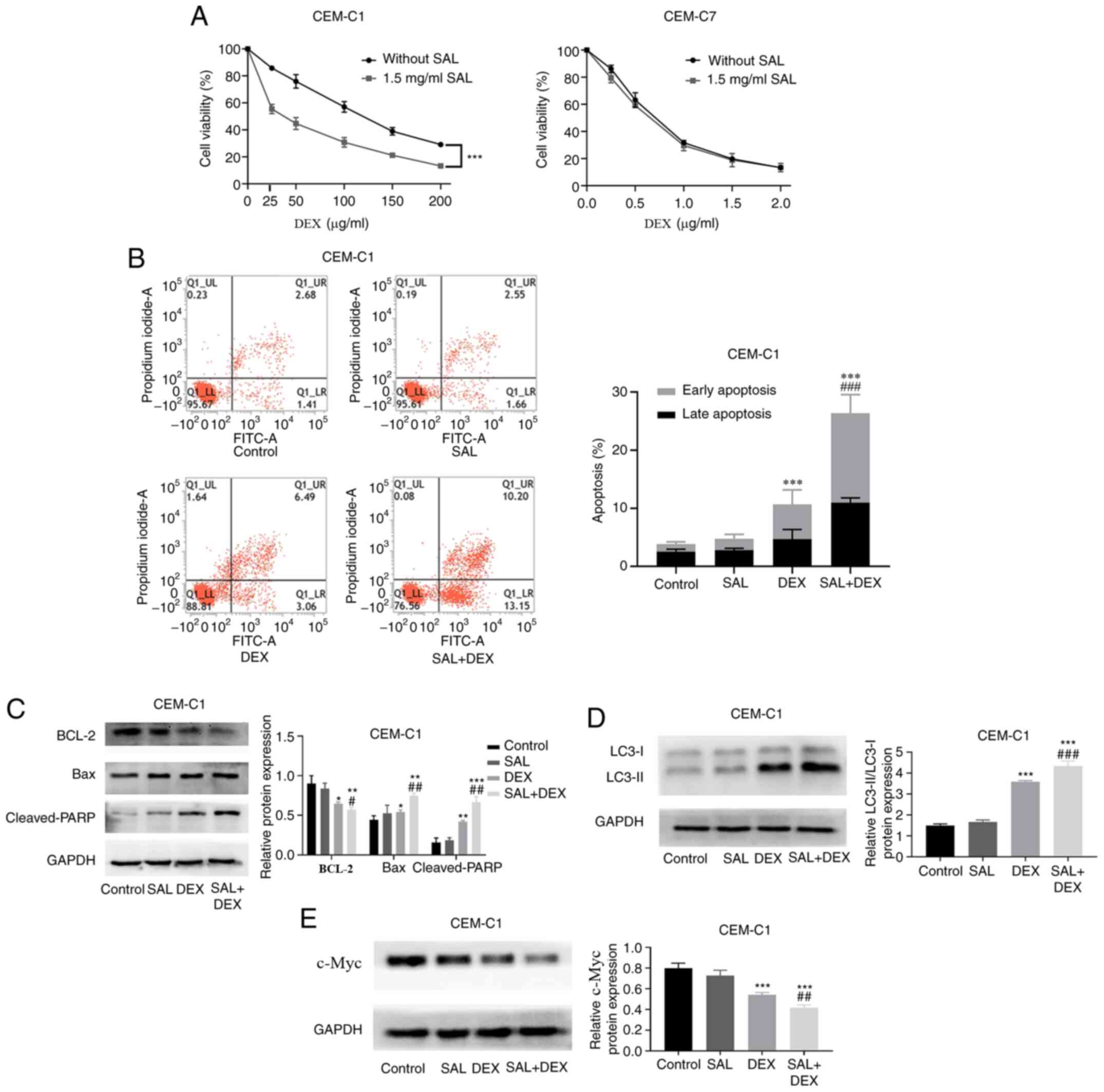
SAL enhances the sensitivity of the
CEM-C1 cells to DEX
To verify the resistance of the CEM-C1 cells to DEX,
the cytotoxic effect of DEX on DEX-sensitive CEM-C7 cells and
DEX-resistant CEM-C1 cells was determined using a CCK-8 assay.
Fig. 3A demonstrated that the
IC50 in the CEM-C1 and CEM-C7 cells, treated with DEX
and without SAL was 111.83±2.87 and 0.67±0.02 µg/ml, respectively,
whereas the RI was 166.92 (data not shown). Our preliminary drug
concentration screening results showed that the cell proliferation
inhibition rate on the CEM-C1 and CEM-C7 cells treated with 1.5
mg/ml SAL was <4% (Fig. S1).
Therefore, 1.5 mg/ml SAL was selected, combined with DEX, to
culture the cells for 48 h. Fig.
S2A indicated that the IC50 in the CEM-C1 cells
treated with DEX + SAL was significantly decreased to 35.59±3.73
µg/ml. The RF was 3.14 (data not shown). In contrast to this
finding, the DEX + SAL group exhibited no significant effect on the
IC50 value in the CEM-C7 cells compared with that in the
cells treated with DEX alone (Fig.
S2B; P>0.05).
To determine whether SAL could enhance the
sensitivity of the CEM-C1 cells to DEX, the CEM-C1 cells were
treated with SAL (1.5 mg/ml), DEX (100 µg/ml) or in combination for
48 h. Flow cytometry analysis showed that a combination of SAL and
DEX increased the apoptotic rate of the CEM-C1 cells from 10.65 to
26.35% compared with that in the DEX only group (Fig. 3B). Subsequently, western blot
analysis showed that the combination treatment induced the
activation of cleaved-PARP and Bax, and decreased the protein
expression of BCL-2 (Fig. 3C).
Notably, in the combination treatment group, there was also a
significant increase in LC3 protein expression level when compared
with that in the DEX or SAL only groups (Fig. 3D). Furthermore, Fig. 3E showed that the DEX alone group
inhibited the protein expression level of c-Myc in the CEM-C1 cells
and the combination of the two drugs was the most effective and
statistically significant. The data suggested that SAL increased
the sensitivity of the CEM-C1 cells to DEX.
