Introduction
Breast cancer is the most common cause of
cancer-associated mortality and the second most frequently
diagnosed malignancy in women, accounting for 0.627 million deaths
and 2.088 million new cases worldwide in 2018 (1,2).
Prevention and early detection interventions have been used and
have significantly contributed to outcome improvements and
increased survival in patients with breast cancer (3). However, extensive evidence has
demonstrated that gene-related risk factors and genetic
alternations serve critical roles in the occurrence and progression
of breast cancer (4), and have
partially contributed to a sharp increase in the mortality rate of
breast cancer in the last 40 years (5). A comprehensive understanding of the
genetics and molecular pathogenesis of breast cancer is therefore
crucial for clinical diagnosis and treatment.
MTHFD2 is a folate-coupled mitochondrial metabolic
enzyme characterized by methylenetetrahydrofolate dehydrogenase and
cyclohydrolase activity (6). This
mitochondrial enzyme has been reported to actively participate in
folate one-carbon metabolism and tetrahydrofolate (THF) cofactor
cycling, which supply important precursors to maintain cell
viability and proliferation (7,8). It
was demonstrated that suppression of MTHFD2 inhibits methylation
reactions (7), decreases protein
synthesis (9) and disrupts redox
homeostasis (6,10), which may ultimately result in
significant changes in cellular metabolic phenotype. Because MTHFD2
expression was reported as a key participant in the metabolic
reprogramming in tumors (7) and a
predictive factor of poor prognosis in patients with various types
of cancer (8,11-13),
including breast cancer (14),
MTHFD2 has received increased attention recently and might be
considered as a potential anticancer target. Numerous studies have
also demonstrated a strong association between tumor cell phenotype
and MTHFD2 expression status (13,15,16).
In addition to the enzymatic activity of MTHFD2 in
metabolic remodeling, certain non-enzymatic activities have been
reported. Sheppard et al (17) demonstrated that MTHFD2 is not only a
mitochondrial enzyme but also a nuclear protein implicated in DNA
synthesis, and that MTHFD2 overexpression confers tumor
cell-sustaining proliferative capacity independently of
dehydrogenase activity, suggesting that MTHFD2 might likely
regulate cell proliferation in a non-enzymatic manner. In
non-small-cell lung cancer, MTHFD2 was also reported to display
non-enzymatic function, where MTHFD2 silencing impaired tumor
proliferation by modulating cell cycle-associated genes (11). Furthermore, it has been demonstrated
that cellular mediators of signal transduction are integrated in
tumor cell metabolic reprogramming to support metabolic autonomy
(18). Subsequently, it could be
hypothesized that MTHFD2 may serve different metabolic regulatory
roles in breast cancer malignancy in a non-enzymatic manner.
AKT activation is known to be involved in the
regulation of metabolic fluxes different from canonical allosteric
mechanisms of pathway regulation, and to modulate complementary
aspects of cellular metabolism (18). The present study examined the impact
of MTHFD2 on the proliferation and colony formation ability of
MCF-7 cells. In addition, the underlying molecular mechanisms of
MTHFD2 in breast cancer carcinogenesis and progression were
investigated.
Materials and methods
Cell culture
MCF-7 cell line and 293T cells were purchased from
the China Center for Type Culture Collection. All cells were
cultured in DMEM medium (Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS and 100 U/ml
penicillin-streptomycin and placed at 37˚C in a humidified
incubator containing 5% CO2. The MCF-7 cell line was
authenticated by STR profiling.
Plasmid construction and
transfection
For overexpressing the candidate gene, PCR-amplified
MTHFD2 cDNA was amplified from MCF-7 cells by Tianyihuiyuan and
inserted into a PHAGE puro retrovirus vector (Addgene, Inc.) with a
Flag tag by BamHI and NotI (Thermo Fisher Scientific,
Inc.) digestion, resulting in MTHFD2-Flag PHAGE puro. 293T cells
(2x105) were transduced with 1.3 µg MTHFD2-Flag PHAGE
puro or 1.3 µg the empty control vector as previously described
(19) for 24 h, as previously
reported. Subsequently, 3x103 MCF-7 cells were infected
with 10 µl filtered virus supernatants following by a dual
puromycin selection (1 µg/ml, 7 days). Western blotting and reverse
transcription quantitative (RT-q) PCR were used to confirm MTHFD2
overexpression. MTHFD2 primer (Tianyihuiyuan) were as follows:
Forward primer GGATCCAGATCAAGCAGGAAGTGCGG; reverse primer
GTCTCACTGTTGATTCCCACACCGGCG.
MTHFD2-deficient MCF-7 cells were generated by
CRISPR/Cas9-mediated genome editing technology. The single guide
RNA (sgRNA) was designed via (http://sam.genome-engineering.org/), and the sequence
of sgRNA targeted MTHFD2 exon 1 is was 5'-GCCACACCTGAGTGTGATCC-3'
as described previously (19).
