Introduction
Reperfusion following acute myocardial infarction is
a double-edged sword. On one hand, reperfusion is the only option
to salvage a dying myocardium (1).
On the other hand, reperfusion can initiate myocardial
ischemia/reperfusion (I/R) injury (MIRI), which produces additional
serious tissue damage and may even become fatal to some patients
receiving revascularization therapy (2).
The mechanisms underlying MIRI are poorly
understood. Recently, complement activation has been found to play
a pivotal role in MIRI (3-5).
As part of the innate immune defense, the complement system
consists of over 30 plasma and cell membrane proteins that are
activated in a sequential manner, producing both local and systemic
manifestations (6). For instance,
excessive complement activation products complement component (C)3a
and C5a are aggravating factors for myocardial necrosis and
inflammatory cell infiltration (7).
Conversely, inhibition of the complement cascade has been shown to
significantly reduce MIRI (8,9).
Therefore, targeting the complement cascade may be a promising
therapeutic strategy for treating I/R injury. Ischemic
postconditioning (IPostC), defined as rapid, intermittent
interruptions of blood flow in early reperfusion, has been shown to
alleviate I/R injury in animal models and patients with acute
myocardial infarction (10-12).
However, the mechanisms of IPostC-related cardioprotection are not
well understood. To the best of our knowledge, the role of critical
novel pathways of I/R injury, such as activation of the complement
system, have not been studied in myocardial IPostC.
MicroRNAs (miRNAs/miR) play a vital role in
regulating ischemic injury (13).
miR-499, a cardiac-enriched miRNA, participates in protection of
the ischemic myocardium (14,15). A
recent study has reported that miR-499 has an antiapoptotic effect
against MIRI (16). However, the
potential role and mechanism of action underlying the
cardioprotective effect of miR-499 in IPostC are poorly understood.
The present study investigated the role of complement activation
and the possible involvement of miR-499 in IPostC of the rat
myocardium during I/R injury.
Materials and methods
Animals
A total of 105 male adult Sprague-Dawley rats (8
weeks old, weighing 240-280 g) were collected from the Laboratory
Animal Center of Guangxi Medical University. The rats were housed
under standard laboratory conditions (temperature, 25±2˚C; relative
humidity, 50±15%; 12 h dark/light cycle) and rats were permitted
ad libitum access to food and water. All animal protocols
were performed in accordance with the Guide for the Care and Use of
Laboratory Animals (National Institutes of Health, USA) and were
approved by the Animal Care and Use Committee of Guangxi Medical
University.
MIRI model
The rat MIRI model was established using a
previously described method (17).
Briefly, the rats were intraperitoneally injected with sodium
pentobarbital (2%, 50 mg/kg) for general anesthesia and
mechanically ventilated using a small animal respirator. A left
parasternal incision was made in the fourth intercostal space to
expose the heart. The left anterior descending coronary artery
(LAD) was temporarily ligated at 2-3 mm below the lower edge of the
left auricle using an 8-0 silk suture. A small plastic tube was
inserted through the ligature to form a snare to enable reperfusion
by reversing occlusion. Cardiac ischemia was induced by tightening
the ligature around the plastic tube for 30 min. Successful
ligation was visually confirmed when the anterior wall of the left
ventricle turned pale and by elevation of the ST segment on
precordial leads of the electrocardiogram. Reperfusion was induced
by loosening the ligation via the plastic tube, which lasted for 2
h. IPostC was performed at the onset of reperfusion with three
cycles of reperfusion for 30 sec, followed by coronary artery
occlusion for 30 sec. Immediately after reperfusion, the rats were
euthanized by cervical dislocation. Death of the rats was confirmed
by the lack of a heart beat and respiration, and then the left
ventricle of rats was harvested for further analysis.
Experimental grouping
The rats were randomly divided into following groups
(n=15 per group): ⅰ) Sham, in which the ligature was passed under
the LAD, but not tied, and maintained for 150 min; ⅱ) I/R, in which
the rats were subjected to 30 min of ischemia followed by 2 h of
reperfusion; ⅲ) IPostC, in which the rats underwent three cycles of
30 sec of reperfusion and 30 sec of ischemia, initiated immediately
at the onset of reperfusion; ⅳ) adeno-associated virus (AAV), in
which the empty AAV vector (1x1012 v.g./rat) was
injected into the tail vein and did not receive any other
treatment; ⅴ) AAV + IPostC, in which the empty AAV vector
(1x1012 v.g./rat) was injected into the tail vein,
followed by the IPostC procedure after 4 weeks; ⅵ) sponge + IPostC,
which received the AAV vector of miR-499-5p-sponge
(AAV-miR-499-5p-sponge, 1x1012 v.g./rat) via tail vein
injection, followed by the IPostC procedure after 4 weeks; ⅶ)
miR-499 + IPostC, which received the AAV vector of miR-499-5p
(AAV-miR-499-5p, 1x1012 v.g./rat) followed by the IPostC
procedure after 4 weeks. In total, 5 of the 105 rats used in this
study were excluded: A total of 2 in the I/R group and 1 in the
IPostC group died due to ventricular fibrillation; 1 in the AAV +
IPostC group died due to cardiogenic shock during reperfusion; and
1 in the sponge + IPostC group died due to viral delivery failure.
The results described are for the remaining 100 rats.
AAV transfection
The AAV-miR-499-5p
(5'-GCTGTTAAGACTTGCAGTGATGTTTAGCTCCTCTCCATGTGAACATCACAGCAAGTCTGTGCTGC-3'),
AAV-miR-499-5p-sponge
(5'-AAACATCACTGCAAGTCTTAATATACAAACATCACTGCAAGTCTTAAACATCAAACATCACTGCAAGTCTTAATCTTCAAAACATCACTGCAAGTCTTAA-3'),
and empty AAV vector (pHBAAV-U6-MCS-CMV-EGFP; Hanbio Biotechnology
Co., Ltd.) as AAV control were constructed using previously
described methods (18). The AAV
vectors were injected through the tail vein at a dose of
1.0x1012 genome copies per rat. Successful transfections
were confirmed by detection of miR-499-5p expression in myocardial
tissue at 4 weeks after injection.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA of ischemic myocardium was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and cDNA was synthesized from 1 µg of total RNA using the
RevertAid First Strand cDNA Synthesis kit (Thermo Scientific, Inc.)
according to the manufacturer's instructions. qPCR was performed in
96-well plates using 2X SYBR Green qPCR ProMix (Guangzhou Yingzan
Biological Technology Co., Ltd.) using an ABI 7300 Real-Time PCR
System (Applied Biosystems). The thermocycling conditions were as
follows: 95˚C for 2 min, 40 cycles of 95˚C for 5 sec, and 60˚C for
30 sec. All reactions were performed in triplicate. U6 was used as
the reference gene. The relative expression of miRNA was determined
using the 2-ΔΔCq method (19). The primer sequences used for RT-qPCR
are listed in Table I.
 | Table IThe primer sequences for reverse
transcription-quantitative PCR. |
Table I
The primer sequences for reverse
transcription-quantitative PCR.
| Gene | Sequence |
|---|
| U6 forward |
5'-CTCGCTTCGGCAGCACA-3' |
| U6 reverse |
5'-AACGCTTCACGAATTTGCGT-3' |
| miR-499
forward |
5'-TTAAGACTTGCAGTGATGTTT-3' |
| miR-499
reverse |
5'-CAGTGCAGGGTCCGAGGTAT-3' |
| miRT Random |
5'-GTCGTATCCAGTGCAGGGTCC |
| Primera |
GAGGTATTCGCACTGGATACGACNNNNN-3' |
Western blotting
Frozen rat myocardial tissue was lysed in
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology) containing 1% phenylmethylsulfonyl fluoride
(Beyotime Institute of Biotechnology) on ice. The protein
concentration was measured using a bicinchoninic acid protein assay
kit (Beyotime Institute of Biotechnology). Normalized protein
samples (30 µg) were resolved by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to PVDF
(EMD Millipore). The membranes were blocked with 5% nonfat milk at
room temperature for 1 h, followed by incubation with primary
antibodies against C3a (Abcam; cat. no. 171080; 1:1,000), C5a
(Abcam; cat. no. 202039; 1:1,000), NF-κB p65 (Cell Signaling
Technology, Inc.; cat. no. 8242; 1:1,000), TNF-α (Cell Signaling
Technology, Inc.; cat. no. 11948; 1:1,000), IL-1β (Abcam; cat. no.