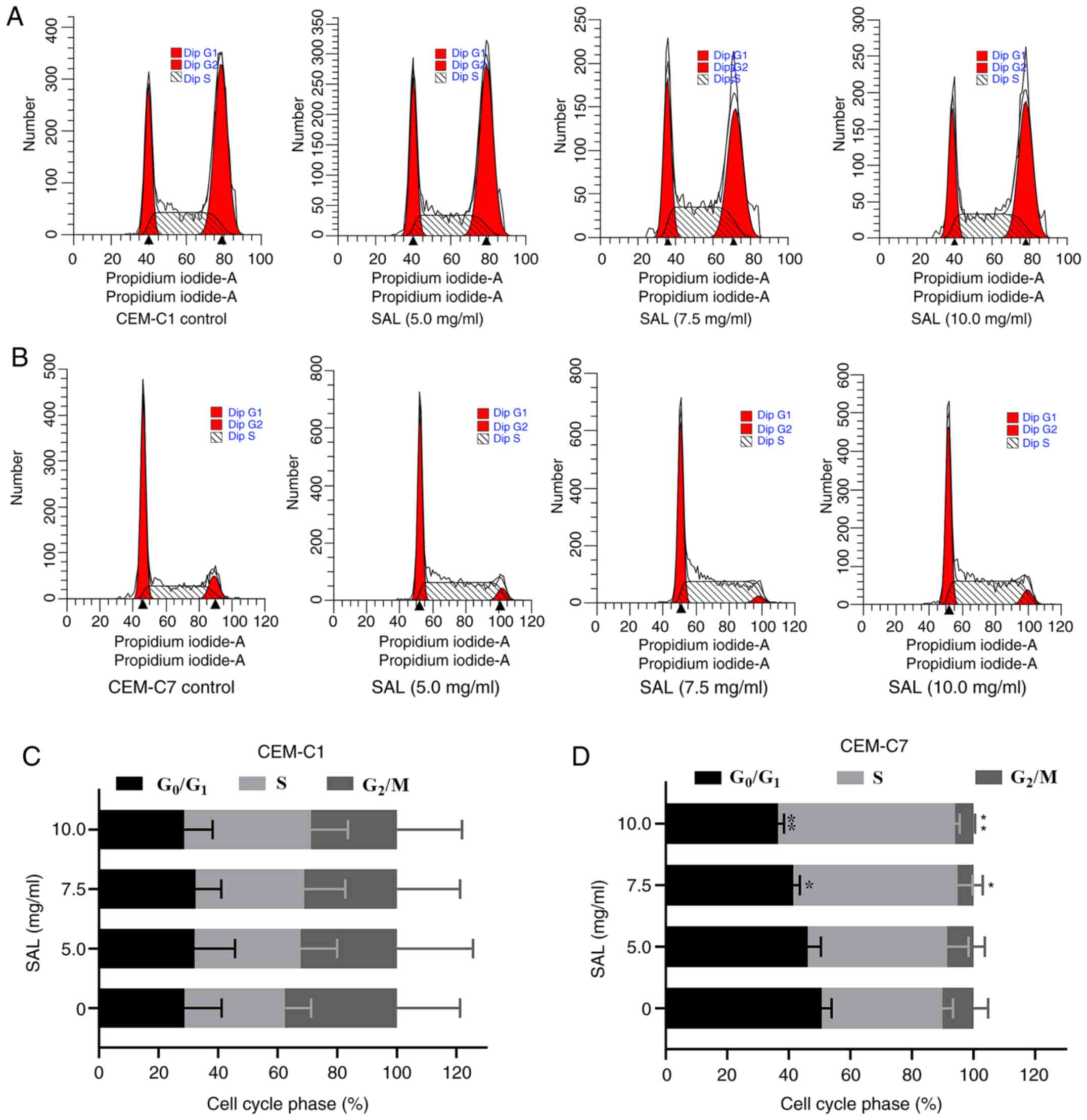
Effect of SAL on the cell cycle in
T-ALL cells
To investigate whether SAL could affect the cell
cycle in the T-ALL cells, the CEM-C1 and CEM-C7 cell lines were
treated with different concentrations of SAL for 48 h and
subsequently stained with PI (Fig.
4A and B). Following an
increase in SAL concentration, the percentage of the cells in the
G0/G1 phase in the CEM-C7 cells was
significantly decreased (F, 11.93; P<0.01), whereas the
percentage of the cells in the S phase was significantly increased
(F, 9.30; P<0.01). No significant change was noted with respect
to the G2/M phase (P>0.05), indicating that SAL
blocked the CEM-C7 cells in the S phase (Fig. 4D). However, SAL exhibited no
significant difference in the cell cycle of the CEM-C1 cells
(P>0.05; Fig. 4C).
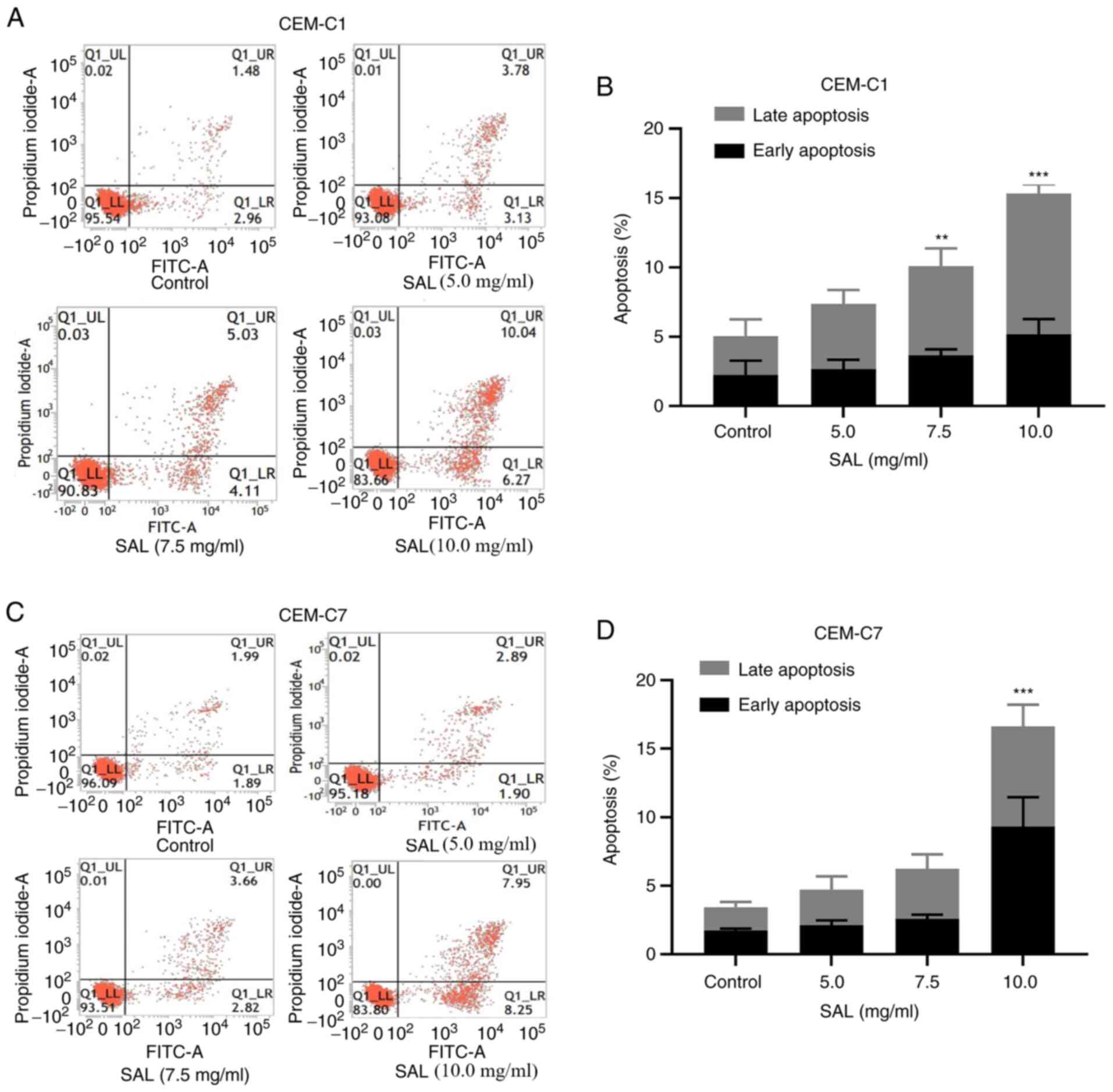
Effect of SAL on the induction of
apoptosis in the T-ALL cells
To investigate whether SAL could induce apoptosis in
the T-ALL cells, the CEM-C1 and CEM-C7 cell lines were treated with
SAL at different concentrations. The results indicated that SAL
could increase the early, late and total apoptotic rate of the
CEM-C1 and CEM-C7 cells (Fig. 5A
and C). Following an increase in
the concentration of SAL, CEM-C1 cells underwent apoptosis. The
total apoptotic rate was significantly increased from 5.06±0.66% in
the control group to 10.18±0.87% in cells treated with 7.5 mg/ml
SAL (P<0.01), whereas treatment with 10.0 mg/ml SAL increased
the total apoptotic rate to 15.34±1.45%, which was significantly
higher compared with that in the control group (P<0.001;
Fig. 5B). In the CEM-C7 cells, the
total apoptotic rate following 10.0 mg/ml SAL treatment was
16.62±3.44%, which was significantly higher compared with that in
the control group 3.43±0.46% (P<0.001; Fig. 5D). This suggested that SAL could
induce apoptosis in the human T-ALL cell lines.