Briefly, 2,500 MCF-7 cells per well were seeded at 6-well plate to
60-70% confluence and were infected with 350 ng MTHFD2 sgRNA or
wild-type Cas9 plasmids (Addgene, Inc.; cat. no. 42230) using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.) for
2 days. The MTHFD2 knockout clones were screened with 1 µg/ml
puromycin for 7 days. MTHFD2 loss was confirmed in MCF-7 cells by
western blotting. Two knockout cell clones, MTHFD2-/-1#
and MTHFD2-/-2#, were chosen for subsequent experiments.
The uninfected cells were used as the control.
RT-qPCR
RNA isolation from MCF-7 cells was performed using
via a High Pure RNA isolation kit [Roche Diagnostics (Shanghai)
Co., Ltd.]. The RNA was reversely transcribed into cDNA using the
SuperScript VILO cDNA kit (Thermo Fisher Scientific, Inc.).
Thermocycling conditions of RT-PCR is 95˚C for 5 min, followed by
40 cycles of 95˚C for 10 sec, 60˚C for 20 sec and 72˚C for 30 sec.
MTHFD2 primers used were as follows: Forward,
5'-GAGCTTTGGAGAAACCAGCC-3'and reverse 5'-CAGAAGAACGAGGACGGAGG-3'.
Quantitative PCR was performed using Brilliant II SYBR-Green
RT-qPCR kit (Merck & Co., Inc.). qPCR thermocycling conditions
were as follows: 94˚C for 5 min; 40 cycles of 95˚C for 20 sec; and
72˚C for 30 sec. Gene expression values were analyzed with
2-ΔΔCq method (20).
Cell proliferation and colony
formation assay
To determine the role of MTHFD2 in breast cancer
malignancy, in vitro functional assays were performed to
compare the effect of MTHFD2 overexpression and MTHFD2 deficiency
in MCF-7 cells.
Cell proliferation was determined using the Cell
Counting Kit-8 (CCK-8; Abmole Bioscience, Inc.). Briefly, cells
were seeded into 96-well plates at the density of 1,000 cells per
well for 1, 3 and 5 days. Cells were incubated with CCK-8 and
incubated at 37˚C for 2 h. Absorbance was read at 450 nm using a
microplate reader.
For the colony formation assay, 2x102
cells were seeded into six-well plates for 14 days at 37˚C and 5%
CO2. Methanol and 0.1% crystal violet were used for
fixation and staining of colonies at room temperature,
respectively. The number of colonies was imaged using a camera
[DSC-HX90; SONY (China), Co. Ltd.] and quantified.
Luciferase assay
Since AKT is an oncogenic checkpoint in cancer cell
metabolism (18), a luciferase
assay was performed to examine the effect of MTHFD2 expression on
the transcriptional activity of AKT. An AKT luciferase reporter
plasmid was purchased from Yeasen Biotech Co., Ltd. and used for
dual-luciferase reporter assays. Briefly, 293T cells were seeded at
the density of 2x105 cells/well in a 24-well plate. Once
the cells had reached 85-90% confluence, they were transfected
using Lipofectamine® 2000 with 200 ng pAKT-luc reporter
plasmids or 10 ng pRL-TK plasmids (internal control) and then
co-transfected with MTHFD2-Flag plasmids (0, 100, 200 and 400 ng).
Two days post-transfection, cells were lysed with
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) and
the luminescence activity of AKT was measured using a
Dual-Luciferase Reporter Assay System (Promega Corporation)
following the manufacturer's protocol. Subsequently, the
luminescence activity of AKT in MTHFD2-/- and normal
MCF-7 cells was analyzed. Firefly luciferase activity was
normalized to Renilla luciferase activity.
Drug treatment
For AKT inhibition studies, MCF-7 cells at 75-85%
confluence were treated with the AKT inhibitor GSK69069 (10 nM;
MedChemExpress) for 24 h at 37˚C (21). Subsequently, a colony formation
assay was performed in MCF-7 cells (Ctrl) and MTHFD2-overexpressing
cells treated or not with GSK690693 (MTHFD2 or MTHFD2 +
GSK69069).
Western blotting
MCF-7 cells were ultrasonically lysed in RIPA buffer
(Sigma-Aldrich; Merck KGaA) and the collected supernatant was
subjected to qualification with BCA methods. Proteins (20 µg) were
separated by 15% SDS-PAGE and transferred onto PVDF membranes.
Membranes were incubated with 5% skim milk for 1 h at room
temperature and with primary antibodies against MTHFD2 (1:1,000;
cat. no. PA5-28169; Thermo Fisher Scientific, Inc.), Flag (1:1,000;
cat. no. A02010; Abbkine Scientific Co, Ltd.), and GAPDH (1:1,000;
cat. no. K106390M; Beijing Solarbio Science & Technology Co.,
Ltd.) at 4˚C for 12 h. Membranes were then incubated with secondary
anti-rabbit or anti-mouse antibodies (1:1,000; cat. nos. A32723 and
A32731; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Bands were detected using HRP Color Development
Solution. (Invitrogen; Thermo Fisher Scientific, Inc.).