200478; 1:1,000), IL-6 (Abcam; cat. no. 208113; 1:1,000) and
β-actin (Abcam; cat. no. 8226; 1:1,000) overnight at 4˚C. The
membranes were washed three times with Tris-buffered saline
containing 0.1% Tween-20 and then incubated with infrared
dye-conjugated secondary antibodies (LI-COR Biosciences; cat. no.
926-32211; 1:10,000) for 1 h at room temperature. Protein bands
were detected using an Odyssey Infrared Imaging System (LI-COR
Biosciences) and the relative intensity of the bands was quantified
using ImageJ v1.8.0 software (National Institutes of Health).
ELISAs
A total of 2 h after reperfusion of the myocardium,
2 ml of venous blood was collected into EDTA-coated tubes. After
centrifugation at 2,000 x g and 4˚C for 10 min, the plasma samples
were collected and stored at -80˚C for further analysis. The plasma
levels of C3a (cat. no. 08510r), C5a (cat. no. 08513r), TNF-α (cat.
no. 11987r), NF-κB p65 (cat. no. 08788r), IL-1β (cat. no.08055r)
and IL-6 (cat. no. 04640r) were detected using commercial ELISA
kits (Cusabio Technology LLC).
Triphenyl tetrazolium chloride (TTC)
staining
The myocardial infarct area was assessed using TTC
staining. Briefly, rat heart tissues were quickly isolated and
washed with cold saline, then immediately frozen at temperature of
-70˚C for 10 min and finally sliced into 2-mm cross-sections from 2
mm below the ligation line. The slices were stained in 2% TTC
solution (Beijing SolarbioScience and Technology Co., Ltd.) at 37˚C
for 20 min and then fixed in 4% paraformaldehyde at room
temperature for 20 min. The infarct size was quantified using
ImageJ software.
TUNEL staining
TUNEL staining was performed on the myocardial
sections to detect apoptotic cardiomyocytes, according to the
manufacturer's instructions (Roche Diagnostics). Myocardial samples
were fixed in 4% paraformaldehyde at 4˚C for 24 h and embedded in
paraffin. The dewaxed myocardial tissue sections were immersed in
3% hydrogen peroxide in methanol for 10 min at room temperature,
and washed 3 times with PBS; then the tissue sections and
proteinase K working solution incubated at 37˚C for 30 min, which
the tissue sections and TdT reaction mixture are incubated at 37˚C
for 2 h. After washing 3 times with PBS, the nuclei were
counterstained with DAPI solution (5 µg/ml) at room temperature for
5 min. A total of 50 µl anti-fade mounting medium was added to
TUNEL-positive cells, which were observed in five randomly selected
visual fields using a fluorescence microscope (magnification,
x200). The index of apoptosis was expressed as follows: Index of
apoptosis=(number of apoptotic cardiomyocytes/total number of
cardiomyocytes) x100%.
Statistical analysis
The data were presented as the mean ± SD and
analyzed using SPSS 13.0 (SPSS Inc.) software. One-way ANOVAs were
performed using post hoc Tukey's multiple comparison tests. A
P-value <0.05 was considered statistically significant.
Results
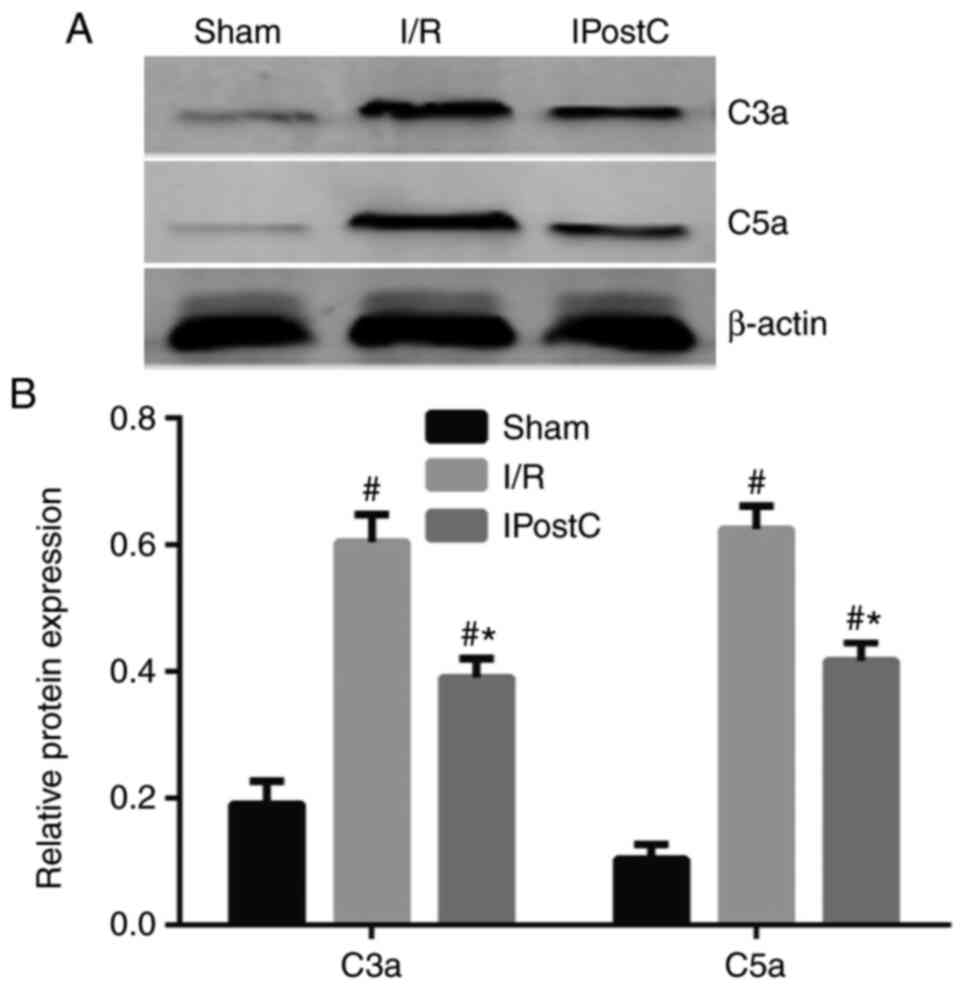
IPostC reduces I/R-induced myocardial
complement activation
Generation of C3a and C5a is commonly involved in
all three complement activation pathways (20). Therefore, the present study examined
the effects of IPostC on complement activation by detecting the
expression of C3a and C5a in the rat myocardium. Western blot
analysis showed that the expression levels of C3a and C5a were
significantly increased in the rat myocardium following I/R injury,
whereas IPostC attenuated the C3a and C5a upregulation following
I/R (Fig. 1).
