Myasthenia gravis (MG) is an autoimmune disease that
is mediated by autoantibodies targeting acetylcholine receptors
(AChRs) at the neuromuscular junction (NMJ), ultimately causing
damage to skeletal muscles (1). The
clinical symptoms of MG include blepharoptosis, muscle weakness,
slurred speech and dysphagia, and may also include paralysis of
respiratory muscles in severe cases (2-4).
Globally, the prevalence of MG is 15-30 per 100,000, with an annual
incidence of >1 per 100,000(5).
The mean duration of the myasthenia crisis course is 42 days and,
if left untreated, fatality rates may reach 80% (6,7).
However, with the advances in medical knowledge, MG-associated
worldwide mortality has been reduced from 70% in the 1930s to 30%
by 1955, and is currently <10% (8).
To date, treatments for MG are limited to the NMJ or
the immune system, and acetylcholinesterase inhibitors are the
first-line treatment of MG (9).
Neostigmine and pyridostigmine are the most commonly used
anticholinesterase drugs in the clinical setting (10). Acetylcholinesterase inhibitors
compensate for the degradation of acetylcholine (Ach) and
pernicious effects on the NMJ by increasing the concentration of
ACh in the synaptic cleft (11).
However, this class of drugs can only alleviate symptoms in a
minority of the patients, and cannot further modify the autoimmune
response (12). Therefore, the
majority of the patients require adjuvant and synergistic therapy.
At present, glucocorticoids are the most effective
immunosuppressant adjuvant for the treatment of MG (5). However, due to serious adverse
reactions caused by high-dose shock therapy over a short period of
time, transient myasthenia aggravation may occur (13). Occasionally, chemical
immunosuppressants, including azathioprine, are used in combination
therapy, which not only enhance the curative effect, but also help
to reduce hormone dosage (14).
Although most patients exhibit good tolerance to azathioprine, the
effect of the drug is slow and, due to the immunosuppression,
patients may be at an increased risk of liver damage and bone
marrow suppression, amongst other adverse effects (15). Moreover, it has been demonstrated
that the levels of choline acetyltransferase in the thymus of
patients with MG and thymic hyperplasia were higher compared with
those in control subjects (16),
which suggested that an abnormal immune tolerance in the thymus
serves an important role in the pathogenesis of MG. Therefore,
thymectomy or thymic radiotherapy is a common therapy used for
patients with MG, and a previous study reported that ~30% of
patients achieved complete and stable remission or pharmacological
remission following this surgery (12). However, thymectomy may also disrupt
self-tolerance and further lead to immune system dysfunction.
Immunomodulatory treatments are promising for the reduction or
elimination of MG symptoms (14).
Plasma exchange can alleviate symptoms by removing the pathogenic
substances in the plasma and reducing the concentration of
autoantibodies (17). Effects may
be observed from 2 to 5 days following treatment (18,19),
but plasma exchange only alleviates MG temporarily by removing
harmful substances from the blood. Additionally, plasma exchange
may be associated with various complications, such as plasma
anaphylaxis (20). Intravenous
immune globulin (IVIG) is often used in the emergency treatment of
severe myasthenia, and has the advantages of being rapid, effective
and relatively low-cost (21).
However, IVIG is associated with potential risks of infection and
thrombosis (22) (Table I).
Although there are a number of treatment options for
MG, ~10% of patients are treatment-intolerant, and up to 80% of
patients do not achieve complete stabilization (23). The onset of MG is associated with
AChR antibody (AChR-Ab), but there is no clear association between
disease severity and antibody titer (24). Moreover, it has been demonstrated
that both neuronal and muscular cells in MG are highly sensitive to
energy deprivation, which significantly affects signal transmission
and the normal physiological function of the NMJ. Therefore, by
investigating mitochondrial biogenesis and energy metabolism
pathways, the current review puts forward the hypothesis that
modulation of mitochondrial biogenesis may represent a viable
therapeutic option MG by regulating energy metabolism, as well as
the dysfunctional NMJ transmission.
Previous studies investigating neuromuscular
diseases mainly focused on immune responses and synapses of the NMJ
(25,26). MG is considered to be a
neuromuscular disease, and dysfunctional transmission at the NMJ
and autoantibody binding appear to be the initial mechanisms of MG
(27). The NMJ is composed of
Schwann cells, which are a highly specialized skeletal muscle cell
membrane, and the axon terminal of the motor nerve (28). Damaging any part of this system may
cause signal transmission disorders (29,30).
The NMJ can be targeted by a variety of autoimmune antibodies,
including AChR-Ab, ryanodine receptor antibody, muscle-specific
receptor tyrosine kinase antibody (MuSK-Ab) and low-density
lipoprotein receptor-related protein 4 antibody (31). These antibodies bind to the
postsynaptic membrane, and attack and destroy postsynaptic
molecules. This damage to postsynaptic structures occurs by
activation of the complement system, enhancing the binding capacity
of the AChR or inhibition of the function of ACh, and subsequently
induces clinical MG symptoms by reducing AChR numbers and
disrupting clustering at muscle tubules (5).
Although the immune mechanisms underlying
neuromuscular diseases, such as MG, has been extensively
investigated, there are relatively few studies on muscle cell
damage and mitochondrial dysfunction. MG autoantibodies do not only
induce AChR destruction directly, but also cause intracellular
mitochondrial changes (32). In a
previous experimental autoimmune MG (EAMG) rat study, it was
observed that both mitochondrial structure and skeletal muscle
function were compromised to varying degrees (15). Mitochondria are the core source of
cellular energy and are essential for the daily activities of
hypermetabolic tissues, such as muscle contraction and utilization
(33). Mitochondrial dysfunction
may affect the normal energy metabolism and, in combination with
specific autoantibodies, aggravate neuromuscular disorders
(34). Kordas et al
(24) demonstrated that the
mitochondrial protein CHCHD10 is required for ATP production, which
facilitates AChR expression and promotes agrin-induced AChR
clustering.
Mitochondrial dysfunction and sensitivity of muscle
cells to energy deprivation are major characteristics of
neuromuscular diseases (35). It
has been demonstrated that mitochondrial dysfunction serves an
important role in muscular dystrophy muscle consumption (36). Excessive reactive oxygen species
(ROS) produced by mitochondrial dysfunction may trigger autophagy,
which results in muscle atrophy from an enhanced catabolism and
decreased protein synthesis in the skeletal muscle (37). Blocking the extracellular ATP/P2X
axis results in enhancement of muscle T regulatory cells and
accelerates the muscular dystrophic process in mdx mice (38). Similarly, mitochondrial dysfunction
has also been indicated to be one of the important pathogenetic
mechanisms involved in amyotrophic lateral sclerosis (ALS)
(39). Abnormal mitochondrial
morphology and dysfunction have been previously identified in
patients with ALS (40). In
addition, it has been reported that resting energy consumption in
patients with ALS was lower compared with that in healthy
individuals using indirect calorimetry (41). Recently, a number of studies have
demonstrated that MG is closely associated with dysfunction of
mitochondria and muscle cells (42,43).
The muscle tissue of patients with MG display abnormal
mitochondrial morphology and function, and the energy levels are
often significantly lower than normal (44,45).
Similarly, abnormally shaped and structured mitochondria on muscle
biopsy, ragged-red fibers, loss of mitochondrial respiratory chain
complexes-1 and muscle aerobic dysfunction were observed in MG
(46,47). These previous findings suggest the
presence of mitochondrial abnormalities in neuromuscular diseases,
such as MG, which may lead to muscular weakness. Therefore, it is
of great significance to further explore the mitochondrial
biogenesis signaling pathway and its link to the energy metabolism
as a potential target for the treatment of MG (Table II).
As aforementioned, mitochondrial dysfunction can
damage the normal function of muscle, leading to the development of
MG. However, the specific role of mitochondria in the pathogenesis
of MG remains unclear. Herein, the current review discusses the
potential mechanisms regulating mitochondrial function in MG from
the perspective of biogenesis and energy metabolism.
The term ‘energy metabolism’ refers to a series of
continuous, cyclic processes in which the energy substance ATP is
produced, transported and utilized by the action of ATP synthase or
ATP hydrolase, respectively (48).