Bioinformatics analysis
MTHFD2 expression data of 1,085 Breast invasive
carcinoma (BRCA) tissues and 291 adjacent normal tissues from the
GEPIA database (22) (http://gepia.cancer-pku.cn/) were analyzed by unpaired
Student's t-test with a cutoff value of **P<0.01. The
overall survival (OS) data for MTHFD2 were also analyzed via GEPIA
using the Kaplan Meier method (cut-off: 50%), plotting the survival
curves on the basis of the MTHFD2 median values.
Statistical analysis
GraphPad Prism version 8 (GraphPad Software, Inc.)
was used for statistical analysis, and data are expressed as the
means ± standard error of the mean. Comparison between two groups
was carried out using Student's t-test, and comparison among
multiple groups was performed using ANOVA followed by Dunnett's
post hoc test. Data were representative of three independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
MTHFD2 expression is increased in
breast cancer tissues and associated with poor prognosis
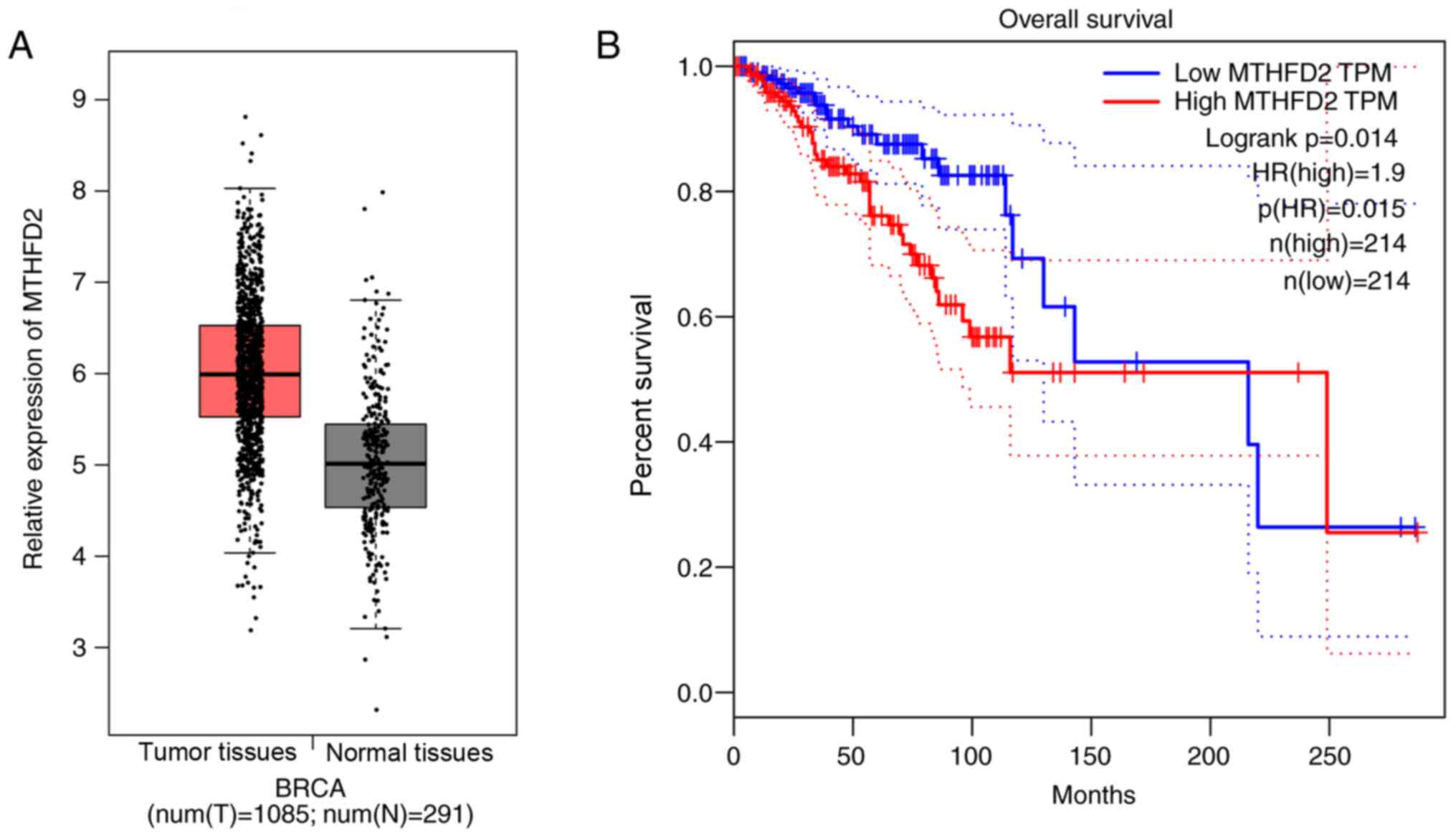
To determine whether MTHFD2 is involved in breast
cancer progression and prognosis, the GEPIA database containing
1,080 breast cancer tissues and 291 adjacent non-tumor tissues was
used to analyze MTHFD2 expression levels. The results demonstrated
that MTHFD2 expression level was higher in breast cancer tissues
compared with adjacent tissues though the difference was not
significant (Fig. 1A). Furthermore,
the results from Kaplan-Meier and Log-rank test analyses were
carried out to evaluate the prognostic value of MTHFD2, and the
results indicated that patients with high MTHFD2 expression had a
worse prognosis compared with patients with low MTHFD2 expression
(Fig. 1B). Taken together, these
findings suggest that MTHFD2 may serve a notable role in breast
cancer progression.