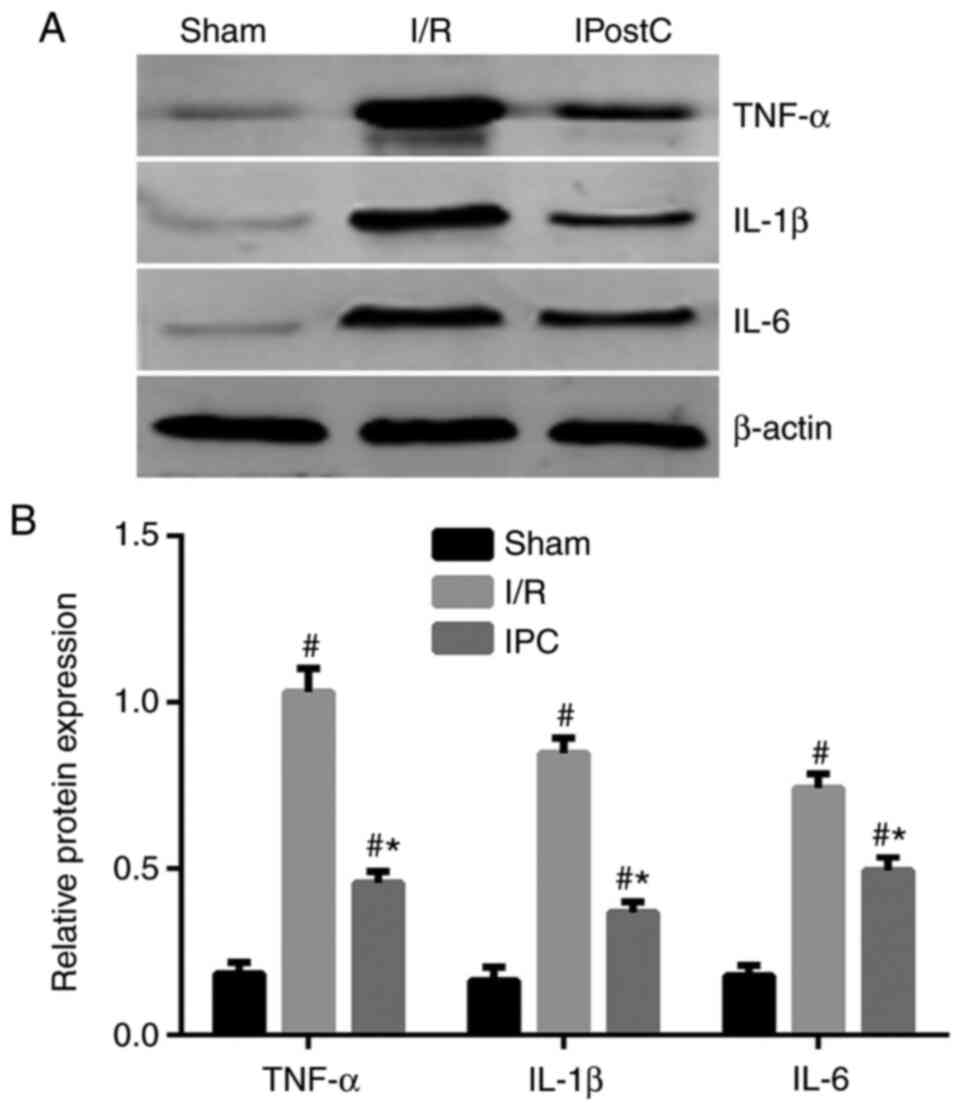
IPostC reduces the I/R-induced
myocardial inflammatory response
Previous studies have demonstrated that activation
of the complement system promotes inflammation in heart, kidney and
brain tissues (21-23).
To determine whether the inhibitory effect of IPostC on the
complement system affects the inflammatory response in the
myocardium the expression of the proinflammatory cytokines TNF-α,
IL-1β and IL-6 was examined. I/R significantly increased the
expression levels of TNF-α, IL-1β and IL-6, as measured by western
blot analyses compared to the sham group. IPostC then significantly
reduced the expression levels of these inflammatory cytokines in
the rat myocardium, compared with the I/R group, although this did
not reach the expression levels observed in the sham group
(Fig. 2).
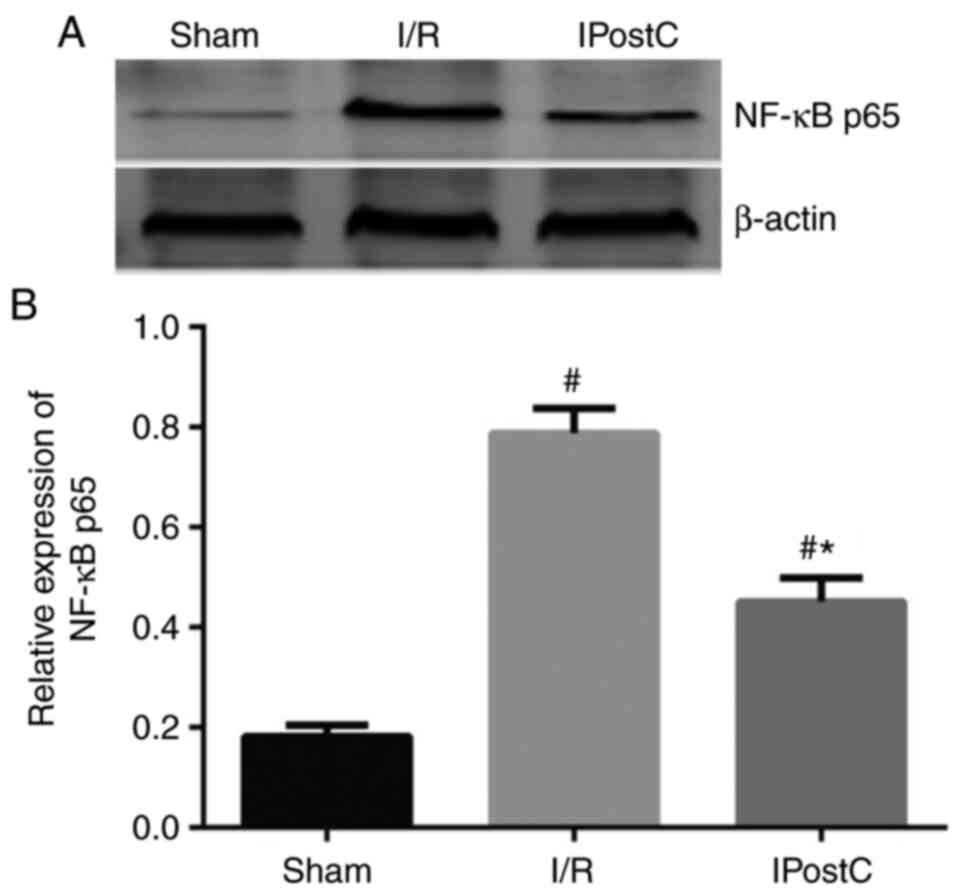
IPostC reduces I/R-induced NF-κB
activation
To explore the underlying mechanism of action behind
the anti-inflammatory effect of IPostC, the present study examined
the expression levels of NF-κB p65, a transcription factor critical
for the activation of the complement system and proinflammatory
cytokines. I/R induced a significant increase in the NF-κB p65
expression levels in the rat myocardium compared with the sham
group, which was partially attenuated by IPostC (Fig. 3).