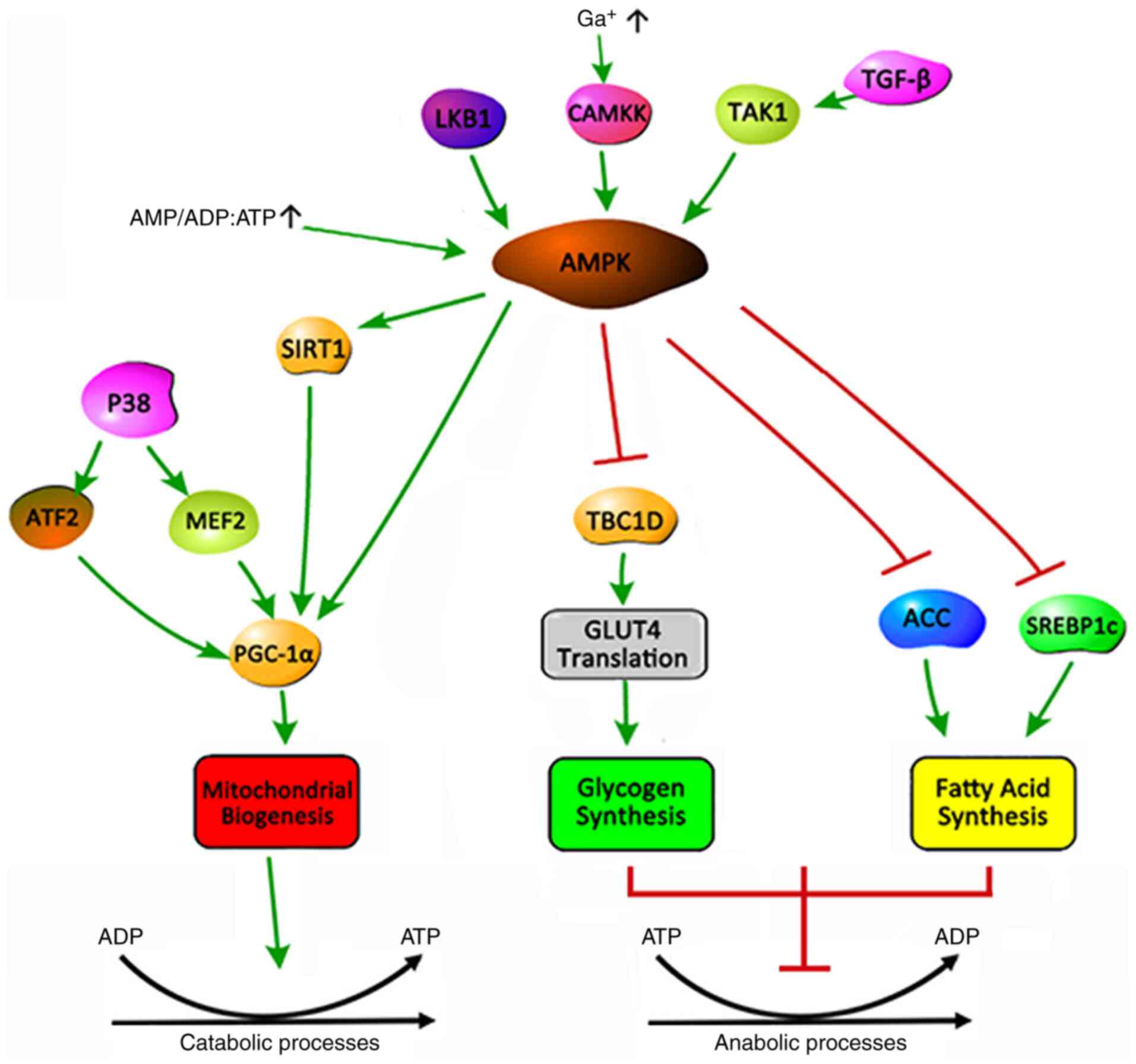
AMP-activated protein kinase (AMPK) is one of the key enzymes of
the mitochondrial energy metabolism (49). AMPK can phosphorylate a variety of
transcription factors included in the energy metabolism and
biogenesis pathways, providing necessary cellular mechanisms for
skeletal muscle plasticity (50).
AMPK is a serine/threonine protein kinase that
regulates cellular energy metabolism and serves a key role in
maintaining the cellular energy balance (51). AMPK is an allotrimer that is
composed of a catalytic α and regulatory β and γ subunits, which
are assembled into a total of 12 possible AMPK complexes (52). In general, AMPK is activated by
upstream kinases, including liver kinase B1,
Ca2+/calmodulin-dependent protein kinase 2 and
transforming growth factor-β-activated kinase 1(53). An increase in the cellular AMP:ATP
and ADP:ATP ratio during low energy state activates AMPK by
inhibiting the synthesis process that consumes ATP to restore
energy balance, while promoting the decomposition process that
produces ATP (Fig. 1) (15). The activation of AMPK triggers a
cellular metabolic transformation, promoting an uptake of lipids
and glucose to balance the energy metabolism of skeletal muscle
(54). The mechanism is that AMPK
inhibits glycogen synthesis by inhibiting the translation of TBC1
domain family member 1 and glucose transporter type 4 (GLUT4), and
fatty acid (FA) synthesis by inhibiting acetyl-coenzyme A
carboxylase and sterol regulatory element-binding protein-1c
(51).
5-Aminoimidazole-4-carboxamide 1-β-D-ribofuranoside (AICAR) acts as
an AMP analogue and has been widely used as an AMPK activator and
stimulant for mitochondrial biogenesis (55). Activating the AMPK signaling pathway
through muscle training or using pharmacological AICAR treatment
may increase the mitochondrial content in cells and tissues
(56). Metformin exerts
anti-inflammatory effects via AMPK. In an EAMG rat model, it was
demonstrated that the oral administration of metformin attenuated
MG severity by correcting the imbalance of different T-cell groups
(57).