MTHFD2 overexpression promotes MCF-7
cell proliferation and clonogenicity
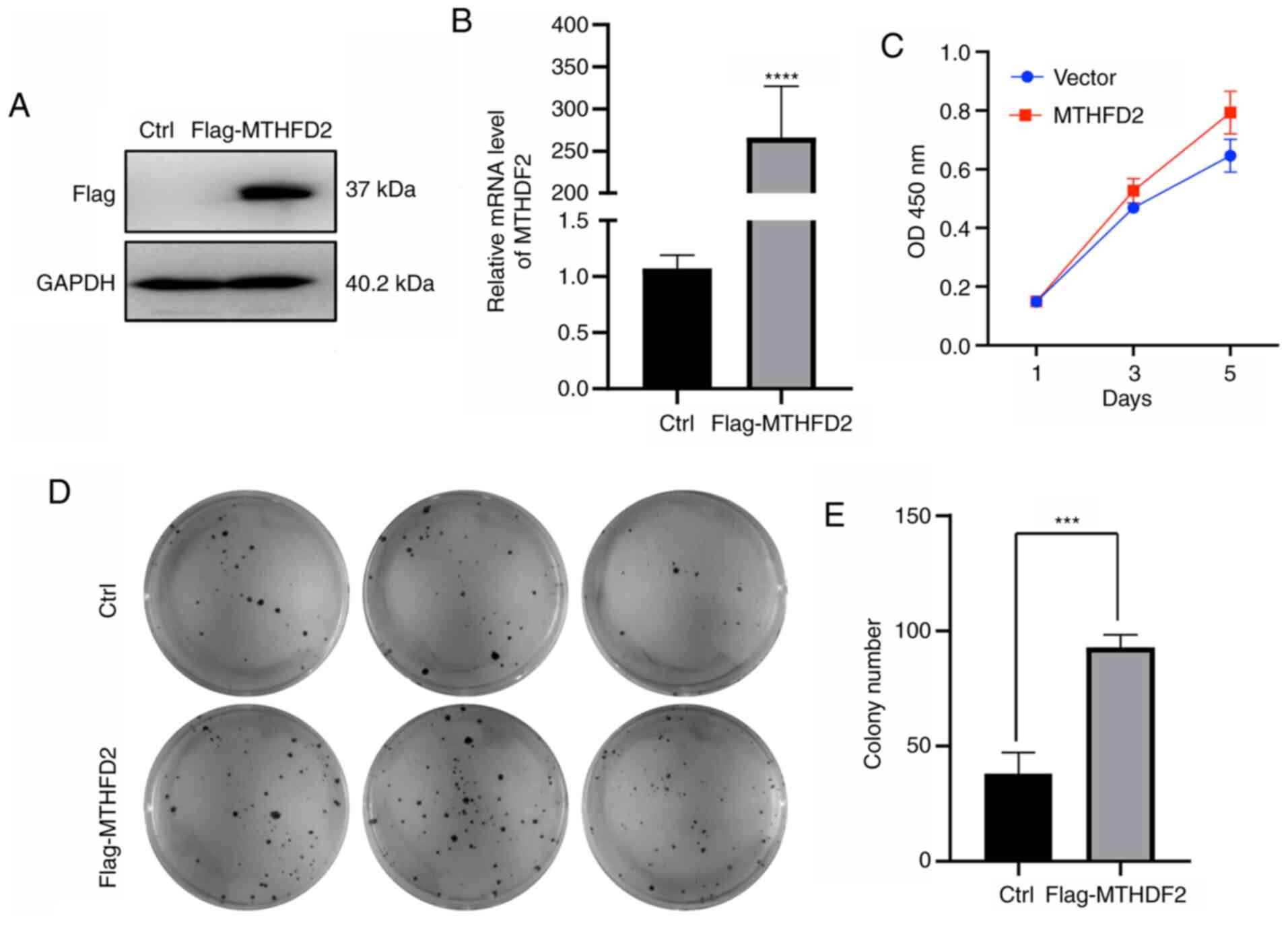
To evaluate the pathogenic role of MTHFD2 in breast
cancer cells, MCF-7 cells stably expressing Flag-MTHFD2 were
established. The results from western blotting and RT-qPCR
confirmed the successful overexpression of MTHFD2 in vitro
(Fig. 2A and B). Furthermore, MCF-7 cells stably
expressing MTHFD2-Flag displayed a higher proliferation rate and
colony formation ability compared with control cells (Fig. 2C-E). These data suggest that MTHFD2
overexpression may promote breast cancer cell proliferation and
clone formation ability in breast cancer.