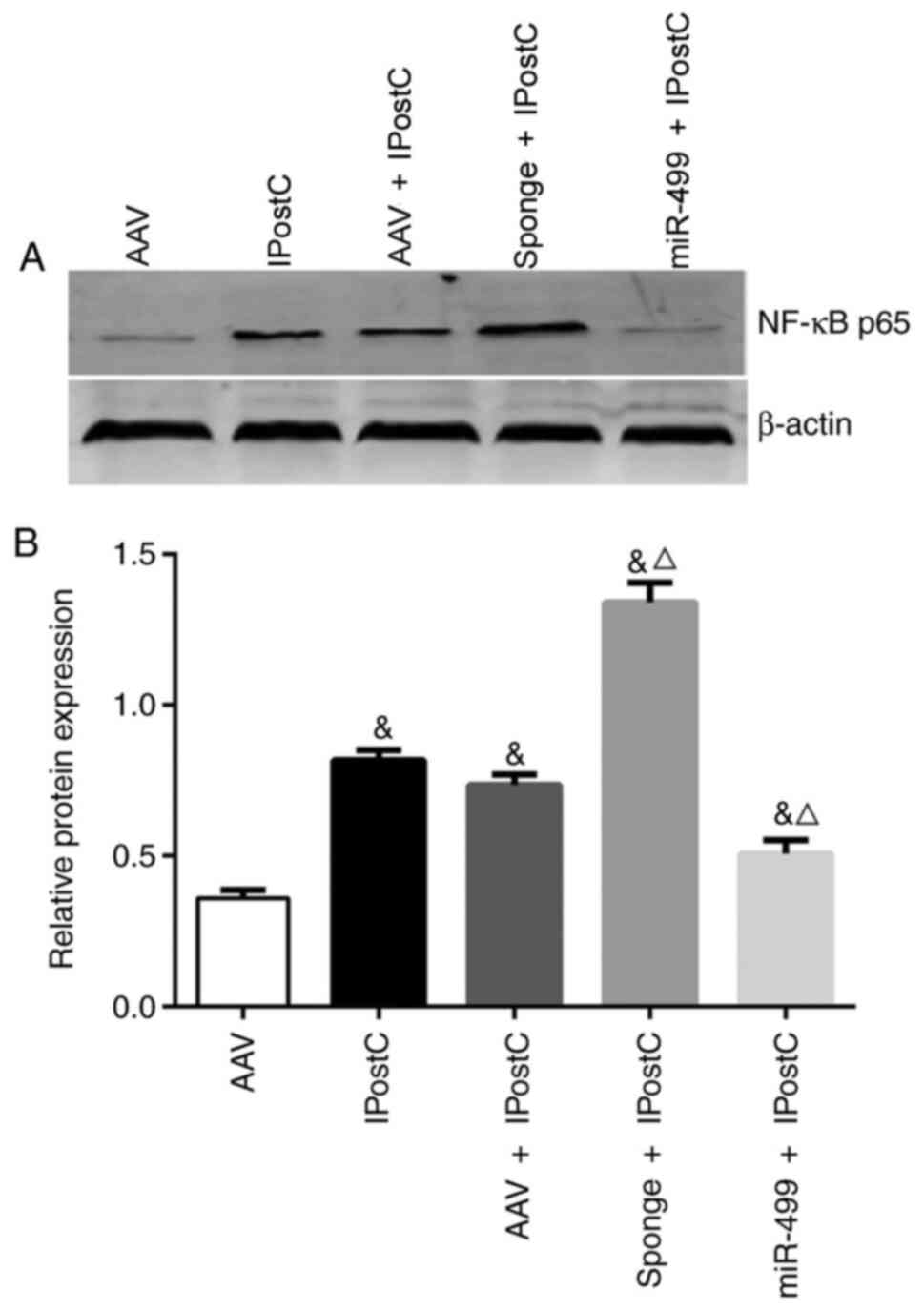
miR-499 reduces the expression of
complement factors and inflammatory cytokines in the IPostC rat
myocardium
Previous studies have suggested that miR-499 has a
protective effect against myocardial ischemia (14,15,24).
In order to further understand the role of miR-499 in myocardial
IPostC, the present study transfected rats with empty AAV,
AAV-miR-499-5p and AAV-miR-499-5p-sponge, and then subjected the
rats to MIRI protocols with or without IPostC. None of the rats
showed behavioral abnormalities following injection of the AAV
vectors. RT-qPCR confirmed the successful AAV transfection of the
agents in the rat myocardium in all animals, treated either with or
without IPostC (Fig. 4A). Compared
with the sham group, the expression of miR-499 was significantly
decreased in the I/R group, but it was significantly increased in
the IPostC group (Fig. 4A).
Notably, in rats receiving IPostC, AAV-miR-499-5p-sponge
significantly increased the expression levels of C3a and C5a
(Fig. 4B) as well as the
proinflammatory cytokines TNF-α, IL-1β and IL-6 (Fig. 4B), compared with the rats receiving
empty AAV. In contrast, AAV-miR-499-5p significantly reversed the
upregulation of complement factors and inflammatory cytokines in
the IPostC rat myocardium (Fig.
4B). Taken together, these results strongly support a
protective effect of miR-499 in the IPostC rat myocardium, in part,
by inhibiting the upregulation of complement factors and
inflammatory cytokines.
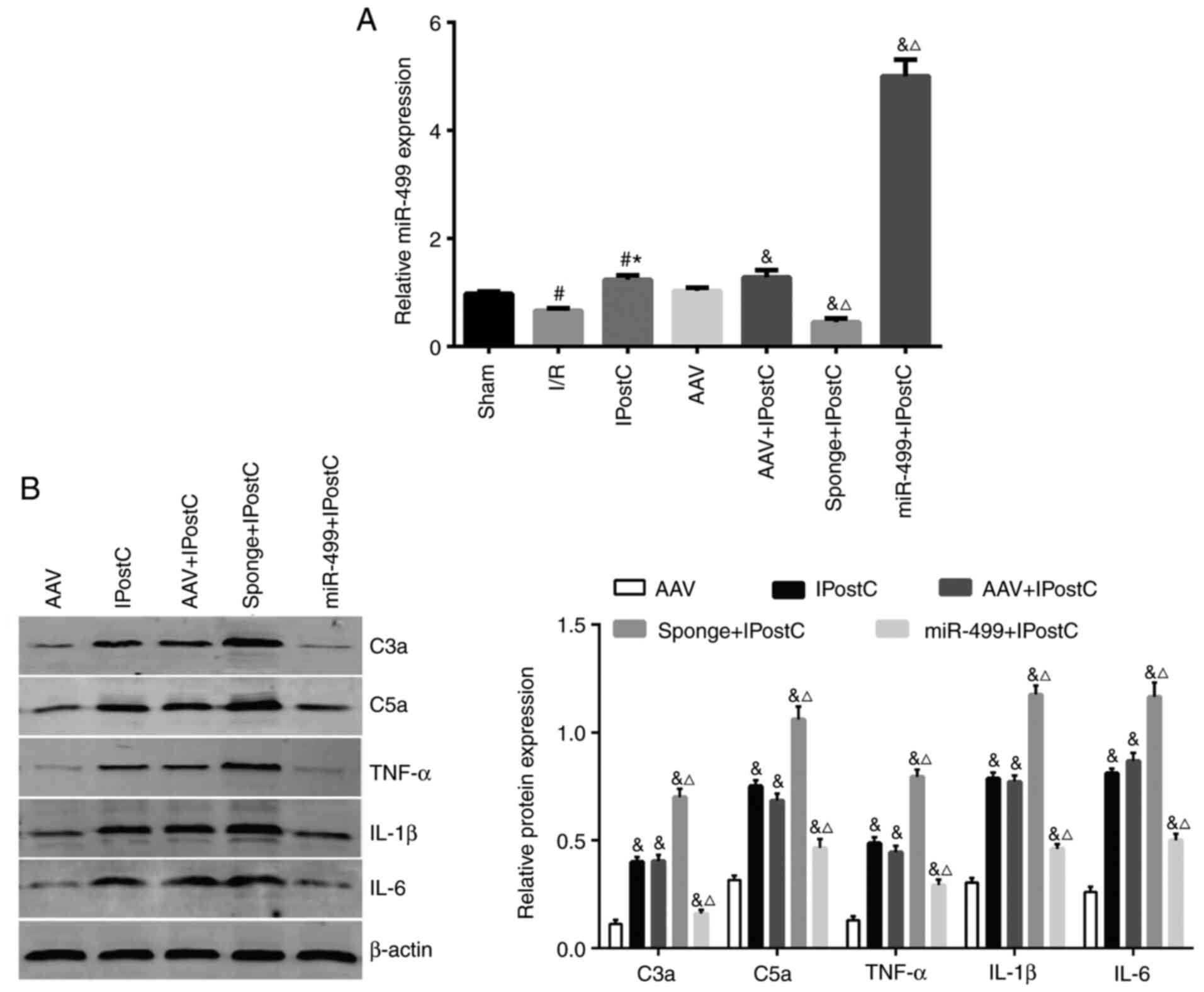 | Figure 4The effects of miR-499 on the
expression of complement factors and inflammatory cytokines in the
rat myocardium. (A) Reverse transcription-quantitative PCR analysis
of miR-499 expression levels in the rat myocardium receiving
transduction of AAV-miR-499-5p, AAV-miR-499-5p-sponge and AAV
control, with and without IPostC. The AAV injection was performed
at 4 weeks prior to the PCR analysis. (B) Western blot analysis of
C3a, C5a, TNF-α, IL-1β and IL-6 expression in the treated rat
myocardium. Quantification of the band intensity is presented
beside the representative blots. Data are presented as the mean ±
SD, n=4. The experiments were repeated three times.