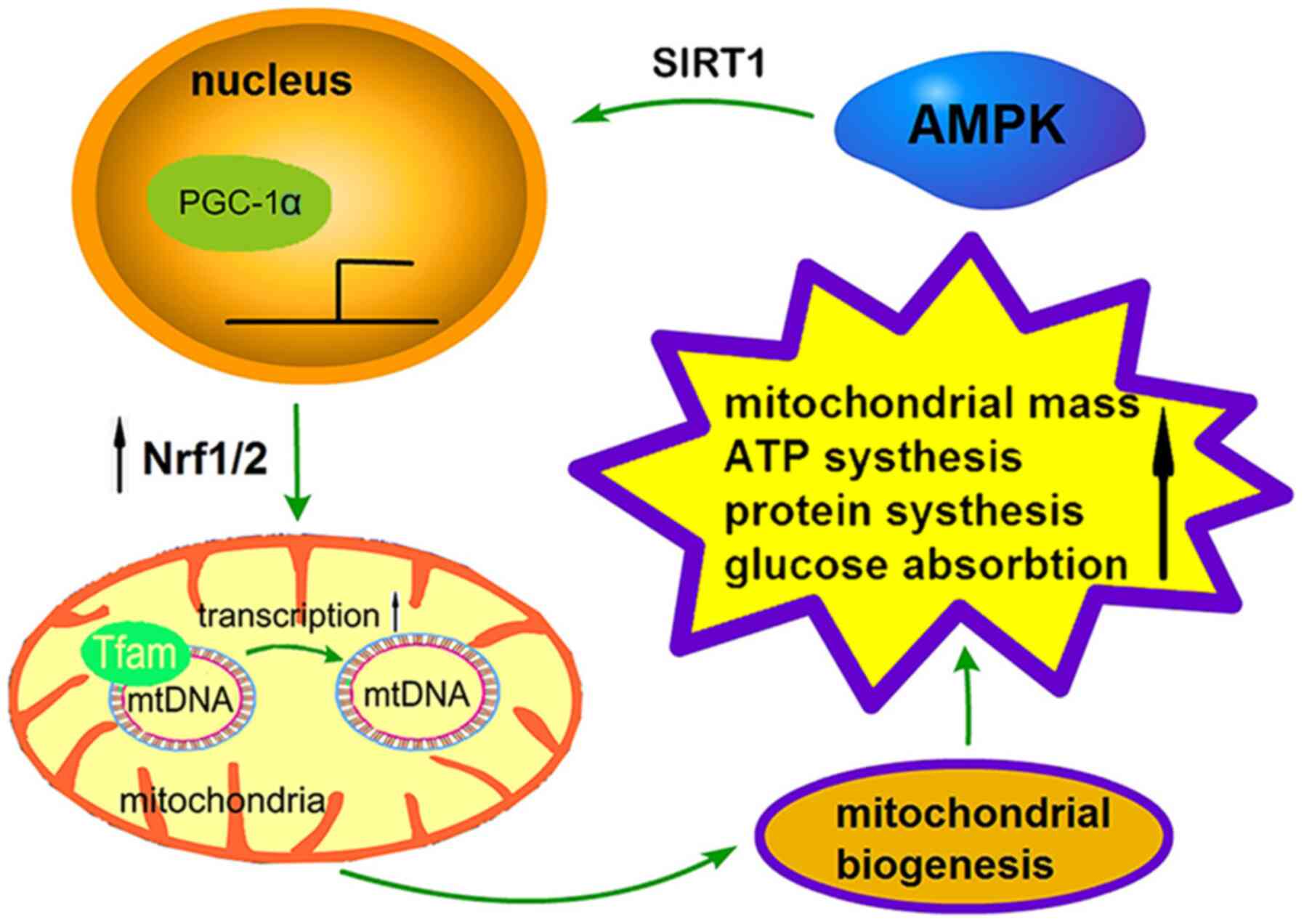
The process of mitochondrial biogenesis is strictly
regulated by a series of upstream energy metabolism pathways,
including AMPK and SIRT1, and the regulation of PGC-1α, nuclear
respiratory factors (Nrfs) and transcription factor A (TFAM) via
downstream effectors (Fig. 2)
(64). AMPK promotes mitochondrial
biogenesis in three ways: i) It increases the activity or
expression of the transcriptional coactivator PGC-1α; ii) it
enhances the deacetylation of AMPK through SIRT; or iii) it
directly phosphorylates itself, which has a positive effect on its
expression levels (65). Recently,
the activation of AMPK has been demonstrated to activate and
facilitate the deacetylation of PGC-1 by phosphorylating
Thr177/Ser538 of PGC-1α (48).
P38-MAPK can directly activate the upstream transcription factors
of the PGC-1α gene, such as activating transcription factor 2 and
myocyte enhancer-binding factor 2, and promote the transcription
and activity of the PGC-1α protein (66). In addition to PGC-1α and SIRT1, AMPK
can phosphorylate a number of different transcription factors,
including Nrf1/2 and TFAM (67).
The synergistic expression of these transcriptional regulators
under AMPK stimulation provides the necessary support for the
metabolism and biogenesis of skeletal muscle.
PGC-1α is a major regulator of diverse metabolic
pathways and mitochondrial biogenesis, and serves an important role
in mitochondrial biogenesis and gene expression (68). As a co-activator with low
transcriptional activation activity, the activity of PGC-1α is
significantly enhanced by its interaction with nuclear receptors,
which can regulate the expression of target genes (69). PGC-1α controls several aspects of
the muscular metabolism during exercise, and it is generally
hypothesized that PGC-1α regulates respiratory function, controls
muscle aerobic metabolism and mitochondrial biogenesis, and
responds to environmental and physiological changes (70,71).
PGC-1α has been identified to serve a key role in regulating
mitochondrial function, and is specialized for the thermogenesis
and differentiation of cell type in brown adipose tissue (72,73).
Forced overexpression of PGC-1α in cardiac myocytes in culture has
been indicated to induce the expression of nuclear and
mitochondrial genes that are involved in multiple mitochondrial
energy transduction/production pathways, increase the number of
cellular mitochondria and stimulate coupled respiration (74). In addition, PGC-1α is the detoxified
cation of ROS that is generated during the mitochondrial
respiration process (75). PGC-1α
knockout mice display marked hyperactivity accompanied by neuronal
degeneration (76). Similarly, the
expression of PGC-1α was also found to be decreased in spinal
muscular atrophy mouse models (77). PGC-1α is expressed at high levels in
tissues with an abundance of mitochondria and an active oxidative
metabolism, allowing for a response to increased energy needs. In
muscle, PGC-1α activates mitochondrial biogenesis, increases FA
oxidation and GLUT4 expression, which, in turn, reduces fat
accumulation and increases insulin sensitivity (75).
Nrfs, which are the downstream nuclear receptors of
PGC-1α, contain the recognition sites of mitochondrial DNA (mtDNA)
promoter and serve a key role in biogenesis (78). As a mtDNA transcription factor, Nrfs
can alleviate MG by regulating the mitochondrial respiratory chain
function and alleviating antioxidant damage (79). Nrf-1 and Nrf-2 belong to the CNC
basic leucine zipper (bZIP) regulatory protein family, are the
major transcription factors in the human genome and powerful
stimulators of the expression of nuclear genes required for
mitochondrial respiratory function (80-82).
Nrf-1 was initially indicated to be associated with the regulation
of genes, serving a role in a wide range of biological functions,
including signal transduction, organelle biogenesis, protein
synthesis and cell growth (83).
Nrf-1 also coordinates synaptic activity and energy metabolism by
regulating excitatory neurotransmission via genes that code for
subunits of the N-methyl-D-aspartate receptor (84). Nrf-1 directly regulates the
expression of nuclear encoded genes related to respiratory chain
expression, assembly and function, or indirectly regulates the
cytochrome oxidase subunit genes that are encoded by mitochondria,
and is associated with the generation of ROS and oxidative stress
(85). Recently, Nrf-2 was
demonstrated to serve an important role in cellular bioenergetics
by controlling substrate availability for mitochondrial respiration
(46). In addition, Nrf-2 regulates
mitochondrial biogenesis through co-activation with PGC-1α and the
expression of the TFAM gene (86).
Nrf-1 and Nrf-2 are involved in the regulation of inflammatory
processes and antioxidant pathways, acting protectively against
ROS-induced toxicity (87). Nrf-2
has been reported to control oxidative stress or exercise-induced
mitochondrial biogenesis by promoting Nrf-1 transcription following
its binding to the antioxidant response element of the Nrf-1
promoter (88).
TFAM is a high mobility family protein factor that
is located in mitochondria but encoded by nuclear genes (89). TFAM can be activated by PGC-1α and
Nrfs to transcribe and regulate mtDNA (90). In a study investigating mtDNA in
patients with MG, mtDNA amplification was performed in 20 muscle
samples of patients with muscle-specific kinase MG, and multiple
mtDNA deletions were identified in 13 patients (91).
The function of TFAM in the maintenance and
transcription initiation of mtDNA can be summarized by the
following three aspects: i) TFAM regulates mtDNA copy number. As a
major regulator of the mtDNA transcriptional machinery, TFAM
directly promotes transcription utilizing the mitochondrial RNA
polymerase (92). In a previous
study, both the mRNA and protein levels of TFAM were assessed
during muscle differentiation, and it was observed that TFAM
protein and mRNA increased two-fold in parallel, corresponding to a
two- to three-fold increase of mitochondrial content (93). Therefore, increasing levels of TFAM
may promote the transcription of mtDNA and the production of
proteins; ii) TFAM maintains mtDNA structural stability. Although
mtDNA is a short molecule, mistakes often occur during replication,
transcription and translation as mitochondria lack a DNA
proofreading and repair system (94). TFAM avoids these errors to a certain
extent. It has been demonstrated that TFAM binds to damaged DNA and
regulates TFAM/DNA affinity by interacting with proteins, leading
to tighter compression of mtDNA and reduced accessibility to
transcription, replication or repair factors (83,95).
TFAM can shape DNA and compensate for the unstable phenotype of
mtDNA, which is the key to maintaining the structure and stability
of mtDNA. iii) TFAM prevents mtDNA damage. The mutation rate of
mtDNA is 10-20 times higher compared with that of nuclear DNA
(96); this is due to its high
superoxide environment, lack of protective histone-like proteins
and poor reparative activity in response to damage. It has been
revealed that TFAM binds and coats mtDNA to protect it from ROS and
degradation, while increasing mitochondrial function (92). The ratio of mtDNA molecules to TFAM
protein molecule in cells is ~1:1,000(97). Furthermore, TFAM acts as a packaging
protein to bind mtDNA, forming mtDNA-protein complexes and
compressing a number of mtDNAs into the nucleoid (86).