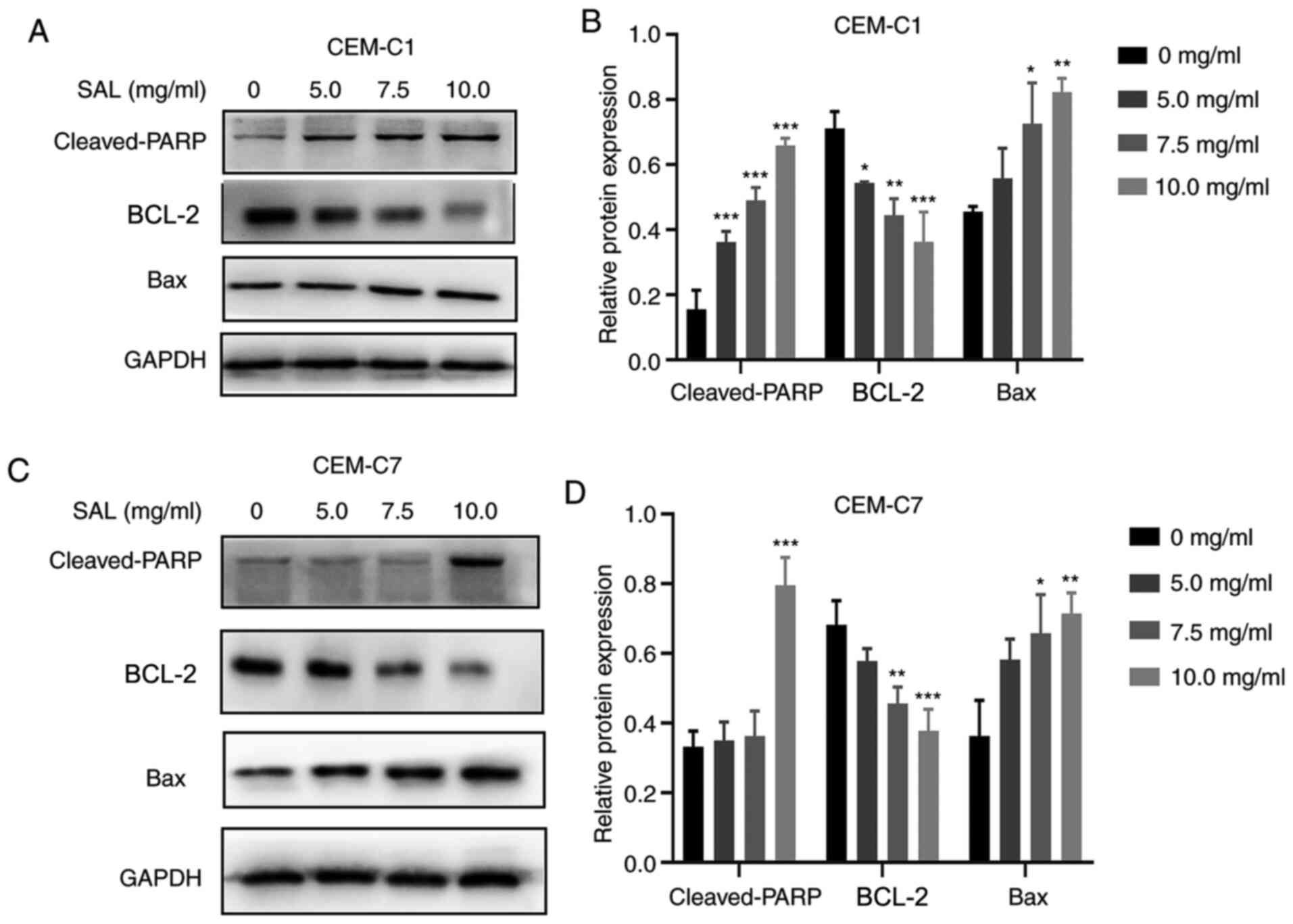
Effect of SAL on the expression level
of apoptosis-associated proteins
To further investigate the molecular mechanism of
SAL in promoting apoptosis of the T-ALL cell lines, the expression
level of the pro-apoptotic and anti-apoptotic proteins was
determined. Western blot analysis indicated that there was an
increase in the expression levels of Bax and cleaved-PARP proteins
following treatment with different concentrations of SAL. There was
also inhibition in the protein expression level of BCL-2 in the
CEM-C1 and CEM-C7 cells, in a dose-dependent manner (Fig. 6).