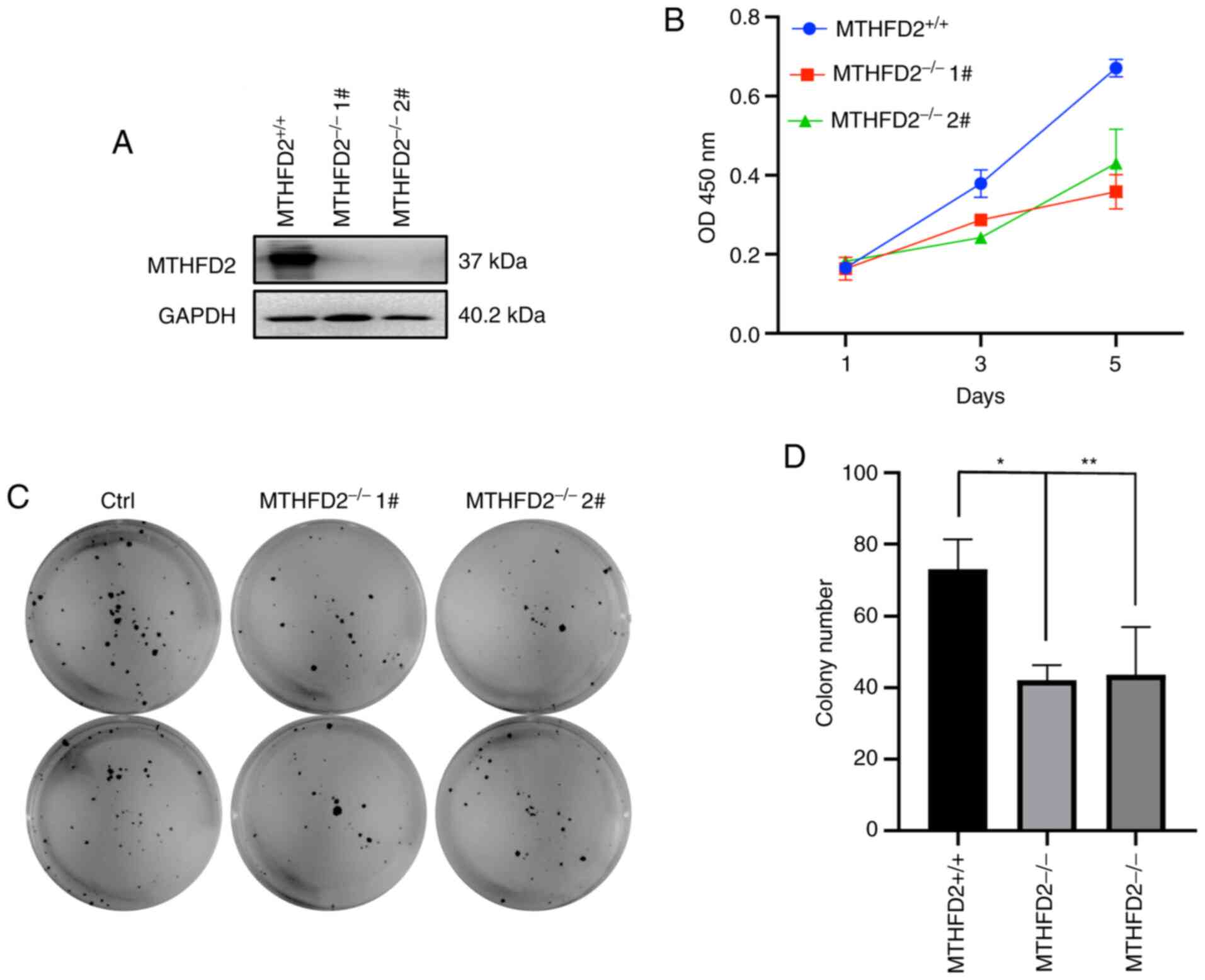
MTHFD2 knockout weakens the
proliferation and colony formation ability of MCF-7 cells
The effect of MTHFD2 knockout on proliferation of
MCF-7 cells was assessed. The efficiency of MTHFD2-knockout by
CRISPR/Cas9-mediated genome was confirmed by western blotting
(Fig. 3A). Furthermore, MTHFD2
deficiency markedly retarded MCF-7 cell proliferation (Fig. 3B), as well as MCF-7 cell colony
formation ability (Fig. 3C and
D). These data suggest that MTHFD2
knockout may inhibit proliferation and viability of breast cancer
in vitro.