Mitochondrial biogenesis based on energy metabolism
is a tightly controlled process (98). When mitochondrial biogenesis is
disrupted, the number of mitochondria and energy metabolism will be
reduced, and mitochondrial gene and protein expression will be
inhibited (99). It has been
demonstrated that deletion of AMPK-α1 and AMPK-α2 in mouse skeletal
muscle can affect FA utilization (100). Overexpression of TFAM in
astrocytes induces mtDNA protection against Aβ1-42 peptide,
ultimately protecting neurons (101), while tissue-specific or partial
deletion of the TFAM protein leads to respiratory chain defects
(102). A previous study of
miR-133a-deficient mice have suggested that the transcription of
the mitochondrial biogenesis regulators PGC-1α, Nrf-1 and TFAM was
reduced in miR-133a-deficient muscle, which was consistent with
lower mitochondrial mass and impaired exercise capacity (103). In summary, these studies indicated
that mitochondrial biogenesis is an indispensable process for
maintaining mitochondrial function and normal muscular activity by
regulating the stability of involved proteins and mtDNA.
It is known that mitochondrial biogenesis, which is
regulated by energy metabolism, is the foundation for maintaining
mitochondrial function and a normal muscular metabolism (104). When failure of mitochondrial
biogenesis and energy metabolism occurs, individuals are prone to
tissue dysfunction and degeneration (105,106). Mitochondrial biogenesis or energy
metabolism disorders have been observed in a number of diseases
[such as epilepsy and Parkinson's disease (PD) involving nerve or
muscle dysfunction (107,108).
The adaptation of mitochondria to exercise is
generally referred to as mitochondrial energy metabolism (109). The physiological function of
mitochondria is to improve metabolic efficiency through oxidative
phosphorylation of ATP, in order to supply energy for the
contraction of muscle cells (104). Muscle contraction utilizes energy
supplied by cells due to ATP decomposition to overcome resistance,
converting chemical energy to mechanical energy (110). However, the mitochondrial energy
metabolism is inevitably accompanied by ROS generation. Excessive
ROS accumulation can cause irreversible oxidative stress damage to
mitochondria and muscles (111).
In recent years, there has been accumulating
evidence that the mitochondrial energy metabolism is crucial for
the maintenance and plasticity of motor neurons, the NMJ and
skeletal muscles (112). A variety of
neurodegenerative diseases, including Alzheimer's disease (AD), PD
and Huntington's disease, are associated with increased ROS
production, mitochondrial dysfunction and apoptosis activation
(113). Mitochondrial dysfunction
and elevated ROS levels are common results in a number of
neurodegenerative diseases and axonopathies, such as multiple
sclerosis (114) and
Charcot-Marie-Tooth disease (115). It is well known that excessive ROS
induces inflammatory cytokine production and T-cell activation
(116). In patients with MG, the
levels of pro-inflammatory cytokines, including interleukin (IL)-6,
IL-17 and interferon-γ, which are secreted by T effector cells, are
increased (117). During the early
stages of MG, exercise intolerance, muscular and nervous system
dysfunction are commonly observed, amongst other clinical symptoms,
which are similar to the clinical observations of mitochondrial
myopathy, respiratory chain and energy metabolism disorders
(118). A disruption of integrity
of the energy metabolism at the NMJ is associated with the
occurrence and development of MG (119). Neurite outgrowth is an energy
consuming process: The energy metabolism provides enough energy to
support protein and membrane synthesis at the NMJ, as well as
intracellular transport (120).
Aging NMJs exhibit abnormal morphology and decreased numbers of
mitochondria, oxidative damage and reduced ATP synthesis (28).
Biogenesis is the general term used to indicate an
assimilation reaction in organisms, and it is a vital regulatory
process of protein synthesis (121). As a semi-autonomous organelle,
mitochondria serve an indispensable role in protein biogenesis
(122). Proteins are the major
effectors of cellular activities, and promote metabolism, maintain
the composition of all cells, including muscle and nerves, and
regulate the physiological functions of the body (123).
Misfolded, conformationally modified proteins can
cause neurodegenerative diseases, such as AD and PD (considered as
protein conformation disorders) (124). It has been demonstrated that
insufficient protein levels make it difficult to maintain normal
motor neuron function and survival (125). Loss of axonal domain proteins in
nerve conduction may have a severe impact on neuromuscular
integrity and health. For example, compromised axonal structural
integrity may lead to pathological changes of peripheral nervous
system myelinated fibers and muscle pathology (126). It has been demonstrated that
impaired mitochondrial function of presynaptic and postsynaptic
cells at the NMJ may cause neuromuscular dysfunction, mainly by
reducing the production of presynaptic ACh and the number of
postsynaptic AChRs (127).
Caveolin-3 is an integral membrane protein that is essential for
the repair of muscle membrane damage and is expressed in skeletal,
cardiac and smooth muscle cells. It has been demonstrated that
partial loss of caveolin-3 expression is a relatively common
occurrence in the muscles of patients with MG. Caveolin-3
overexpression induced after NMJ defects in MG muscles exerts a
protective effect on skeletal myotubes (128). It has been indicated that
complement-dependent lysis of the post-synaptic membrane, receptor
internalization, as well as direct interference with the binding
capacity of ACh to the AChR, may cause muscular damage in patients
with MG (129). The normal
function of the low-density lipoprotein receptor-related protein 4
and clustering of proteins serve a key role in the proper formation
and maintenance of the NMJ (130).
These findings suggest that the abnormalities of
mitochondrial biogenesis and the energy metabolism are closely
associated with the pathogenesis of MG. Regulation of mitochondrial
function and the restoration of the function of related targets in
the mitochondrial biogenesis pathway may represent a novel
therapeutic approach to MG and similar muscle contraction
disorders.
Although recent studies have investigated MG, the
majority of these studies have focused on immunity (22,131),
while relatively few studies to date assessed the link between MG
and mitochondrial function. As the powerhouse of the cell,
mitochondria are the main energy source for skeletal muscle cells,
enabling various biological processes and providing muscle movement
support, and are key organelles for the maintenance of signal
transmission and normal physiological function. Mitochondrial
dysfunction directly affects the energy metabolism of skeletal
muscle, impairing ACh biogenesis and NMJ signal transmission,
ultimately accelerating the progression of MG. Therefore, it may be
inferred that the mitochondrial function pathway may represent a
promising target for the development of neuromuscular drugs for the
treatment of MG.
Mitochondria and bioenergetic dysfunction are
increasingly considered to be key components of neuromuscular
diseases. In a number of neuromuscular diseases, it has been
demonstrated that nerves and muscles are highly sensitive to energy
deprivation, which has an adverse impact on recovery (35,132).
The mitochondrial biogenesis pathway involves multiple targets,
including AMPK, SIRT1, PGC-1a, NRFs and TFAM, each of which serves
an important regulatory role in neuromuscular function (133). Dysfunction of these factors may be
involved in the development of neuromuscular diseases, such as MG,
which are sensitive to energy deprivation and motor
dysfunction.
Although the mitochondrial biogenesis pathway
exhibits potential for the treatment of neuromuscular diseases, no
specific pharmacological activator has yet been developed for the
treatment of related diseases. A large number of researchers are
currently investigating this approach in order to identify more
effective treatment strategies. In summary, the biogenesis pathway
represents a reasonable and feasible potential therapeutic target
for neuromuscular diseases.
Not applicable.
Funding: The current study was supported by the National Science
Foundation of China (grant no. 81473568) and Science Program for
Overseas Scholar of Guangzhou University of Chinese Medicine (grant
no. XH20160106).
Not applicable.
LK, HP and YS designed the present study. LK, QL and
TC prepared the draft of the manuscript; JS and WJ reviewed and
edited the manuscript; AJ produced the figures; QL, HP and YS are
responsible for text layout. All authors read and approved the
final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Cataneo AJM, Felisberto G Jr and Cataneo
DC: Thymectomy in nonthymomatous myasthenia gravis-systematic
review and meta-analysis. Orphanet J Rare Dis.