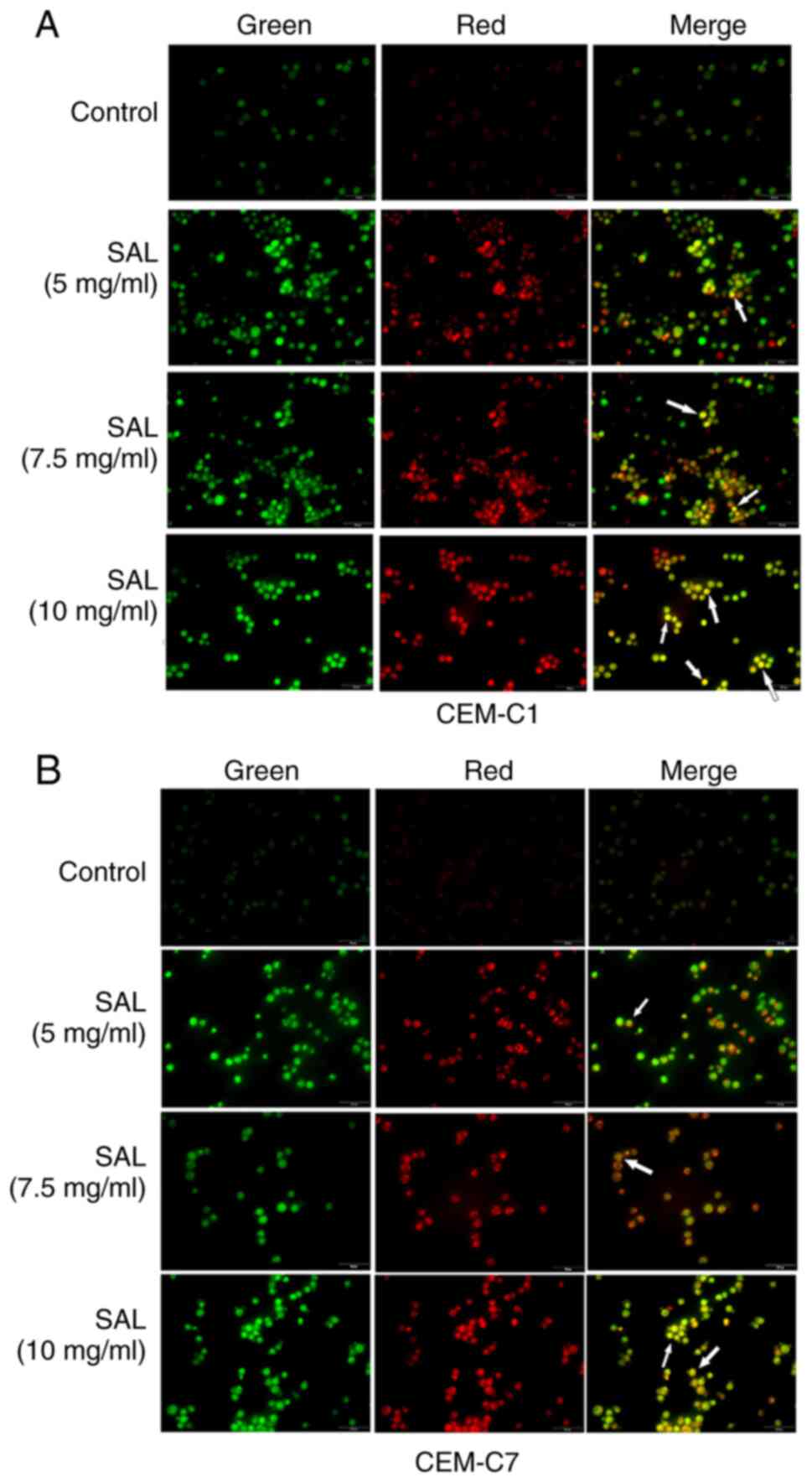
SAL induces autophagy in the T-ALL
cells
Autophagy is characterized by the formation of
acidic autophagy vesicles in the cells and can be determined using
acridine orange staining (29).
Acridine orange is a fluorescent dye used for detecting the
structure of acid vesicles that produces green fluorescence
following binding to the nucleoli and the cytoplasm, and red
fluorescence following binding to autophagic lysosomes (30). The results of acridine orange
staining indicated that the number of orange fluorescent organelles
in the CEM-C1 and CEM-C7 cells, corresponding to the number of
acidic autophagy vesicles, was notably increased compared with that
in the control group. This suggested that SAL promoted autophagy in
the human T-ALL cell lines (Fig. 7A
and B).
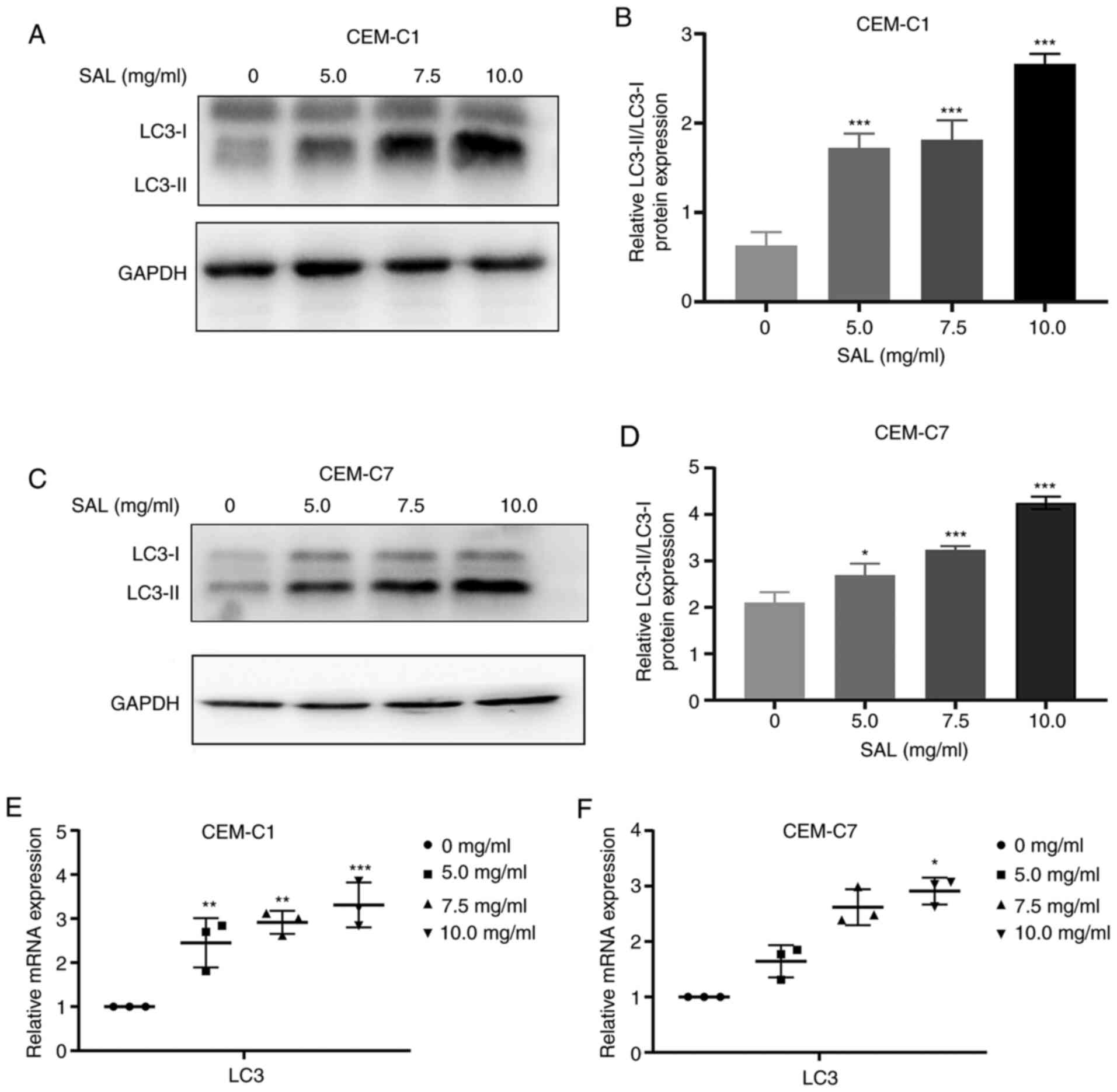
Effect of SAL on autophagy-related
protein expression levels
During the process of autophagy, LC3 is the membrane
component of the autophagosome extension and LC3 is converted from
LC3-I to LC3-II (31). Therefore,
LC3-II can be used to quantify the number of intracellular
autophagosomes (32). The results
of western blot analysis showed that compared with that in the
control group, the expression levels of the LC3-II protein in the
CEM-C1 and CEM-C7 cells was significantly increased, and the
protein expression ratio of LC3-II/LC3-I was also increased (F,
77.64 and 73.88, respectively, with 10.0 mg/ml SAL; both
P<0.001; Fig. 8A-D). The mRNA
expression level of LC3 was also found to be upregulated (F, 19.11
and 37.49, with 10.0 mg/ml SAL; P<0.05; Fig. 8E and F). This suggested that SAL could induce
autophagy in the human T-ALL cell lines, CEM-C1 and CEM-C7.