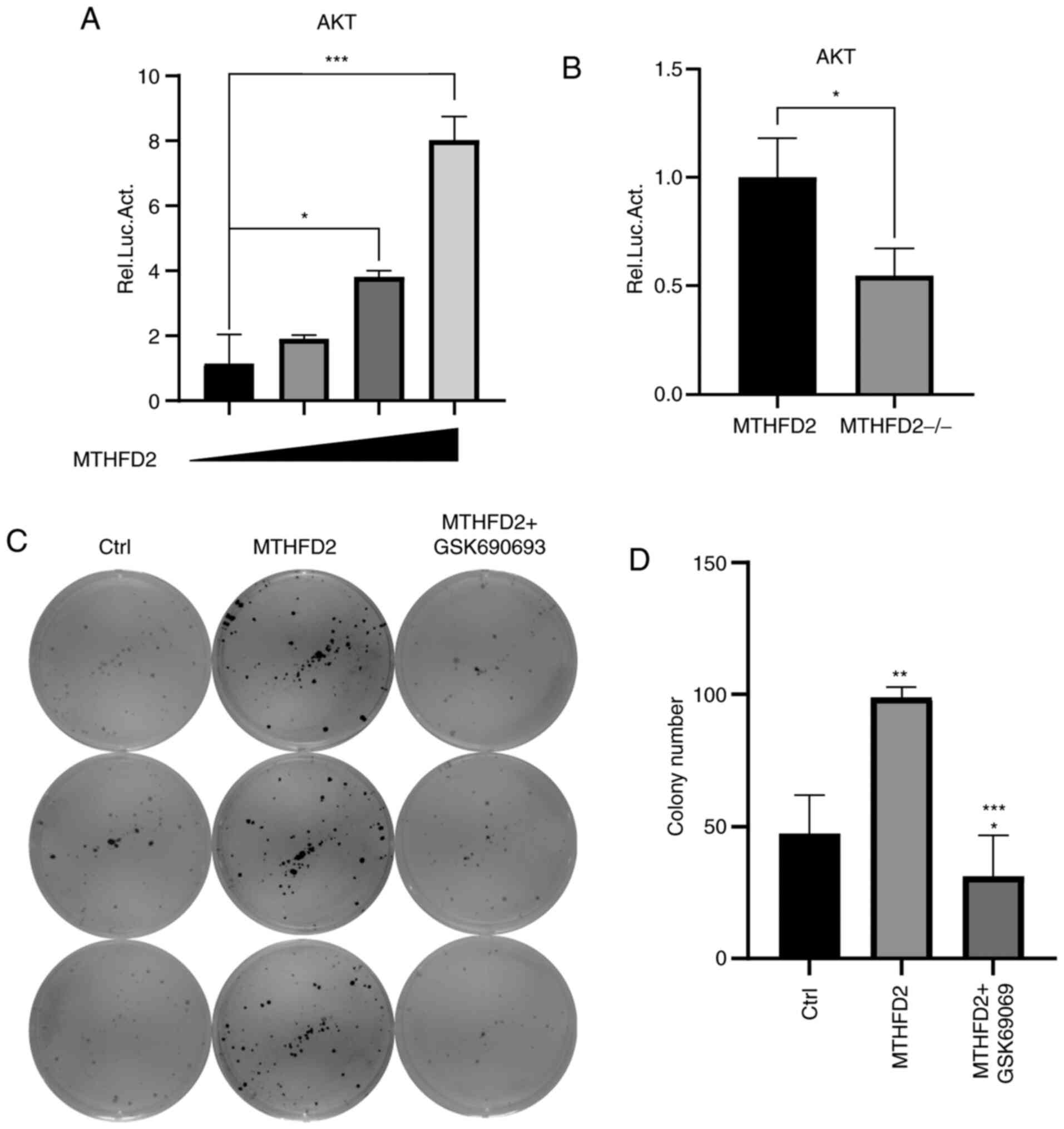
MTHFD2 activates the AKT signaling
pathway
Since AKT is involved in oncogenic signaling and
cancer metabolism (18), the
relative luciferase activity of AKT modulated by MTHFD2 was
quantified using dual-luciferase assays. The results demonstrated
that AKT transcriptional activity was enhanced in an MTHFD2
expression-dependent manner (Fig.
4A), suggesting that AKT may be a direct regulator of MTHFD2 in
breast cancer cells. The transcriptional activity of AKT was also
evaluated in normal and MTHFD2-deficient MCF-7 cells. The results
demonstrated that MTHFD2 knockout yielded an inhibitory effect on
the AKT-mediated luciferase reporter expression. Subsequently, an
AKT inhibitor, GSK69069, was used to inhibit AKT signaling in order
to verify whether suppressing AKT signaling could counteract MCF-7
cell clonogenicity caused by MTHFD2 overexpression. As presented in
Fig. 4C and D, the colony formation ability increase
following MTHFD2-overexpression was abolished when cells were
treated with GSK69069. These results indicate that MTHFD2 may
stimulate MCF-7 cell proliferation and colony formation ability via
the AKT signaling pathway.