13(99)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Barnett C, Bril V, Kapral M, Kulkarni A
and Davis AM: Development and validation of the myasthenia gravis
impairment index. Neurology. 87:879–886. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Gwathmey KG and Burns TM: Myasthenia
gravis. Semin Neurol. 35:327–339. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang Z and Yan YP: Immunopathogenesis in
myasthenia gravis and neuromyelitis optica. Front Immunol.
8(1785)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gilhus NE, Skeie GO, Romi F, Lazaridis K,
Zisimopoulou P and Tzartos S: Myasthenia gravis-autoantibody
characteristics and their implications for therapy. Nat Rev Neurol.
12:259–268. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Juel VC: Myasthenia gravis: Management of
myasthenic crisis and perioperative care. Semin Neurol. 24:75–81.
2004.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Thomas CE, Mayer SA, Gungor Y, Swarup R,
Webster EA, Chang I, Brannagan TH, Fink ME and Rowland LP:
Myasthenic crisis: Clinical features, mortality, complications, and
risk factors for prolonged intubation. Neurology. 48:1253–1260.
1997.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mantegazza R and Antozzi C: When
myasthenia gravis is deemed refractory: Clinical signposts and
treatment strategies. Ther Adv Neurol Disord.
11(1756285617749134)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mehndiratta MM, Pandey S and Kuntzer T:
Acetylcholinesterase inhibitor treatment for myasthenia gravis.
Cochrane Database Syst Rev. 16(CD006986)2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Watanabe G, Yuki T, Sugaya R, et al:
Effectiveness of treatment based on the simultaneous administration
of pyridostigmine, prednisolone, calcineurin inhibitor, and
intravenous immunoglobulin (PPCI therapy) in patients with
myasthenia gravis. Eur J Neurol. 25:143. 2018.
|
|
11
|
Luo J and Lindstrom J: AChR-specific
immunosuppressive therapy of myasthenia gravis. Biochem Pharmacol.
97:609–619. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Binks S, Vincent A and Palace J:
Myasthenia gravis: A clinical-immunological update. J Neurol.
263:826–834. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang L, Xi J, Zhang S, Wu H, Zhou L, Lu J,
Zhang T and Zhao C: Effectiveness and safety of tacrolimus therapy
for myasthenia gravis: A single arm meta-analysis. J Clin Neurosci.
63:160–167. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Gilhus NE and Verschuuren JJ: Myasthenia
gravis: Subgroup classification and therapeutic strategies. Lancet
Neurol. 14:1023–1036. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Song JW, Lei XW, Jiao W, Song Y, Chen W,
Li J and Chen Z: Effect of Qiangji Jianli decoction on
mitochondrial respiratory chain activity and expression of
mitochondrial fusion and fission proteins in myasthenia gravis
rats. Sci Rep. 8(8623)2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li YL, Li L and Li JM: Proteomic analysis
of 11000 bands in thymic hyperplasia tissues of patients with
myasthenia gravis. J Zhengzhou Univ. 43:291–295. 2012.
|
|
17
|
Guptill JT, Juel VC, Massey JM, Anderson
AC, Chopra M, Yi JS, Esfandiari E, Buchanan T, Smith B, Atherfold
P, et al: Effect of therapeutic plasma exchange on immunoglobulins
in myasthenia gravis. Autoimmunity. 49:472–479. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Alipour-Faz A, Shojaei M, Peyvandi H,
Ramzi D, Oroei M, Ghadiri F and Peyvandi M: A comparison between
IVIG and plasma exchange as preparations before thymectomy in
myasthenia gravis patients. Acta Neurol Belg. 117:245–249.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Newsom-Davis J, Wilson SG, Vincent A and
Ward CD: Long-term effects of repeated plasma exchange in
myasthenia gravis. Lancet. 1:464–468. 1979.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Guptill JT, Oakley D, Kuchibhatla M,
Guidon AC, Hobson-Webb LD, Massey JM, Sanders DB and Juel VC: A
retrospective study of complications of therapeutic plasma exchange
in myasthenia. Muscle Nerve. 47:170–176. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Furlan JC, Barth D, Barnett C and Bril V:
Cost-minimization analysis comparing intravenous immunoglobulin
with plasma exchange in the management of patients with myasthenia
gravis. Muscle Nerve. 53:872–876. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Mantegazza R, Bernasconi P and Cavalcante
P: Myasthenia gravis: From autoantibodies to therapy. Curr Opin
Neurol. 31:517–525. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Breiner A, Widdifield J, Katzberg HD,
Barnett C, Bril V and Tu K: Epidemiology of myasthenia gravis in
Ontario, Canada. Neuromuscul Disord. 26:41–46. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kordas G, Lagoumintzis G, Sideris S,
Poulas K and Tzartos SJ: Direct proof of the in vivo pathogenic
role of the AChR autoantibodies from myasthenia gravis patients.
PLoS One. 9(e108327)2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sala TP, Crave JC, Duracinsky M, Lepira
Bompeka F, Tadmouri A, Chassany O and Cherin P: Efficacy and
patient satisfaction in the use of subcutaneous immunoglobulin
immunotherapy for the treatment of auto-immune neuromuscular
diseases. Autoimmun Rev. 17:873–881. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Khedraki A, Reed EJ, Romer SH, Wang Q,
Romine W, Rich MM, Talmadge RJ and Voss AA: Depressed synaptic
transmission and reduced vesicle release sites in Huntington's
disease neuromuscular junctions. J Neurosci. 37:8077–8091.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Verschuuren J, Strijbos E and Vincent A:
Neuromuscular junction disorders. Handb Clin Neurol. 133:447–466.
2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gonzalez-Freire M, de Cabo R, Studenski SA
and Ferrucci L: The neuromuscular junction: Aging at the crossroad
between nerves and muscle. Front Aging Neurosci.
6(208)2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang SJ, Li XX, Yu Y, Chiu AP, Lo LH, To
JC, Rowlands DK and Keng VW: Schwann cell-specific PTEN and EGFR
dysfunctions affect neuromuscular junction development by impairing
agrin signaling and autophagy. Biochem Biophys Res Commun.
515:50–56. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu W, Klose A, Forman S, Paris ND,
Wei-LaPierre L, Cortés-Lopéz M, Tan A, Flaherty M, Miura P, Dirksen
RT and Chakkalakal JV: Loss of adult skeletal muscle stem cells
drives age-related neuromuscular junction degeneration. Elife.
6(e26464)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Pasnoor M, Dimachkie MM, Farmakidis C and
Barohn RJ: Diagnosis of myasthenia gravis. Neurol Clin. 36:261–274.
2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Özkök E, Durmuş H, Yetimler B, Taşlı H,
Trakas N, Ulusoy C, Lagoumintzis G, Tzartos S and Tüzün E: Reduced
muscle mitochondrial enzyme activity in MuSK-immunized mice. Clin
Neuropathol. 34:359–363. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chang DT and Reynolds IJ: Mitochondrial
trafficking and morphology in healthy and injured neurons. Prog
Neurobiol. 80:241–268. 2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Sorrentino V, Menzies KJ and Auwerx J:
Repairing mitochondrial dysfunction in disease. Annu Rev Pharmacol.
58:353–389. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chaturvedi RK, Calingasan NY, Yang L,
Hennessey T, Johri A and Beal MF: Impairment of PGC-1alpha
expression, neuropathology and hepatic steatosis in a transgenic
mouse model of Huntington's disease following chronic energy
deprivation. Hum Mol Genet. 19:3190–3205. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Theilen NT, Kunkel GH and Tyagi SC: The
role of exercise and TFAM in preventing skeletal muscle atrophy. J
Cell Physiol. 232:2348–2358. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Gorgey AS, Witt O, O'Brien L, Cardozo C,
Chen Q, Lesnefsky EJ and Graham ZA: Mitochondrial health and muscle
plasticity after spinal cord injury. Eur J Appl Physiol.
119:315–331. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Klinge CM: Estrogenic control of
mitochondrial function and biogenesis. J Cell Biochem.