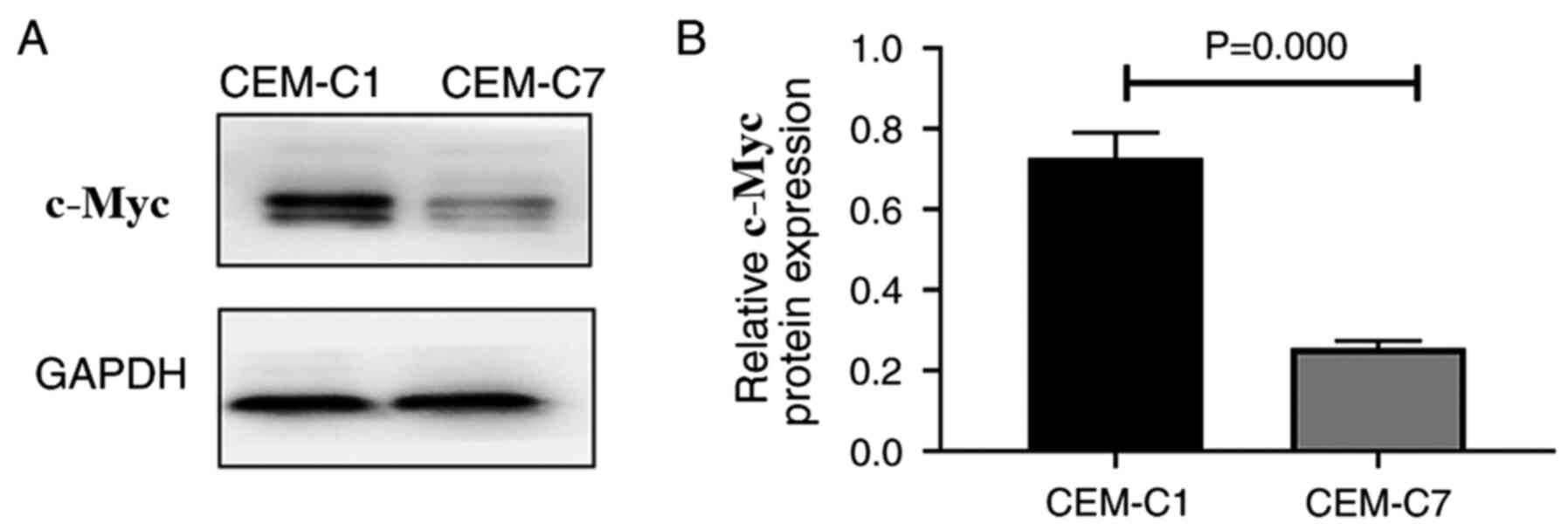
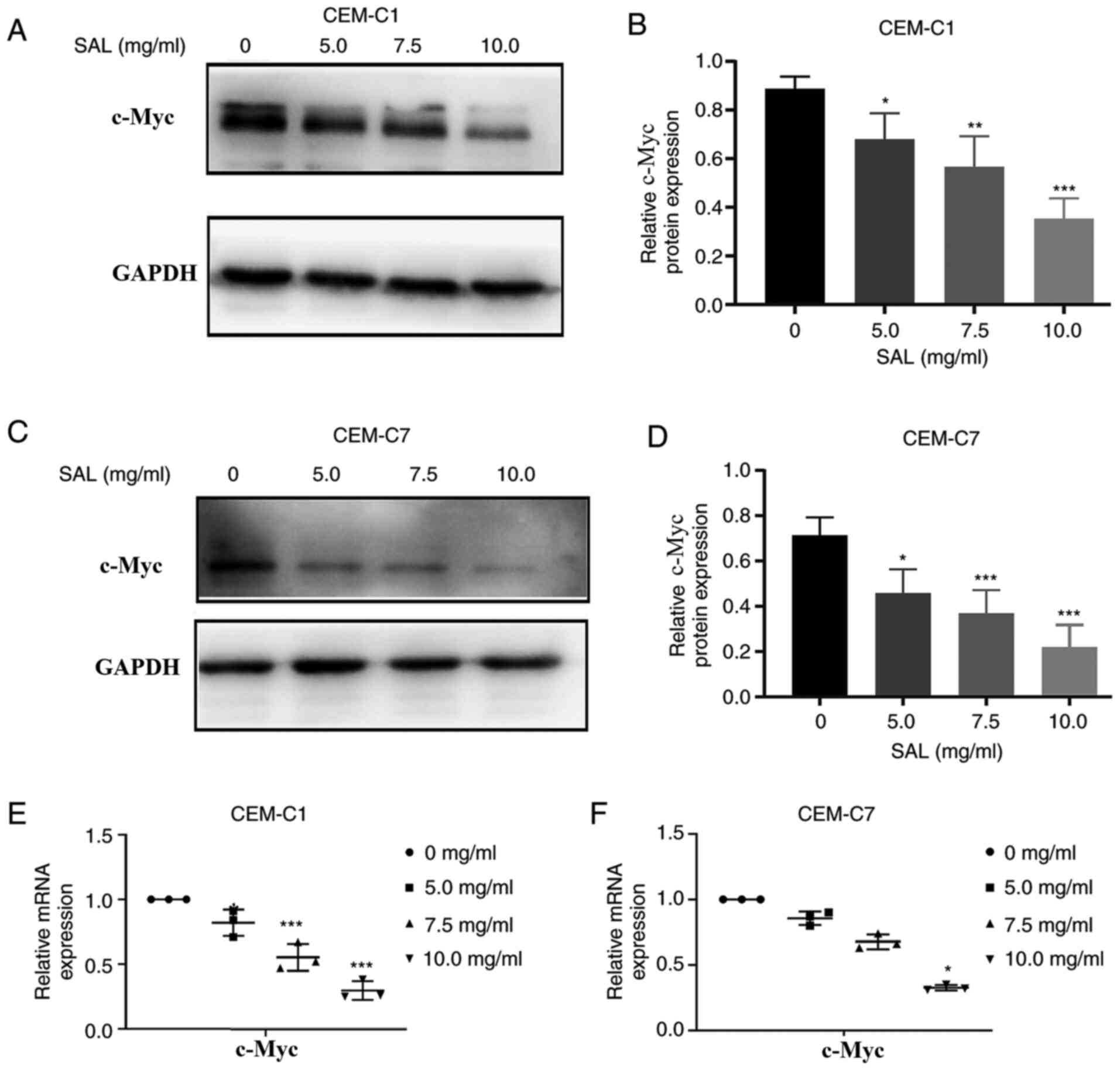
Protein expression of c-Myc in the
DEX-resistant CEM-C1 cells
To investigate the role of c-Myc in the
DEX-resistant CEM-C1 cells, western blot analysis was used to
detect the expression levels of the c-Myc protein in the CEM-C1 and
CEM-C7 cells. The results indicated that the CEM-C1 cells expressed
higher c-Myc protein levels compared with that in the CEM-C7 cells
(Fig. 9A and B). High expression of c-Myc may reduce the
sensitivity of the CEM-C1 cells to DEX, indicating that c-Myc could
play an important role in the occurrence and development of tumor
drug resistance.