#P<0.05 vs. Sham; *P<0.05 vs. I/R;
&P<0.05 vs. AAV; ΔP<0.05 vs. AAV +
IPostC. AAV, adeno-associated virus; C, complement component; I/R,
ischemia/reperfusion; IPostC, ischemic postconditioning; miR,
microRNA. |
miR-499 inhibits NF-κB activation in
the rat myocardium treated with IPostC
The present study further examined the effect of
miR-499 on NF-κB activation during IPostC. The expression of NF-κB
p65 was not affected by the AAV (Fig.
5; AAV + IPostC). However, transfection with
AAV-miR-499-5p-sponge significantly increased NF-κB p65 expression
(Fig. 5; sponge + IPostC vs.
IPostC), whereas transduction with AAV-miR-499-5p significantly
inhibited NF-κB p65 expression (Fig.
5; miR-499 + IPostC vs. IPostC). Together, these results
further indicated a protective role of miR-499 in myocardial
IPostC, in part by inhibiting NF-κB p65 pathway activation.
miR-499 reduces circulating
inflammatory cytokines in rats treated with IPostC
To further assess the effects of miR-499 on the
systemic inflammatory status during IPostC, the present study
transfected rats with empty AAV, AAV-miR-499-5p and
AAV-miR-499-5p-sponge and then measured the plasma levels of
circulating complement factors and proinflammatory cytokines,
including C3a, C5a, TNF-α, IL-1β, IL-6 and NF-κB p65, using ELISAs.
There were no significant differences in the plasma measurements
between the IPostC and AAV + IPostC groups (Table II). In comparison with the IPostC
and AAV + IPostC groups, the plasma levels of C3a, C5a, TNF-α,
IL-1β, IL-6 and NF-κB p65 were all significantly higher in the rats
receiving AAV-miR-499-5p-sponge and they were all significantly
lower in the rats receiving AAV-miR-499-5p (Table II).
| Table IIThe plasma levels of complement
factors and inflammatory cytokines in rats (mean ± SD). |
Table II
The plasma levels of complement
factors and inflammatory cytokines in rats (mean ± SD).
| Group | C3a (µg/ml) | C5a (ng/ml) | TNF-α (ng/l) | IL-1β (ng/ml) | IL-6 (pg/ml) | NF-κB p65
(pg/ml) |
|---|
| Sham | 887.5±67.6 | 116.4±7.8 | 276.4±5.7 | 25.9±1.2 | 70.3±4.1 | 644.4±26.6 |
| I/R |
1941.0±47.1a |
235.3±11.0a |
540.3±6.9a |
60.1±1.5a |
181.8±4.5a |
1282.0±26.6a |
| IPostC |
1333.9±69.8a,b |
180.6±8.8a,b |
355.7±13.2a,b |
36.2±1.1a,b |
130.1±7.7a,b |
813.4±20.0a,b |
| AAV + IPostC |
1366.7±51.2a,b |
188.3±9.5a,b |
360.0±14.4a,b |
40.6±1.6a,b |
131.0±8.1a,b |
849.6±19.2a,b |
| Sponge +
IPostC |
1735.0±42.6c,d |
212.3±9.4c,d |
445.1±7.4c,d |
46.9±1.3c,d |
161.8±3.5c,d |
975.5±15.8c,d |
| miR-499 +
IPostC |
1085.6±64.4c,d |
154.8±9.7c,d |
308.3±5.9c,d |
31.5±0.7c,d |
89.3±3.3c,d |
742.4±17.8c,d |
miR-499 reduces I/R-induced
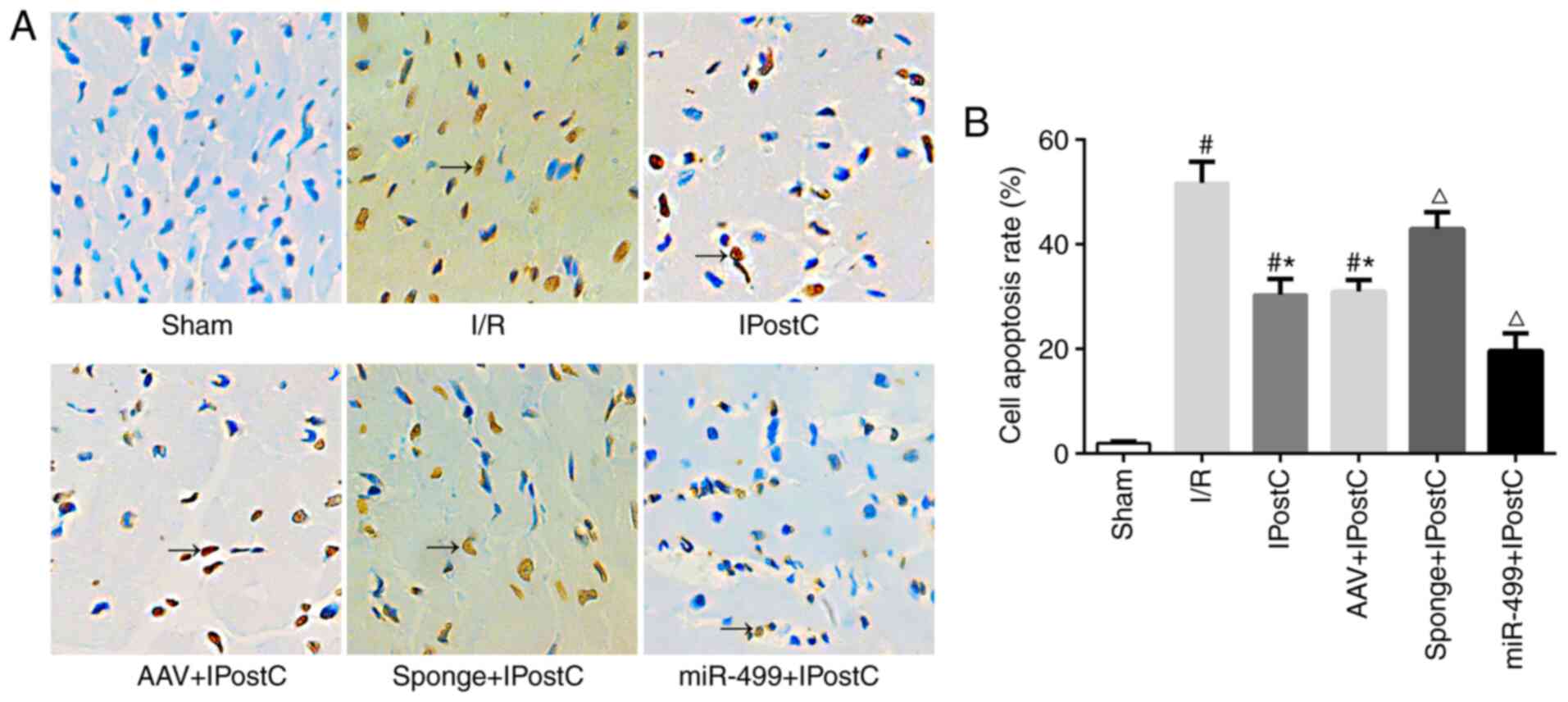
cardiomyocyte apoptosis during IPostC
The present study assessed cell apoptosis in the rat
myocardium using TUNEL assays. Compared with the sham group, a
significantly higher number of TUNEL-positive cells was detected in
the I/R group (Fig. 6; brown nuclei
in I/R vs. Sham). IPostC treatment produced significantly fewer
apoptotic cells than the I/R group (Fig. 6; IPostC vs. I/R). Transfection of
the empty AAV had a minimal effect on cell apoptosis (Fig. 6; IPostC vs. AAV + IPostC). In
contrast, AAV-miR-499-5p further reduced the number of apoptotic
myocardiocytes in the IPostC rats (Fig.