Discussion
High expression of MTHFD2 has been considered as a
prognostic indicator in breast cancer, and results from MTHFD2
silencing in vitro have highlighted its metabolic role in
breast cancer malignancy (20).
However, a previous study reported that there is no association
between tumor cell proliferation and the enzymatic activity of
MTHFD2(13); however, MTHFD2 may
still be involved in signal transduction related to cell
proliferation (17). In the present
study, the high expression of MTHFD2 in breast cancer tissues, and
its prognostic role in patients with breast cancer, was verified
using the GEPIA database. Furthermore, this study demonstrated that
MTHFD2 may exert its oncogenic roles by modulating the AKT
signaling pathway. These findings may enrich the theoretical basis
of MTHFD2-targeted therapy.
In recent years, MTHFD2 expression has been
demonstrated to be increased in various types of cancer (7,11),
such as renal cell carcinoma, colorectal cancer, and its
overexpression is considered as an independent prognostic signature
in patients with breast cancer (14). In silico gene expression analysis
carried out by Lehtinen et al (23) demonstrated that MTHFD2
overexpression is associated with metastasis of patients with
breast cancer. Immunohistochemistry staining performed by Liu et
al (14) also confirmed the
patients with breast cancer patients and high expression of MTHFD2
experienced an unfavorable clinical outcome. Similarly to these two
previous studies, the present study demonstrated that MTHFD2
expression was significantly increased in breast cancer tissues
compared with adjacent normal tissues, according to the GEPIA
database. In addition, OS analysis highlighted the potential of
MTHFD2 as a prognostic biomarker for patients with breast
cancer.
To further evaluate the oncogenic role of MTHFD2,
functional assays were performed using MCF-7 cells. The results
from the present study demonstrated that MTHFD2 overexpression
increased MCF-7 cell proliferation and colony formation ability,
whereas MTHFD2 knockout had an inhibitory effect on MCF-7 cell
proliferation and colony formation ability. These results suggest
that MTHFD2 may act as an oncogenic modulator in breast cancer
cells, which is consistent with the results of a previous study on
colorectal cancer (13). Lehtinen
et al (23) also reported
that RNAi-mediated silencing of MTHFD2 decreased migration and
aggressiveness, resulting in decreased breast cancer stem cell
properties in vivo. Koufaris et al (24) demonstrated that MTHFD2-knockdown
MCF-7 cells presented with decreased proliferation and a slight
decrease in their colony formation ability compared with parental
cells after 72 h. Similarly, the present study demonstrated that
MTHFD2-deficient MCF-7 cells showed no significant colony formation
after 3 days (data not shown). In the present study, there was a
difference in colony formation ability between MTHFD2-deficient and
normal MCF-7 cells after day 14. Off-target effects of the shRNA is
also the possible reason to explain no impact on the colony
formation shMTHFD2 caused. In addition, a significant difference in
colon cancer xenograft tumor growth was observed between mice
injected with MTHFD2-knockdown or control HCT-116 cells; however,
no effects of MTHFD2-knockdown were detected in HCT-116 cell
proliferation in vitro (24), which was not the case in the present
study. This difference may be due to the choice of cell line.
It has been established that MTHFD2 functions as a
critical enzyme in folate one-carbon metabolism and regulates
numerous physiological processes in tumor cells, including
nucleotide metabolism and amino acid interconversion (25). Its enzymatic role was also supported
by a recent study demonstrating that MTHFD2 confers redox cofactor
preference to mitochondrial NADPH, which is advantageous for
defense against redox equilibrium failure and which therefore
supports cell proliferation (6).
In addition to the enzymatic role of MTHFD2,
increasing attention has been paid to the non-enzymatic functions
of MTHFD2. The data from RNA profiling of NCI-H1299 cells following
MTHFD2 silencing indicated that cycle-related genes, such as CCNA2,
are overexpressed, implying that MTHFD2 might have no metabolic
function (13). Furthermore,
overexpression of MTHFD2 shows no reliance on its enzymatic
activity to trigger tumor cell proliferative phenotypes in breast
cancer cells (17). In addition,
the nuclear localization of MTHFD2 has been demonstrated,
suggesting that MTHFD2 may be a nuclear protein likely to be
involved in the regulation of apoptosis and transcriptional events
in order to facilitate tumor cell proliferation (17). These findings are supported by
studies reporting that signal transduction pathways must
participate in metabolic reprogramming to meet biosynthetic
requirements and support tumor cell proliferation (26,27).
Furthermore, it has been reported that Pyrroline-5-carboxylate
reductase 1, which is a mitochondrial metabolic enzyme, can
regulate tumor cell phenotype by altering signaling pathways
(28). The PI3K/AKT signaling
pathway has a crucial role in cellular metabolism (25). In the present study, a luciferase
reporter gene assay was therefore performed to examine the effect
of MTHFD2 on the changes in transcriptional activity of AKT. As
expected, the AKT signaling pathway was markedly activated upon
MTHFD2 overexpression, indicating that MTHFD2 may regulate MCF-7
cell proliferation via the AKT signaling pathway.