SAL overcomes DEX-resistance in the
CEM-C1 cells by downregulating c-Myc protein and mRNA
expression
Various studies have shown that high mRNA expression
of c-Myc has been associated with drug resistance in pancreatic
cancer and HPV-negative neck squamous cell carcinoma cells
(33,34). It has also been shown that
downregulation of c-Myc mRNA expression using siRNA could improve
the efficacy of DEX in treatment of ALL (35). To investigate the mechanism in which
the CEM-C1 cells could overcome DEX resistance following treatment
with SAL, the CEM-C1 and CEM-C7 cells were treated with different
concentrations of SAL for 48 h, and the protein and mRNA expression
levels of c-Myc were determined. Western blot analysis indicated
that the c-Myc protein expression level was decreased in a
dose-dependent manner, in both cells, compared with that in the
control group (F, 21.74 and 18.58, with 10.0 mg/ml SAL; P<0.001;
Fig. 10A-D). The RT-qPCR results
indicated that the c-Myc mRNA expression levels were also decreased
in a dose-dependent manner compared with that in the control group
(F, 43.14 and 161.0, with 10.0 mg/ml SAL; P<0.05; Fig. 10E and F). This suggested that SAL could reduce
DEX resistance in the human T-ALL, CEM-C1 cells by downregulating
c-Myc protein and mRNA expression.
Discussion
ALL is one of the most common malignancies, with the
highest incidence rate in children, accounting for ~80% of leukemia
cases. ALL is five times more common than acute myeloid leukemia
(36). ALL can be divided into
B-ALL and T-ALL. DEX is a synthetic GC, which has been used to
treat patients with T-ALL (37). At
present, resistance to DEX is one of the important reasons leading
to treatment failure or recurrence. Therefore, it is important to
clarify the mechanism of DEX resistance and overcome it.
Tumor cells are characterized by unrestricted
proliferation. The two main pathways of tumor cell death are
apoptosis and autophagy. Cell apoptosis and autophagy have been
associated with tumorigenesis and cancer prevention (38). A previous study has shown that
dysregulation of apoptosis promoted the survival of malignant cells
and reduced the sensitivity of tumor cells to specific drugs in
leukemia (39). Autophagy is an
important intracellular process that causes the degradation of
unnecessary or damaged cytoplasmic contents to maintain metabolism
and homeostasis (40). Autophagy
exhibits a dual function by promoting cell survival and cell death,
and has been associated with tumorigenesis, metastasis and drug
resistance (41). The induction of
apoptosis and autophagy is an effective antitumor therapy strategy
(42,43). Long et al (44) demonstrated that by promoting the
induction of autophagy and apoptosis, this process could increase
the sensitivity to GC treatment in human acute lymphoblastic
leukemia cells.
SAL has been reported to have a wide range of
pharmacological functions, including anti-tumor activity, that
SAL-based activation of apoptosis and autophagy are the major
mechanisms responsible for the anti-cancer activity of this
compound (45). A previous study
has shown that SAL induced apoptosis and autophagy in human colon
cancer cells by inhibiting the PI3K/Akt/mTOR pathway (46). The therapeutic effect of SAL on a
variety of tumors has been confirmed, including colorectal cancer
(12), gastric cancer (47), bladder cancer (14), ovarian cancer (15), breast cancer (48) and Wilms' tumor (17); however, its role in promoting T-ALL
apoptosis and autophagy and its molecular mechanism are not clear.
In the present study, the protein expression levels of
cleaved-PARP, Bax and LC3 were increased, while BCL-2 protein
expression level was decreased in the CEM-C1 and CEM-C7 cells
following treatment with SAL. This indicated that SAL could be a
potential treatment for T-ALL. It was also found that DEX could
induce apoptosis and autophagy in the CEM-C1 cells. In addition,
when 1.5 mg/ml SAL (cell inhibition rate, <4%) was combined with
DEX, the induction of apoptosis and autophagy was significantly
increased (P<0.01) compared with that in the DEX group.
Previous studies have shown that DEX resistance was
associated with upregulation of the oncogene c-Myc mRNA expression
(10,49). In a separate study, Bhadri et
al (50) demonstrated that
in vivo DEX treatment in a DEX-sensitive ALL xenograft
caused significant repression of c-Myc mRNA expression. In the
present study, it was found that the CEM-C1 cells exhibited a
higher protein expression level of c-Myc compared with that in the
CEM-C7 cells. Long et al (51) demonstrated that imatinib-resistant
K562/G cells exhibited high protein expression level of c-Myc
compared with that in the parental K562 cells, and the c-Myc
inhibitor 10058-F4 was found to reverse resistance caused by high
expression level of c-Myc. It has also been shown that c-Myc
inhibitors can produce synergistic anti-cancer effects with
vincristine and sensitize pre-B-ALL cells to the anti-tumor effects
of this chemotherapeutic drug by inducing apoptosis and autophagy
(52). Sayyadi et al
(53) demonstrated that c-Myc
inhibition, using 10058-F4, increased the sensitivity of acute
promyelocytic leukemia cells to arsenic trioxide. The results from
the present study demonstrated that SAL could reduce c-Myc protein
and mRNA expression levels. Notably, the combination treatment of
SAL with DEX resulted in a more significant inhibition of c-Myc
expression compared with that in the DEX group. Therefore, future
studies should combine c-Myc inhibitors with SAL to verify their
effects on apoptosis and autophagy, and the sensitivity to T-ALL
cells to DEX.
In summary, the present study demonstrated the
reversal effect of SAL on DEX resistance in the CEM-C1 cell line
and confirmed that SAL exhibited an optimal effect on inhibiting
proliferation, and induced apoptosis and autophagy in both the
CEM-C1 and CEM-C7 cells. The CEM-C1 cells were more sensitive to
SAL. SAL may overcome the resistance of the CEM-C1 cells to DEX by
downregulating c-Myc protein and mRNA expression level. DEX
resistance is a challenging problem for T-ALL chemotherapy. This
provides a new treatment strategy for overcoming drug resistance
and new evidence for clarifying the molecular mechanism of
T-ALL-associated DEX resistance. The data further suggested that
c-Myc may be a target for treating T-ALL resistance to DEX.
Supplementary Material
SAL (1.5 mg/ml) does not induce
cytotoxicity in CEM-C1 and CEM-C7 cells. CEM-C1 and CEM-C7 cells
were treated with 1.5 mg/ml SAL for 48 h. Cell viability was
detected using the Cell Counting Kit-8 assay. Data are presented as
the mean ± SD. SAL, salidroside.