105:1342–1351. 2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Fukunaga K, Shinoda Y and Tagashira H: The
role of SIGMAR1 gene mutation and mitochondrial dysfunction in
amyotrophic lateral sclerosis. J Pharmacol Sci. 127:36–41.
2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Walczak J, Dębska-Vielhaber G, Vielhaber
S, Szymański J, Charzyńska A, Duszyński J and Szczepanowska J:
Distinction of sporadic and familial forms of ALS based on
mitochondrial characteristics. FASEB J. 33:4388–4403.
2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Jésus P, Fayemendy P, Nicol M, Lautrette
G, Sourisseau H, Preux PM, Desport JC, Marin B and Couratier P:
Hypermetabolism is a deleterious prognostic factor in patients with
amyotrophic lateral sclerosis. Eur J Neurol. 25:97–104.
2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Europa TA, Nel M and Heckmann JM: A review
of the histopathological findings in myasthenia gravis: Clues to
the pathogenesis of treatment-resistance in extraocular muscles.
Neuromuscul Disord. 29:381–387. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wu H, She S, Liu Y, Xiong W, Guo Y, Fang
H, Chen H and Li J: Protective effect of Sijunzi decoction on
neuromuscular junction ultrastructure in autoimmune myasthenia
gravis rats. J Tradit Chin Med. 33:669–673. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Vercauteren K, Gleyzer N and Scarpulla RC:
PGC-1-related coactivator complexes with HCF-1 and NRF-2beta in
mediating NRF-2(GABP)-dependent respiratory gene expression. J Biol
Chem. 283:12102–12111. 2008.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Taherzadeh-Fard E, Saft C, Akkad DA,
Wieczorek S, Haghikia A, Chan A, Epplen JT and Arning L: PGC-1alpha
downstream transcription factors NRF-1 and TFAM are genetic
modifiers of Huntington disease. Mol Neurodegener.
6(32)2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Finsterer J, Oberman I and Reitner A:
Respiratory chain complex-I defect mimicking myasthenia. Metab
Brain Dis. 17:41–46. 2002.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Shichijo K, Mitsui T, Kunishige M, Kuroda
Y, Masuda K and Matsumoto T: Involvement of mitochondria in
myasthenia gravis complicated with dermatomyositis and rheumatoid
arthritis: A case report. Acta Neuropathol. 109:539–542.
2005.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kjøbsted R, Hingst JR, Fentz J, Foretz M,
Sanz MN, Pehmøller C, Shum M, Marette A, Mounier R, Treebak JT, et
al: AMPK in skeletal muscle function and metabolism. FASEB J.
32:1741–1777. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zhang MH, Fang XS, Guo JY and Jin Z:
Effects of AMPK on apoptosis and energy metabolism of gastric
smooth muscle cells in rats with diabetic gastroparesis. Cell
Biochem Biophys. 77:165–177. 2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Garcia-Carrizo F, Nozhenko Y, Palou A and
Rodriguez AM: Leptin effect on acetylation and phosphorylation of
Pgc1α in muscle cells associated with Ampk and Akt activation in
high-glucose medium. J Cell Physiol. 231:641–649. 2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Tamás P, Hawley SA, Clarke RG, Mustard KJ,
Green K, Hardie DG and Cantrell DA: Regulation of the energy sensor
AMP-activated protein kinase by antigen receptor and Ca2+ in T
lymphocytes. J Exp Med. 203:1665–1670. 2006.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Martignago S, Fanin M, Albertini E,
Pegoraro E and Angelini C: Muscle histopathology in myasthenia
gravis with antibodies against MuSK and AChR. Neuropathol Appl
Neurobiol. 35:103–110. 2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Willows R, Sanders MJ, Xiao B, Patel BR,
Martin SR, Read J, Wilson JR, Hubbard J, Gamblin SJ and Carling D:
Phosphorylation of AMPK by upstream kinases is required for
activity in mammalian cells. Biochem J. 474:3059–3073.
2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Ke R, Xu Q, Li C, Luo L and Huang D:
Mechanisms of AMPK in the maintenance of ATP balance during energy
metabolism. Cell Biol Int. 42:384–392. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Inata Y, Kikuchi S, Samraj RS, Hake PW,
O'Connor M, Ledford JR, O'Connor J, Lahni P, Wolfe V, Piraino G and
Zingarelli B: Autophagy and mitochondrial biogenesis impairment
contribute to age-dependent liver injury in experimental sepsis:
dysregulation of AMP-activated protein kinase pathway. FASEB J.
32:728–741. 2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Melser S, Lavie J and Benard G:
Mitochondrial degradation and energy metabolism. Biochim Biophys
Acta. 1853:2812–2821. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Cui Y, Chang L, Wang C, Han X, Mu L, Hao
Y, Liu C, Zhao J, Zhang T, Zhang H, et al: Metformin attenuates
autoimmune disease of the neuromotor system in animal models of
myasthenia gravis. Int Immunopharmacol. 75(105822)2019.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Nillni EA: The metabolic sensor Sirt1 and
the hypothalamus: Interplay between peptide hormones and
pro-hormone convertases. Mol Cell Endocrinol. 438:77–88.
2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Xu YH, Song QQ, Li C, Hu YT, Song BB, Ye
JM, Rao Y and Huang ZS: Bouchardatine suppresses rectal cancer in
mice by disrupting its metabolic pathways via activating the
SIRT1-PGC-1α-UCP2 axis. Eur J Pharmacol. 854:328–337.
2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Jang SY, Kang HT and Hwang ES:
Nicotinamide-induced mitophagy: Event mediated by high
NAD+/NADH ratio and SIRT1 protein activation. J Biol
Chem. 287:19304–19314. 2012.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Snyder-Warwick AK, Satoh A, Santosa KB,
Imai S and Jablonka-Shariff A: Hypothalamic Sirt1 protects terminal
Schwann cells and neuromuscular junctions from age-related
morphological changes. Aging Cell. 17(e12776)2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Wu B, Feng JY, Yu LM, Wang YC, Chen YQ,
Wei Y, Han JS, Feng X, Zhang Y, Di SY, et al: Icariin protects
cardiomyocytes against ischaemia/reperfusion injury by attenuating
sirtuin 1-dependent mitochondrial oxidative damage. Br J Pharmacol.
175:4137–4153. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Li Y, Xu S, Li J, Zheng L, Feng M, Wang X,
Han K, Pi H, Li M, Huang X, et al: SIRT1 facilitates hepatocellular
carcinoma metastasis by promoting PGC-1α-mediated mitochondrial
biogenesis. Oncotarget. 7:29255–29274. 2016.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Johnson ML, Robinson MM and Nair KS:
Skeletal muscle aging and the mitochondrion. Trends Endocrinol
Metab. 24:247–256. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Salt IP and Hardie DG: AMP-activated
protein kinase an ubiquitous signaling pathway with key roles in
the cardiovascular system. Circ Res. 120:1825–1841. 2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Akimoto T, Pohnert SC, Li P, Zhang M,
Gumbs C, Rosenberg PB, Williams RS and Yan Z: Exercise stimulates
Pgc-1alpha transcription in skeletal muscle through activation of
the p38 MAPK pathway. J Biol Chem. 280:19587–19593. 2005.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Ljubicic V, Burt M and Jasmin BJ: The
therapeutic potential of skeletal muscle plasticity in Duchenne
muscular dystrophy: Phenotypic modifiers as pharmacologic targets.
FASEB J. 28:548–568. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Abrahan C and Ash JD: The potential use of
PGC-1α and PGC-1β to protect the retina by stimulating
mitochondrial repair. Adv Exp Med Biol. 854:403–409.
2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Shu JT, Xu WJ, Zhang M, Song WT, Shan YJ,
Song C, Zhu WQ, Zhang XY and Li HF: Transcriptional co-activator
PGC-1α gene is associated with chicken skeletal muscle fiber types.
Genet Mol Res. 13:895–905. 2014.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Jiang SN, Teague AM, Tryggestad JB and
Chernausek SD: Role of microRNA-130b in placental PGC-1α/TFAM
mitochondrial biogenesis pathway. Biochem Biophys Res Commun.