6; miR-499 + IPostC vs. IPostC), whereas AAV-miR-499-5p-sponge
significantly increased the number of TUNEL-positive cells in the
IPostC myocardium (Fig. 6; sponge +
IPostC vs. IPostC). Taken together, these results indicated a
protective role of miR-499 against apoptosis in the IPostC rat
myocardium.
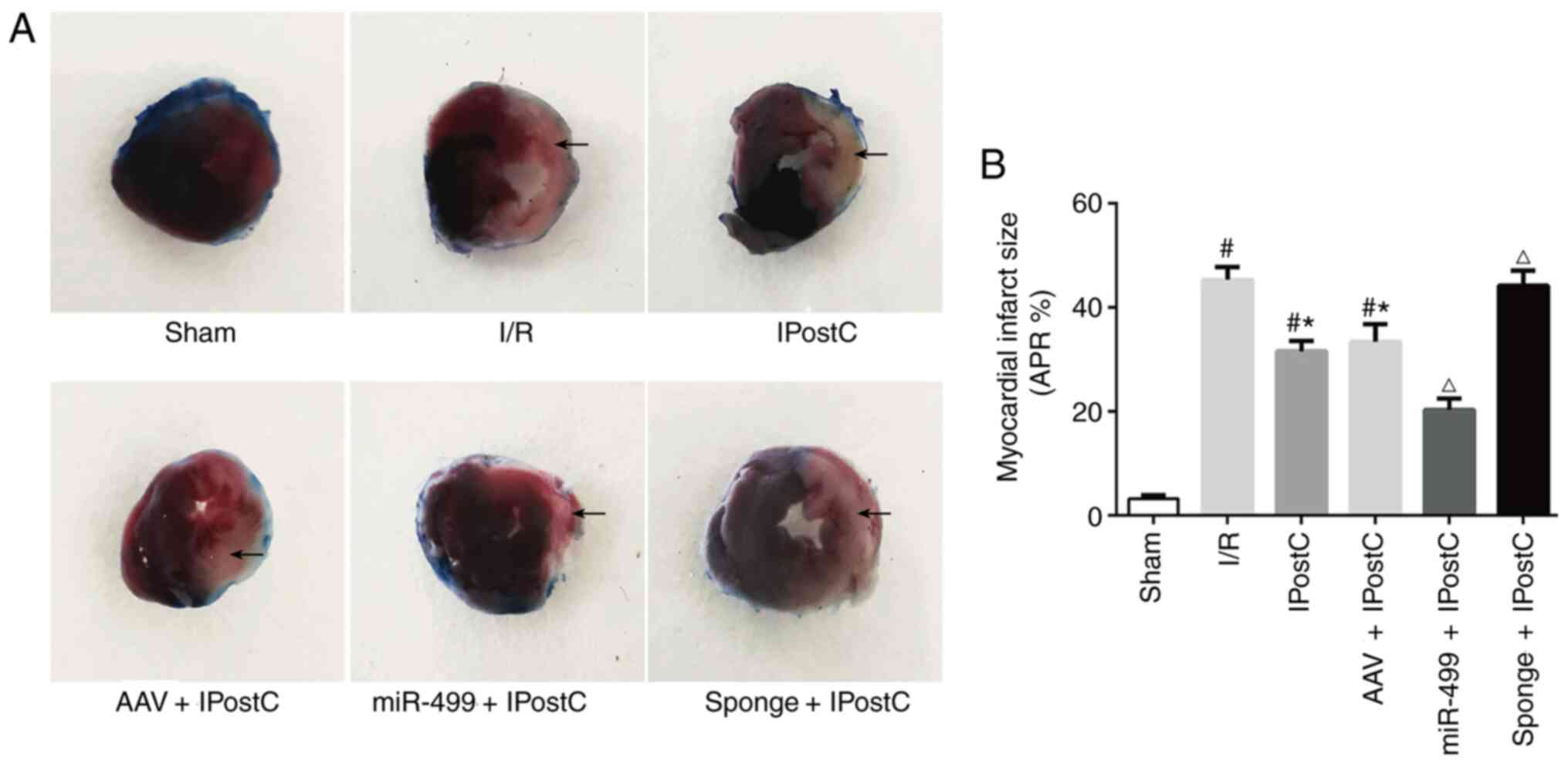
miR-499 reduces the rat myocardial
infarct size during IPostC
Finally, the present study assessed the role of
miR-499 on IPostC by measuring the myocardial infarct size. There
was virtually no visible myocardial infarction in the sham group
(Fig. 7). In contrast, a large
infarct zone was visible on the left ventricle of the I/R group
(Fig. 7; I/R). Interestingly,
IPostC significantly reduced the infarct myocardium area, with or
without introduction of AAV compared with the I/R group (Fig. 7; IPostC vs. I/R; AAV + IPostC vs.
I/R). Notably, overexpression of the miR-499 further reduced the
myocardial infarct zone following IPostC (Fig. 7; IPostC and AAV + IPostC vs. miR-499
+ IPostC). Similarly, inhibition of miR-499 abolished the
protective effect of IPostC on the myocardium and resulted in an
increased infarct size (Fig. 7;
IPostC and AAV + IPostC vs. sponge + IPostC).
Discussion
The major finding of the present study was that
IPostC attenuated I/R-induced myocardial injury in a
miR-499-dependent manner. IPostC effectively reduced I/R-induced
complement activation, local and systemic inflammation,
cardiomyocyte apoptosis and the myocardial infarct size. To the
best of our knowledge, the present study is the first to
demonstrate that miR-499 is an essential regulator of
IPostC-mediated protection against I/R-induced myocardial injury,
which is in part through local and systemic inhibition of
complement activation, inflammation and NF-κB signaling. Taken
together, these results revealed a novel mechanism of action for
miRNA-mediated IPostC protection.
Complement activation plays an important role in
MIRI. Hill and Ward (25) first
reported C3 deposition in an infarcted myocardium and revealed the
relevance of complement activation in MIRI. In addition, Yasojima
et al (26) have shown that
endogenous C3 produced by the heart contributes to the degree of
MIRI. Furthermore, it has been shown that suppressing specific
components of the complement cascade protects against MIRI
(27-30)
and that ischemic preconditioning attenuates MIRI by inhibiting
complement activation (31).
However, to the best of our knowledge, the effect of IPostC on the
complement system has not been reported. The present study found
that the local and circulating levels of C3a and C5a were higher in
the I/R group and lower in the IPostC group, cementing a critical
role of complement factors in IPostC-induced cardioprotection.
Inflammatory responses represent a major
pathological process leading to MIRI (32). An unchecked local inflammatory
response has been observed in tissues subjected to I/R, whereas
IPostC appears to inhibit the inflammatory response by reducing the
expression or activation of local inflammatory mediators, or by
decreasing inflammatory cytokine release (33,34).
Approaches that aim to inhibit these inflammatory responses
following I/R could potentially lead to the attenuation of MIRI.