Effects of DEX and DEX+SAL on the
IC50 in CEM-C1 and CEM-C7 cells. (A) CEM-C1 and (B)
CEM-C7 cells. The data are presented as the mean ± SD.
***P<0.001. ns, not significant; DEX, dexamethasone;
SAL, salidroside.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Basic Research Project
of Sichuan Province (grant no. 2019YJ0690), Luzhou Science and
Technology Plan Project (grant nos. 2019-RCW-96 and 2019-RCM-98)
and the Major Science and Technology Projects in Sichuan Province
(grant no. 2019YFS0531).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WJL designed and conceived the current study. YNN
and YZ performed the experiments, analyzed the data and drafted and
wrote the manuscript. FFZ, SLL, DWR and XQ contributed to analysis
and interpretation of data, drafted the manuscript and revised it
critically for important intellectual content. YNN, YZ and WJL
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhou R, Mo W, Wang S, Zhou W, Chen X and
Pan S: miR-141-3p and TRAF5 network contributes to the progression
of T-cell acute lymphoblastic leukemia. Cell Transplant. 28
(Suppl-1):S59–S65. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Qin X, Zhang MY and Liu WJ: Application of
minimal residual disease monitoring in pediatric patients with
acute lymphoblastic leukemia. Eur Rev Med Pharmacol Sci.
22:6885–6895. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dufinck K, Goossens S, Peirs S, Wallaert
A, Van Loocke W, Matthijssens F, Pieters T, Milani G, Lammens T,
Rondou P, et al: Novel biological insights in T-cell acute
lymphoblastic leukemia. Exp Hematol. 43:625–639. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bongiovanni D, Tosello V, Saccomani V,
Dalla-Santa S, Amadori A, Zanovello P and Piovan E: Crosstalk
between Hedgehog pathway and the glucocorticoid receptor pathway as
a basis for combination therapy in T-cell acutelymphoblastic
leukemia. Oncogene. 39:6544–6555. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lin KT and Wang LH: New dimension of
glucocorticoids in cancer treatment. Steroids. 111:84–88.
2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Scheijen B: Molecular mechanisms
contributing to glucocorticoid resistance in lymphoid malignancies.
Cancer Drug Resist. 2:647–664. 2019.
|
|
7
|
Verbeke D, Demeyer S, Prieto C, de-Bock
CE, De-Bie J, Gielen O, Jacobs K, Mentens N, Verhoeven BM,
Uyttebroeck A, et al: The XPO1 inhibitor KPT-8602 synergizes with
dexamethasone in acutelymphoblastic leukemia. Clin Cancer Res.
26:5747–5758. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Jing D, Bhadri VA, Beck D, Thoms JA, Yakob
NA, Wong JW, Knezevic K, Pimanda JE and Lock RB: Opposing
regulation of BIM and BCL2 controls glucocorticoid-induced
apoptosis of pediatric acute lymphoblastic leukemia cells. Blood.
125:273–283. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Roderick JE, Gallagher KM, Murphy LC,
O'Connor KW, Tang K, Zhang B, Brehm M, Greiner DL, Yu J, Zhu LJ, et
al: Prostaglandin E2 stimulates cAMP signaling and re-sensitizes
human leukemia cells to glucocorticoid-induced cell death. Blood:
Aug 5, 2020 (Epub ahead of print).
|
|
10
|
Toscan CE, Jing D, Mayoh C and Lock RB:
Reversal of glucocorticoid resistance in paediatric acute
lymphoblastic leukaemia is dependent on restoring BIM expression.
Br J Cancer. 122:1769–1781. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Meyer LK, Huang BJ, Delgado-Martin C, Roy
RP, Hechmer A, Wandler AM, Vincent TL, Fortina P, Olshen AB, Wood
BL, et al: Glucocorticoids paradoxically facilitate steroid
resistance in T-cell acute lymphoblastic leukemias and thymocytes.
J Clin Invest. 130:863–876. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Shi X, Zhao W, Yang Y, Wu S and Lv B:
Salidroside could enhance the cytotoxic effect of L-OHP on
colorectal cancer cells. Mol Med Rep. 17:51–58. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Qi Z, Tang T, Sheng L, Ma Y, Liu Y, Yan L,
Qi S, Ling L and Zhang Y: Salidroside inhibits the proliferation
and migration of gastric cancer cells via suppression of
Src-associated signaling pathway activation and heat shock protein
70 expression. Mol Med Rep. 18:147–156. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li T, Xu K and Liu Y: Anticancer effect of
salidroside reduces viability through autophagy/PI3K/Akt and MMP-9
signaling pathways in human bladder cancer cells. Oncol Lett.
16:3162–3168. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yu G, Li N, Zhao Y, Wang W and Feng XL:
Salidroside induces apoptosis in human ovarian cancer SKOV3 and
A2780 cells through the p53 signaling pathway. Oncol Lett.
15:6513–6518. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhao G, Shi A, Fan Z and Du Y: Salidroside
inhibits the growth of human breast cancer in vitro and in vivo.
Oncol Rep. 33:2553–2560. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li H, Huang D and Hang S: Salidroside
inhibits the growth, migration and invasion of Wilms' tumor cells
through down-regulation of miR-891b. Life Sci. 222:60–68.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Qin Y, Liu HJ, Li M, Zhai DH, Tang YH,
Yang L, Qiao KL, Yang JH, Zhong WL, Zhang Q, et al: Salidroside
improves the hypoxic tumor microenvironment and reverses the drug
resistance of platinum drugs via HIF-1α signaling pathway.
EBioMedicine. 38:25–36. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pelengaris S, Khan M and Evan G: c-MYC:
More than just a matter of life and death. Nat Rev Cancer.
2:764–776. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Fauriat C and Olive D: AML drug
resistance: c-Myc comes into play. Blood. 123:3528–3530.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ji W, Zhang W, Wang X, Shi Y, Yang F, Xie
H, Zhou W, Wang S and Guan X: c-myc regulates the sensitivity of
breast cancer cells to palbociclib via c-myc/miR-29b-3p/CDK6 axis.
Cell Death Dis. 11(760)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Pyko IV, Nakada M, Sabit H, Teng L,
Furuyama N, Hayashi Y, Kawakami K, Minamoto T, Fedulau AS and
Hamada J: Glycogen synthase kinase 3β inhibition sensitizes human
glioblastoma cells to temozolomide by affecting O6-methylguanine
DNA methyltransferase promoter methylation via c-Myc signaling.
Carcinogenesis. 34:2206–2217. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tan Y, Sementino E, Chernoff J and Testa
JR: Targeting MYC sensitizes malignant mesothelioma cells to PAK
blockage-induced cytotoxicity. Am J Cancer Res. 7:1724–1737.
2017.PubMed/NCBI
|
|
24
|
Ge JC, Yu WD, Li JH, Ma HB, Wang PY, Zhou
YH, Wang Y, Zhang J and Shi GW: USP16 regulates
castration-resistant prostate cancer cell proliferation by
deubiquitinating and stablizing c-Myc. J Exp Clin Cancer Res.