487:607–612. 2017.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Felszeghy S, Viiri J, Paterno JJ, Hyttinen
JMT, Koskela A, Chen M, Leinonen H, Tanila H, Kivinen N, Koistinen
A, et al: Loss of NRF-2 and PGC-1α genes leads to retinal pigment
epithelium damage resembling dry age-related macular degeneration.
Redox Biol. 20:1–12. 2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Du H, Zhou C, Wu H, Shan T, Wu Z, Xu B and
Zhang Y: Effects of electroacupuncture on PGC-1 α expression in
brown adipose tissue. Evid Based Complement Alternat Med.
2013(625104)2013.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Cooper MP, Uldry M, Kajimura S, Arany Z
and Spiegelman BM: Modulation of PGC-1 coactivator pathways in
brown fat differentiation through LRP130. J Biol Chem.
283:31960–31967. 2008.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Lehman JJ, Barger PM, Kovacs A, Saffitz
JE, Medeiros DM and Kelly DP: Peroxisome proliferator-activated
receptor gamma coactivator-1 promotes cardiac mitochondrial.
biogenesis. 106:847–856. 2000.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Zhang Q and Liang XC: Effects of
mitochondrial dysfunction via AMPK/PGC-1 α signal pathway on
pathogenic mechanism of diabetic peripheral neuropathy and the
protective effects of Chinese medicine. Chin J Integr Med.
25:386–394. 2019.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Jones AW, Yao Z, Vicencio JM,
Karkucinska-Wieckowska A and Szabadkai G: PGC-1 family coactivators
and cell fate: Roles in cancer, neurodegeneration, cardiovascular
disease and retrograde mitochondria-nucleus signalling.
Mitochondrion. 12:86–99. 2012.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Xiang Z, Valenza M, Cui L, Leoni V, Jeong
HK, Brilli E, Zhang J, Peng Q, Duan W, Reeves SA, et al:
Peroxisome-proliferator-activated receptor gamma coactivator 1 α
contributes to dysmyelination in experimental models of
Huntington's disease. J Neurosci. 31:9544–9553. 2011.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Wang Y, Zhao X, Lotz M, Terkeltaub R and
Liu-Bryan R: Mitochondrial biogenesis is impaired in osteoarthritis
chondrocytes but reversible via peroxisome proliferator-activated
receptor γ coactivator 1α. Arthritis Rheumatol. 67:2141–2153.
2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Koh JH, Hancock CR, Terada S, Higashida K,
Holloszy JO and Han DH: PPARβ is essential for maintaining normal
levels of PGC-1α and mitochondria and for the increase in muscle
mitochondria induced by exercise. Cell Metab. 25:1176–1185 e5.
2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Hsieh PF, Liu SF, Hung TJ, Hung CY, Liu
GZ, Chuang LY, Chen MF, Wang JL, Shi MD, Hsu CH, et al: Elucidation
of the therapeutic role of mitochondrial biogenesis transducers
NRF-1 in the regulation of renal fibrosis. Exp Cell Res. 349:23–31.
2016.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Lanza IR and Nair KS: Regulation of
skeletal muscle mitochondrial function: Genes to proteins. Acta
Physiol (Oxf). 199:529–547. 2010.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Ramachandran B, Yu GS and Gulick T:
Nuclear respiratory factor 1 controls myocyte enhancer factor 2A
transcription to provide a mechanism for coordinate expression of
respiratory chain subunits. J Biol Chem. 283:11935–11946.
2008.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Matsuda T, Kanki T, Tanimura T, Kang D and
Matsuura ET: Effects of overexpression of mitochondrial
transcription factor A on lifespan and oxidative stress response in
Drosophila melanogaster. Biochem Biophys Res Commun. 430:717–721.
2013.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Thirupathi A and Pinho RA: Effects of
reactive oxygen species and interplay of antioxidants during
physical exercise in skeletal muscles. J Physiol Biochem.
74:359–367. 2018.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Brandt N, Dethlefsen MM, Bangsbo J and
Pilegaard H: PGC-1α and exercise intensity dependent adaptations in
mouse skeletal muscle. PLoS One. 12(e0185993)2017.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Wu KLH, Wu CW, Chao YM, Hung CY and Chan
JYH: Impaired Nrf2 regulation of mitochondrial biogenesis in
rostral ventrolateral medulla on hypertension induced by systemic
inflammation. Free Radic Biol Med. 97:58–74. 2016.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Hu Q, Ren J, Li G, Wu J, Wu X, Wang G, Gu
G, Ren H, Hong Z and Li J: The mitochondrially targeted antioxidant
MitoQ protects the intestinal barrier by ameliorating mitochondrial
DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis.
9(403)2018.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Bernard K, Logsdon NJ, Miguel V, Benavides
GA, Zhang J, Carter AB, Darley-Usmar VM and Thannickal VJ: NADPH
oxidase 4 (Nox4) suppresses mitochondrial biogenesis and
bioenergetics in lung fibroblasts via a nuclear factor
erythroid-derived 2-like 2 (Nrf2)-dependent pathway. J Biol Chem.
292:3029–3038. 2017.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kang I, Chu CT and Kaufman BA: The
mitochondrial transcription factor TFAM in neurodegeneration:
Emerging evidence and mechanisms. FEBS Lett. 592:793–811.
2018.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Piao Y, Kim HG, Oh MS and Pak YK:
Overexpression of TFAM, NRF-1 and myr-AKT protects the
MPP(+)-induced mitochondrial dysfunctions in neuronal cells.
Biochim Biophys Acta. 1820:577–585. 2012.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Rostedt Punga A, Ahlqvist K, Bartoccioni
E, Scuderi F, Marino M, Suomalainen A, Kalimo H and Stålberg EV:
Neurophysiological and mitochondrial abnormalities in MuSK antibody
seropositive myasthenia gravis compared to other immunological
subtypes. Clin Neurophysiol. 117:1434–1443. 2006.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Kunkel GH, Chaturvedi P and Tyagi SC:
Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail Rev.
21:499–517. 2016.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Ruzzenente B, Rötig A and Metodiev MD:
Mouse models for mitochondrial diseases. Hum Mol Genet.
25:R115–R122. 2016.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Li H, Slone J, Fei L and Huang T:
Mitochondrial DNA variants and common diseases: A mathematical
model for the diversity of age-related mtDNA mutations. Cells.
8(608)2019.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Lezza AMS: Mitochondrial transcription
factor A (TFAM): One actor for different roles. Front Biol.
7:30–39. 2012.
|
|
96
|
Xu S, Zhong M, Zhang L, Wang Y, Zhou Z,
Hao Y, Zhang W, Yang X, Wei A, Pei L and Yu Z: Overexpression of
Tfam protects mitochondria against beta-amyloid-induced oxidative
damage in SH-SY5Y cells. FEBS J. 276:3800–3809. 2009.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Kang D, Kim SH and Hamasaki N:
Mitochondrial transcription factor A (TFAM): Roles in maintenance
of mtDNA and cellular functions. Mitochondrion. 7:39–44.
2007.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Dong J, Zhao J, Zhang M, Liu G, Wang X,
Liu Y, Yang N, Liu Y, Zhao G, Sun J, et al: β3-Adrenoceptor impairs
mitochondrial biogenesis and energy metabolism during rapid atrial
pacing-induced atrial fibrillation. J Cardiovasc Pharmacol Ther.
21:114–126. 2016.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Tao L, Wang L, Yang X, Jiang X and Hua F:
Recombinant human glucagon-like peptide-1 protects against chronic
intermittent hypoxia by improving myocardial energy metabolism and
mitochondrial biogenesis. Mol Cell Endocrinol. 481:95–103.
2019.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Jeon SM: Regulation and function of AMPK
in physiology and diseases. Exp Mol Med. 48(e245)2016.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro
M, Iradi A, Obrador E, Ortega A, Mauricio MD, Vila JM and Valles
SL: Astrocytes protect neurons from Aβ1-42 peptide-induced
neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and
SIRT-1. Int J Med Sci. 12:48–56. 2015.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Hood DA, Tryon LD, Carter HN, Kim Y and
Chen CCW: Unravelling the mechanisms regulating muscle
mitochondrial biogenesis. Biochem J. 473:2295–2314. 2016.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Nie Y, Sato Y, Wang C, Yue F, Kuang S and
Gavin TP: Impaired exercise tolerance, mitochondrial biogenesis,
and muscle fiber maintenance in miR-133a-deficient mice. FASEB J.