Previous studies have shown that IPostC inhibits inflammation in
renal tissues following I/R (35-37).
The present study demonstrated a significant inflammatory response
in rat cardiomyocytes following I/R injury and that IPostC
significantly attenuated the inflammatory responses, partially due
to the effect of miR-499 on reducing the expression of
proinflammatory mediators in the IPostC rat myocardium.
The importance of complement activation is
highlighted by its interaction with the inflammatory response and
the NF-κB pathway, which together profoundly determine the outcome
of I/R-induced tissue injury (38).
Activated C3a and C5a attract proinflammatory leukocytes and
promote the release of inflammatory cytokines during I/R (39). Once stimulated, such an inflammatory
process tends to be self-propagative, resulting in sustained
apoptosis and necrosis following MIRI (40). Recently, a correlation between
complement activation and the NF-κB signaling pathway has been
reported (38,41). NF-κB belongs to a family of
transcription factors that plays a key role in regulating
inflammatory responses and cell survival (42). Prior work has shown that IPostC
inhibits I/R-induced inflammation and organ injury through NF-κB
signaling (35,43). However, the exact role of the
complement-NF-κB interaction in myocardial IPostC is unclear. The
present results showed, for the first time, that IPostC
simultaneously blocked complement activation and NF-κB signaling,
supporting the notion that inhibition of both complement and NF-κB
signaling may be involved in IPostC-induced cardioprotection.
miR-499 is most abundantly expressed in the heart
and plays important roles in myocardial infarction and MIRI
(44,45). It has been shown that the miR-499
expression levels correlate with cardiomyocyte apoptosis and the
severity of the infarction induced by I/R (15). For example, reperfusion reduces the
expression levels of miR-499 in canine left ventricles and the
miR-499 expression levels are negatively correlated with troponin T
and creatine kinase-muscle/brain levels (24). However, the role of miR-499 in
IPostC was unknown until a report in 2016(16). In agreement with this previous
report, the present study found that MIRI resulted in a reduction
of miR-499, which was correlated with increased cardiomyocyte
apoptosis and an increased myocardial infarct size. Moreover, the
converse changes were found in the IPostC myocardium. The present
mechanistic studies revealed that IPostC led to an increased
miR-499 level and that by manipulating the miR-499 level, critical
pathways such as complement activation, inflammation and NF-κB
signaling were affected. This resulted in significantly altered
outcomes of myocardial protection following IPostC. Therefore,
miR-499 plays a central role in IPostC-induced
cardioprotection.
A primary limitation of the present study is that
the effects of IPostC and miR-499 on cardiac function in
ischemia-reperfusion rats were not evaluated. Previous studies have
shown that IPostC improves the longitudinal contractile function of
the reperfused myocardium in patients with acute myocardial
infarction (46) and that miR-499
improves left ventricular function in a rat model of IPostC
(16). Considering that
cardiomyocyte apoptosis and myocardial necrosis are closely related
to cardiac function, it is speculated that miR-499 may improve
cardiac function in ischemia-reperfusion rats. Future functional
studies using ultrasound or electrocardiography to evaluate the
effect of miR-499 on live rats during IPostC are recommended.
In conclusion, miR-499 regulated IPostC-mediated
protection against I/R-induced myocardial injury, in part by
inhibiting the activation of local and systemic C3a and C5a;
inflammation; and NF-κB signaling. The present data provided
mechanistic evidence which could support the development of novel
therapeutics aimed at harnessing IPostC-mediated cardioprotection
against MIRI.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81560068) and the
Natural Science Foundation of Guangxi Province (grant no.
2015GXNSFAA139198).
Availability of data and materials
The datasets generated and analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
ZH and YH designed and performed experiments,
analyzed data and co-wrote the paper. QJL, HW and XYZ performed
experiments. RHT and GQZ designed experiments; provided research
funding; and edited and revised the manuscript. All authors have
read and provided final approval of the manuscript. ZH and RHT
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal protocols were performed in accordance
with the Guide for the Care and Use of Laboratory Animals (National
Institutes of Health, USA) and were approved by the Animal Care and
Use Committee of Guangxi Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ibanez B, James S, Agewall S, Antunes MJ,
Bucciarelli-Ducci C, Bueno H, Caforio ALP, Crea F, Goudevenos JA,
Halvorsen S, et al: 2017 ESC guidelines for the management of acute
myocardial infarction in patients presenting with ST-segment
elevation: The Task Force for the management of acute myocardial
infarction in patients presenting with ST-segment elevation of the
European Society of Cardiology (ESC). Eur Heart J. 39:119–177.
2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/reperfusion injury following acute
myocardial infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017(7018393)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tang K, Cheng Y, Wu S, Liu L and Cheng L:
Protective effect of C5 shRNA on myocardial ischemia-reperfusion
injury in rats. Can J Physiol Pharmacol. 90:1394–1402.
2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Frangogiannis NG: The inflammatory
response in myocardial injury, repair, and remodelling. Nat Rev
Cardiol. 11:255–265. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chun N, Haddadin AS, Liu J, Hou Y, Wong
KA, Lee D, Rushbrook JI, Gulaya K, Hines R, Hollis T, et al:
Activation of complement factor B contributes to murine and human
myocardial ischemia/reperfusion injury. PLoS One.
12(e0179450)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bardhan M and Kaushik R: Physiology,
complement cascade. In: StatPearls, Treasure Island, FL, 2021.
|
|
7
|
Busche MN and Stahl GL: Role of the
complement components C5 and C3a in a mouse model of myocardial
ischemia and reperfusion injury. Ger Med Sci.
8(Doc20)2010.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Fu J, Lin G, Wu Z, Ceng B, Wu Y, Liang G,
Qin G, Li J, Chiu I and Liu D: Anti-apoptotic role for C1 inhibitor
in ischemia/reperfusion-induced myocardial cell injury. Biochem
Biophys Res Commun. 349:504–512. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pavlov VI, Tan YS, McClure EE, La Bonte
LR, Zou C, Gorsuch WB and Stahl GL: Human mannose-binding lectin
inhibitor prevents myocardial injury and arterial thrombogenesis in
a novel animal model. Am J Pathol. 185:347–355. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hao M, Zhu S, Hu L, Zhu H, Wu X and Li Q:
Myocardial ischemic postconditioning promotes autophagy against
ischemia reperfusion injury via the activation of the
nNOS/AMPK/mTOR Pathway. Int J Mol Sci. 18(614)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Khan AR, Binabdulhak AA, Alastal Y, Khan
S, Faricy-Beredo BM, Luni FK, Lee WM, Khuder S and Tinkel J:
Cardioprotective role of ischemic postconditioning in acute
myocardial infarction: A systematic review and meta-analysis. Am
Heart J. 168:512–521.e4. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Aslan G, Gul HF, Tektemur A and Sahna E:
Ischemic postconditioning reduced myocardial ischemia-reperfusion
injury: The roles of melatonin and uncoupling protein 3. Anatol J
Cardiol. 23:19–27. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li Y, Lu J, Bao X, Wang X, Wu J, Li X and
Hong W: MiR-499-5p protects cardiomyocytes against ischaemic injury
via anti-apoptosis by targeting PDCD4. Oncotarget. 7:35607–35617.
2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang JX, Jiao JQ, Li Q, Long B, Wang K,
Liu JP, Li YR and Li PF: miR-499 regulates mitochondrial dynamics
by targeting calcineurin and dynamin-related protein-1. Nat Med.
17:71–78. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Zhu J, Yao K, Wang Q, Guo J, Shi H, Ma L,
Liu H, Gao W, Zou Y and Ge J: Ischemic postconditioning-regulated
miR-499 protects the rat heart against ischemia/reperfusion injury
by inhibiting apoptosis through PDCD4. Cell Physiol Biochem.