40(59)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yi XL, Lou LP, Wang J, Xiong J and Zhou S:
Honokiol antagonizes doxorubicin resistance in human breast cancer
via miR-188-5p/FBXW7/c-Myc pathway. Cancer Chemother Pharmacol: Feb
5, 2021 (Epub ahead of print).
|
|
26
|
Monga J, Subramani D, Bharathan A and
Ghosh J: Pharmacological and genetic targeting of 5-lipoxygenase
interrupts c-Myc oncogenic signaling and kills
enzalutamide-resistant prostate cancer cells via apoptosis. Sci
Rep. 10(6649)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sheng Q, Zhang Y, Wang Z, Ding J, Song Y
and Zhao W: Cisplatin-mediated down-regulation of miR-145
contributes to up-regulation of PD-L1 via the c-Myc transcription
factor in cisplatin-resistant ovarian carcinoma cells. Clin Exp
Immunol. 200:45–52. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hirasawa M and Kurita-Ochiai T:
Porphyromonasgingivalis induces apoptosis and autophagy via ER
stress in human umbilical vein endothelial cells. Mediators
Inflamm. 2018(1967506)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
31
|
Zhang Y, Zhang Y, Jin XF, Zhou XH, Dong
XH, Yu WT and Gao WJ: The role of astragaloside IV against cerebral
ischemia/reperfusion injury: Suppression of apoptosis via promotion
of P62-LC3-autophagy. Molecules. 24(1838)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kim D, Hwang HY, Kim JY, Lee JY, Yoo JS,
Marko-Varga G and Kwon HJ: FK506, an immunosuppressive drug,
induces autophagy by binding to the V-ATPase catalytic subunit a in
neuronal cells. J Proteome Res. 16:55–64. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jin X, Fang R, Fan P, Zeng L, Zhang B, Lu
X and Liu T: PES1 promotes BET inhibitors resistance and cells
proliferation through increasing c-Myc expression in pancreatic
cancer. J Exp Clin Cancer Res. 38(463)2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Robinson AM, Rathore R, Redlich NJ, Adkins
DR, VanArsdale T, Van Tine BA and Michel LS: Cisplatin exposure
causes c-Myc-dependent resistance to CDK4/6 inhibition in
HPV-negative head and neck squamous cell carcinoma. Cell Death Dis.
10:867–879. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lv M, Wang Y, Wu W, Yang S, Zhu H, Hu B,
Chen Y, Shi C, Zhang Y, Mu Q and Ouyang G: C-Myc inhibitor 10058-F4
increases the efficacy of dexamethasone on acute lymphoblastic
leukaemia cells. Mol Med Rep. 18:421–428. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Huang HP, Liu WJ, Guo QL and Bai YQ:
Effect of silencing HOXA5 gene expression using RNA interference on
cell cycle and apoptosis in Jurkat cells. Int J Mol Med.
37:669–678. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Capria S, Molica M, Mohamed S, Bianchi S,
Moleti ML, Trisolini SM, Chiaretti S and Testi AM: A review of
current induction strategies and emerging prognostic factors in the
management of children and adolescents with acute lymphoblastic
leukemia. Expert Rev Hematol. 13:755–769. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yu Y, Yu X, Ma J, Tong Y and Yao J:
Effects of NVP-BEZ235 on the proliferation, migration, apoptosis
and autophagy in HT-29 human colorectal adenocarcinoma cells. Int J
Oncol. 49:285–293. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Vazanova A, Jurecekova J, Balharek T,
Marcinek J, Stasko J, Dzian A, Plank L, Zubor P, Racay P and Hatok
J: Differential mRNA expression of the main apoptotic proteins in
normal and malignant cells and its relation to in vitro resistance.
Cancer Cell Int. 18(33)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Klionsky DJ, Abdel-Aziz AK, Abdelfatah S,
Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu
YP, Acevedo-Arozena A, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy (4rd edition).
Autophagy. 17:1–382. 2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Fu Y, Zhang Y, Gao M, Quan L, Gui R and
Liu J: Alisertib induces apoptosis and autophagy through targeting
the AKT/mTOR/AMPK/p38 pathway in leukemic cells. Mol Med Rep.
14:394–398. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanismsof apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838.
2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Long SL, Ren DW, Zhong FF, Niu YN, Qin X,
Mu D and Liu WJ: Reversal of glucocorticoid resistance in acute
lymphoblastic leukemia cells by miR-145. PeerJ.
8(e9337)2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Magani SKJ, Mupparthi SD, Gollapalli BP,
Shukla D, Tiwari AK, Gorantala J, Yarla NS and Tantravahi S:
Salidroside-can it be a multifunctional drug. Curr Drug Metab.
21:512–524. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fan XJ, Wang Y, Wang L and Zhu M:
Salidroside induces apoptosis and autophagy in human colorectal
cancer cells through inhibition of PI3K/Akt/mTOR pathway. Oncol
Rep. 36:3559–3567. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhang ZD, Yang W, Ma F, Ma Q, Zhang B,
Zhang YL, Liu YQ, Liu HX and Hua YW: Enhancing the chemotherapy
effect of Apatinib on gastric cancer by co-treating with
salidroside to reprogram the tumor hypoxia micro-environment and
induce cell apoptosis. Drug Deliv. 27:691–702. 2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Yu X, Sun LL, Tan LJ, Wang M, Ren XL, Pi
JX, Jiang MM and Li N: Preparation and characterization of
PLGA-PEG-PLGA nanoparticles containing salidroside and tamoxifen
for breast cancer therapy. AAPS PharmSciTech. 21(85)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Beesley AH, Firth MJ, Ford J, Weller RE,
Freitas JR, Perera KU and Kees UR: Glucocorticoid resistance in
T-lineage acute lymphoblastic leukaemia is associated with a
proliferative metabolism. Br J Cancer. 100:1926–1936.
2009.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Bhadri VA, Cowley MJ, Kaplan W, Trahair TN
and Lock RB: Evaluation of the NOD/SCID xenograft model for
glucocorticoid-regulated gene expression in childhood B-cell
precursor acute lymphoblastic leukemia. BMC Genomics.
12(565)2011.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Long ZJ, Fang ZG, Pan XN, Fan RF and Lin
DJ: Inhibition of c-Myc by 10058-F4 overcomes imatinib resistance
in chronic myeloid leukemia cells. Chin J Pathophysiol.
30:1590–1594. 2014.
|
|
52
|
Sheikh-Zeineddini N, Safaroghli-Azar A,
Salari S and Bashash D: C-Myc inhibition sensitizes pre-B ALL cells
to the anti-tumor effect of vincristine by altering apoptosis and
autophagy: Proposing a probable mechanism of action for 10058-F4.
Eur J Pharmacol. 870(172821)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Sayyadi M, Safaroghli-Azar A,
Pourbagheri-Sigaroodi A, Abolghasemi H, Anoushirvani AA and Bashash
D: c-Myc inhibition using 10058-F4 increased the sensitivity of
acute promyelocytic leukemia cells to arsenic trioxide via blunting
PI3K/NF-κB axis. Arch Med Res. 51:636–644. 2020.PubMed/NCBI View Article : Google Scholar
|