30:3745–3758. 2016.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Nirwane A and Majumdar A: Understanding
mitochondrial biogenesis through energy sensing pathways and its
translation in cardio-metabolic health. Arch Physiol Biochem.
124:194–206. 2018.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Dhar SS and Wong-Riley MTT: Coupling of
energy metabolism and synaptic transmission at the transcriptional
level: Role of nuclear respiratory factor 1 in regulating both
Cytochrome c oxidase and NMDA glutamate receptor subunit genes. J
Neurosci. 29:483–492. 2009.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Huang DD, Fan SD, Chen XY, Yan XL, Zhang
XZ, Ma BW, Yu DY, Xiao WY, Zhuang CL and Yu Z: Nrf2 deficiency
exacerbates frailty and sarcopenia by impairing skeletal muscle
mitochondrial biogenesis and dynamics in an age-dependent manner.
Exp Gerontol. 119:61–73. 2019.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Chuang YC, Chen SD, Jou SB, Lin TK, Chen
SF, Chen NC and Hsu CY: Sirtuin 1 regulates mitochondrial
biogenesis and provides an endogenous neuroprotective mechanism
against seizure-induced neuronal cell death in the hippocampus
following status epilepticus. Int J Mol Sci.
20(3588)2019.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Van Laar VS, Arnold B, Howlett EH,
Calderon MJ, St Croix CM, Greenamyre JT, Sanders LH and Berman SB:
Evidence for compartmentalized axonal mitochondrial biogenesis:
Mitochondrial DNA replication increases in distal axons as an early
response to Parkinson's disease-relevant stress. J Neurosci.
38:7505–7515. 2018.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Osborne B, Cooney GJ and Turner N: Are
sirtuin deacylase enzymes important modulators of mitochondrial
energy metabolism? Biochim Biophys Acta. 1840:1295–1302.
2014.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Jarmuszkiewicz W and Szewczyk A:
Energy-dissipating hub in muscle mitochondria: Potassium channels
and uncoupling proteins. Arch Biochem Biophys. 664:102–109.
2019.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Dan Dunn J, Alvarez LA, Zhang X and
Soldati T: Reactive oxygen species and mitochondria: A nexus of
cellular homeostasis. Redox Biol. 6:472–485. 2015.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Rahman S and Hanna MG: Diagnosis and
therapy in neuromuscular disorders: Diagnosis and new treatments in
mitochondrial diseases. J Neurol Neurosurg Psychiatry. 80:943–953.
2009.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Cabezas R, Baez-Jurado E, Hidalgo-Lanussa
O, Echeverria V, Ashrad GM, Sahebkar A and Barreto GE: Growth
factors and neuroglobin in astrocyte protection against
neurodegeneration and oxidative stress. Mol Neurobiol.
56:2339–2351. 2019.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Niemann A, Huber N, Wagner KM, Somandin C,
Horn M, Lebrun-Julien F, Angst B, Pereira JA, Halfter H, Welzl H,
et al: The Gdap1 knockout mouse mechanistically links redox control
to charcot-marie-tooth disease. Brain. 137:668–682. 2014.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Li Y, Zhao X, Hu Y, Sun H, He Z, Yuan J,
Cai H, Sun Y, Huang X and Kong W and Kong W: Age-associated decline
in Nrf2 signaling and associated mtDNA damage may be involved in
the degeneration of the auditory cortex: Implications for central
presbycusis. Int J Mol Med. 42:3371–3385. 2018.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Li X, Fang P, Yang WY, Chan K, Lavallee M,
Xu K, Gao T, Wang H and Yang X: Mitochondrial ROS, uncoupled from
ATP synthesis, determine endothelial activation for both
physiological recruitment of patrolling cells and pathological
recruitment of inflammatory cells. Can J Physiol Pharmacol.
95:247–252. 2017.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Danikowski KM, Jayaraman S and Prabhakar
BS: Regulatory T cells in multiple sclerosis and myasthenia gravis.
J Neuroinflammation. 14(117)2017.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Tarnopolsky M, Brady L and MacNeil L:
Myasthenia graves-like symptoms associated with rare mitochondrial
mutation (m.5728T>C). Mitochondrion. 47:139–140. 2019.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Wang X and Rich MM: Homeostatic synaptic
plasticity at the neuromuscular junction in myasthenia gravis. Ann
NY Acad Sci. 1412:170–177. 2018.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Lanser AJ, Rezende RM, Rubino S, Lorello
PJ, Donnelly DJ, Xu H, Lau LA, Dulla CG, Caldarone BJ, Robson SC
and Weiner HL: Disruption of the ATP/adenosine balance in
CD39-/- mice is associated with handling-induced
seizures. Immunology. 152:589–601. 2017.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Zhang Y and Xu H: Translational regulation
of mitochondrial biogenesis. Biochem Soc Trans. 44:1717–1724.
2016.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Doan KN, Ellenrieder L and Becker T:
Mitochondrial porin links protein biogenesis to metabolism. Curr
Genet. 65:899–903. 2019.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Jeffery CJ: Enzymes, pseudoenzymes, and
moonlighting proteins: Diversity of function in protein
superfamilies. FEBS J, Jun 13, 2020 (Online ahead of print).
|
|
124
|
Askanas V, Engel WK and Nogalska A:
Pathogenic considerations in sporadic inclusion-body myositis, a
degenerative muscle disease associated with aging and abnormalities
of myoproteostasis. J Neuropathol Exp Neurol. 71:680–693.
2012.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Arnold W, McGovern VL, Sanchez B, Li J,
Corlett KM, Kolb SJ, Rutkove SB and Burghes AH: The neuromuscular
impact of symptomatic SMN restoration in a mouse model of spinal
muscular atrophy. Neurobiol Dis. 87:116–123. 2016.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Saifetiarova J, Liu X, Taylor AM, Li J and
Bhat MA: Axonal domain disorganization in Caspr1 and Caspr2 mutant
myelinated axons affects neuromuscular junction integrity, leading
to muscle atrophy. J Neurosci Res. 95:1373–1390. 2017.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Wu H, She S, Liu Y, Xiong W, Guo Y, Fang
H, Chen H and Li J: Protective effect of Sijunzi decoction on
neuromuscular junction ultrastructure in autoimmune myasthenia
gravis rats. J Tradit Chin Med. 33:669–673. 2013.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Attia M, Maurer M, Robinet M, Le Grand F,
Fadel E, Le Panse R, Butler-Browne G and Berrih-Aknin S: Muscle
satellite cells are functionally impaired in myasthenia gravis:
Consequences on muscle regeneration. Acta Neuropathol. 134:869–888.
2017.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Iwasa K, Furukawa Y, Yoshikawa H and
Yamada M: Caveolin-3 is aberrantly expressed in skeletal muscle
cells in myasthenia gravis. J Neuroimmunol. 301:30–34.
2016.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Rivner MH, Pasnoor M, Dimachkie MM, Barohn
RJ and Mei L: Muscle-specific tyrosine kinase and myasthenia gravis
owing to other antibodies. Neurol Clin. 36:293–310. 2018.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Beecher G, Putko BN, Wagner AN and Siddiqi
ZA: Therapies directed against b-cells and downstream effectors in
generalized autoimmune myasthenia gravis: Current status. Drugs.
79:353–364. 2019.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Valbuena GN, Rizzardini M, Cimini S,
Siskos AP, Bendotti C, Cantoni L and Keun HC: Metabolomic analysis
reveals increased aerobic glycolysis and amino acid deficit in a
cellular model of amyotrophic lateral sclerosis. Mol Neurobiol.
53:2222–2240. 2016.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Lysenko EA, Popov DV, Vepkhvadze TF,
Lednev EM and Vinogradova OL: Effect of combined aerobic and
strength exercise on regulation of mitochondrial biogenesis,
protein synthesis and degradation in human skeletal muscle. Fiziol
Cheloveka. 42:58–69. 2016.PubMed/NCBI(In Russian).
|