39:2364–2380. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhong GQ, Tu RH, Zeng ZY, Li QJ, He Y, Li
S, He Y and Xiao F: Novel functional role of heat shock protein 90
in protein kinase C-mediated ischemic postconditioning. J Surg Res.
189:198–206. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Fan JJ, Gao B, Song AQ, Zhu YJ, Zhou J, Li
WZ, Yin YY and Wu WN: Spinal cord NLRP1 inflammasome contributes to
dry skin induced chronic itch in mice. J Neuroinflammation.
17(122)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Haddad A and Wilson AM: Biochemistry,
complement. In: StatPearls, Treasure Island, FL, 2021.
|
|
21
|
Shahini N, Michelsen AE, Nilsson PH,
Ekholt K, Gullestad L, Broch K, Dahl CP, Aukrust P, Ueland T,
Mollnes TE, et al: The alternative complement pathway is
dysregulated in patients with chronic heart failure. Sci Rep.
7(42532)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Mocco J, Mack WJ, Ducruet AF, Sosunov SA,
Sughrue ME, Hassid BG, Nair MN, Laufer I, Komotar RJ, Claire M, et
al: Complement component C3 mediates inflammatory injury following
focal cerebral ischemia. Circ Res. 99:209–217. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Danobeitia JS, Ziemelis M, Ma X, Zitur LJ,
Zens T, Chlebeck PJ, Van Amersfoort ES and Fernandez LA: Complement
inhibition attenuates acute kidney injury after
ischemia-reperfusion and limits progression to renal fibrosis in
mice. PLoS One. 12(e0183701)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Qin H, Chen GX, Liang MY, Rong J, Yao JP,
Liu H and Wu ZK: The altered expression profile of microRNAs in
cardiopulmonary bypass canine models and the effects of mir-499 on
myocardial ischemic reperfusion injury. J Transl Med.
11(154)2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hill JH and Ward PA: The phlogistic role
of C3 leukotactic fragments in myocardial infarcts of rats. J Exp
Med. 133:885–900. 1971.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yasojima K, Kilgore KS, Washington RA,
Lucchesi BR and McGeer PL: Complement gene expression by rabbit
heart: Upregulation by ischemia and reperfusion. Circ Res.
82:1224–1230. 1998.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kassimatis T, Qasem A, Douiri A, Ryan EG,
Rebollo-Mesa I, Nichols LL, Greenlaw R, Olsburgh J, Smith RA, Sacks
SH and Drage M: A double-blind randomised controlled investigation
into the efficacy of Mirococept (APT070) for preventing ischaemia
reperfusion injury in the kidney allograft (EMPIRIKAL): Study
protocol for a randomised controlled trial. Trials.
18(255)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vo AA, Zeevi A, Choi J, Cisneros K, Toyoda
M, Kahwaji J, Peng A, Villicana R, Puliyanda D, Reinsmoen N, et al:
A phase I/II placebo-controlled trial of C1-inhibitor for
prevention of antibody-mediated rejection in HLA sensitized
patients. Transplantation. 99:299–308. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Thielmann M, Marggraf G, Neuhäuser M,
Forkel J, Herold U, Kamler M, Massoudy P and Jakob H:
Administration of C1-esterase inhibitor during emergency coronary
artery bypass surgery in acute ST-elevation myocardial infarction.
Eur J Cardiothorac Surg. 30:285–293. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Smith PK, Carrier M, Chen JC, Haverich A,
Levy JH, Menasché P, Shernan SK, Van de Werf F, Adams PX, Todaro TG
and Verrier E: Effect of pexelizumab in coronary artery bypass
graft surgery with extended aortic cross-clamp time. Ann Thorac
Surg. 82:781–789. 2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tanhehco EJ, Yasojima K, McGeer PL, McGeer
EG and Lucchesi BR: Preconditioning reduces myocardial complement
gene expression in vivo. Am J Physiol Heart Circ Physiol.
279:H1157–H1165. 2000.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Blancke F, Claeys MJ, Jorens P, Vermeiren
G, Bosmans J, Wuyts FL and Vrints CJ: Systemic inflammation and
reperfusion injury in patients with acute myocardial infarction.
Mediators Inflamm. 2005:385–389. 2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kong Y, Rogers MR and Qin X: Effective
neuroprotection by ischemic postconditioning is associated with a
decreased expression of RGMa and inflammation mediators in ischemic
rats. Neurochem Res. 38:815–825. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wu H, Lei S, Yuan J, Liu X, Zhang D, Gu X,
Zhang L and Xia Z: Ischemic postconditioning downregulates Egr-1
expression and attenuates postischemic pulmonary inflammatory
cytokine release and tissue injury in rats. J Surg Res.
181:204–212. 2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chen H, Wang L, Xing BZ, Liu XH, Chen ZY,
Weng XD, Qiu T and Liu L: Ischemic postconditioning attenuates
inflammation in rats following renal ischemia and reperfusion
injury. Exp Ther Med. 10:513–518. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chen R, Zeng Z, Zhang YY, Cao C, Liu HM,
Li W, Wu Y, Xia ZY, Ma D and Meng QT: Ischemic postconditioning
attenuates acute kidney injury following intestinal
ischemia-reperfusion through Nrf2-regulated autophagy,
anti-oxidation, and anti-inflammation in mice. FASEB J.
34:8887–8901. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Guo Q, Du X, Zhao Y, Zhang D, Yue L and
Wang Z: Ischemic postconditioning prevents renal ischemia
reperfusion injury through the induction of heat shock proteins in
rats. Mol Med Rep. 10:2875–2881. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lin Z, Lin H, Li W, Huang Y and Dai H:
Complement component C3 promotes cerebral ischemia/reperfusion
injury mediated by TLR2/NFκB activation in diabetic mice. Neurochem
Res. 43:1599–1607. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Peng Q, Li K, Smyth LA, Xing G, Wang N,
Meader L, Lu B, Sacks SH and Zhou W: C3a and C5a promote renal
ischemia-reperfusion injury. J Am Soc Nephrol. 23:1474–1485.
2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Gorsuch WB, Chrysanthou E, Schwaeble WJ
and Stahl GL: The complement system in ischemia-reperfusion
injuries. Immunobiology. 217:1026–1033. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Liu M, Wang H, Zhang J, Yang X, Li B, Wu C
and Zhu Q: NF-κB signaling pathway-enhanced complement activation
mediates renal injury in trichloroethylene-sensitized mice. J
Immunotoxicol. 15:63–72. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Liu T, Zhang L, Joo D and Sun SC: NF-κB
signaling in inflammation. Signal Transduct Target Ther.
2(17023)2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kin H, Wang NP, Mykytenko J, Reeves J,
Deneve J, Jiang R, Zatta AJ, Guyton RA, Vinten-Johansen J and Zhao
ZQ: Inhibition of myocardial apoptosis by postconditioning is
associated with attenuation of oxidative stress-mediated nuclear
factor-kappa B translocation and TNF alpha release. Shock.
29:761–768. 2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Adachi T, Nakanishi M, Otsuka Y, Nishimura
K, Hirokawa G, Goto Y, Nonogi H and Iwai N: Plasma microRNA 499 as
a biomarker of acute myocardial infarction. Clin Chem.
56:1183–1185. 2010.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sluijter JP, van Mil A, van Vliet P, Metz
CH, Liu J, Doevendans PA and Goumans MJ: MicroRNA-1 and -499
regulate differentiation and proliferation in human-derived
cardiomyocyte progenitor cells. Arterioscler Thromb Vasc Biol.
30:859–868. 2010.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yang Z, Zhou Q, Fang Z, Yu L, Zhou J and
Zhao B: Ischemic postconditioning improves longitudinal contractile
function of the reperfused myocardium in patients with anterior
wall acute myocardial infarction. Zhong Nan Da Xue Xue Bao Yi Xue
Ban. 44:1397–1405. 2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|