AKT is an important molecule that senses numerous
extracellular signals after being activated by various signal
transduction cascades, including Bcr-Abl, Her2/neu and Ras
(29). AKT activation has numerous
effects to drive carcinogenesis of a number of types of cancers
(29). Previous studies have
reported that the AKT signaling pathway is a critical pathway by
which tumor cell metabolism supports the metabolic autonomy of
tumor cells (30,31). It has been demonstrated that the AKT
oncogene is responsible for the shift toward aerobic glycolysis,
which is a distinct feature of metallic remodeling in tumor cells
(29). The switch to aerobic
glycolysis in tumor cells causes a high glycolytic rate and
benefits cell survival by controlling mitochondrial homeostasis and
preventing the activation of Bax (32). Increased AKT-induced metabolic
alternations can also ensure a constant glycolysis and lactate
production for both bioenergetics and biosynthesis to support tumor
cell proliferation (33). In
addition, AKT signaling can stimulate intracellular expression of
nutrition transporter-associated proteins to maintain enough
cell-autonomous nutrient uptake and capture (34). MTHFD2-mediated AKT signaling could
therefore successfully regulate cell metabolism and signaling
transduction, supporting therefore tumor cell
self-renewal/proliferation (35).
The findings from the present study strongly support the
non-enzymatic role of nuclear MTHFD2 and explain why mutant MTHFD2,
which lacks enzymatic activity, can still promote tumor cell
proliferation. In the present study, activation of the AKT
signaling pathway was decreased in MTHFD2 knockout MCF-7 cells. In
addition, cell treatment with an AKT inhibitor inhibited the
colonic formation ability caused by MTHFD2 overexpression. These
results suggest that MTHFD2 overexpression activates the AKT
signaling pathway, enabling cancer cells to override the action of
some limiting factors of cell proliferation in order to support
biosynthesis metabolic phenotypes. However, AKT is not the only
signaling pathway that is involved in the integrated metabolic
reprogramming of tumor cells. For example, the Hippo pathway
cascade has been demonstrated to be involved in cancer metabolic
reprogramming (36). Future work
will investigate the complex signaling networks mediated by MHTFD2
in breast cancer.
In summary, the present study demonstrated that the
MTHFD2 expression level was increased in breast cancer tissues
compared with adjacent normal tissues, and described its role on
the stimulation of tumor cell proliferation via the AKT signaling
pathway. This study demonstrated that MTHFD2 may increase
proliferation of breast cancer cells via the AKT signaling pathway,
complementing the suggestion of its enzymatic role in metabolic
remodeling. These findings provide a theoretical basis for the use
of MTHFD2 as a potential druggable target for breast cancer
therapy. However, the lack of data from different cell lines is the
main limitation of the present study. Future investigation is
required to further determine the role of MTHFD2 in breast
cancer.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the Guangdong Medical
Science and Technology Research Fund Project, China (grant no.
C2019133).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JH, YQ and CL drafted the manuscript and revised it
critically for important intellectual content, acquired, analyzed
and interpreted the data. XH has been involved in drafting the
manuscript and interpreting the data. FZ made substantial
contributions to conception and design of the present study. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mattiuzzi C and Lippi G: Current cancer
epidemiology. J Epidemiol Glob Health. 9:217–222. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
DeSantis CE, Ma J, Gaudet MM, Newman LA,
Miller KD, Goding Sauer A, Jemal A and Siegel RL: Breast cancer
statistics, 2019. CA Cancer J Clin. 69:438–451. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ito H and Matsuo K: Molecular
epidemiology, and possible real-world applications in breast
cancer. Breast Cancer. 23:33–38. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ghoncheh M, Pournamdar Z and Salehiniya H:
Incidence and mortality and epidemiology of breast cancer in the
world. Asian Pac J Cancer Prev. 17:43–46. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shin M, Momb J and Appling DR: Human
mitochondrial MTHFD2 is a dual redox cofactor-specific
methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate
cyclohydrolase. Cancer Metab. 5(11)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Green NH, Galvan DL, Badal SS, Chang BH,
LeBleu VS, Long J, Jonasch E and Danesh FR: MTHFD2 links RNA
methylation to metabolic reprogramming in renal cell carcinoma.
Oncogene. 38:6211–6225. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nilsson R, Jain M, Madhusudhan N, Sheppard
NG, Strittmatter L, Kampf C, Huang J, Asplund A and Mootha VK:
Metabolic enzyme expression highlights a key role for MTHFD2 and
the mitochondrial folate pathway in cancer. Nat Commun.
5(3128)2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mattaini KR, Sullivan MR and Vander Heiden
MG: The importance of serine metabolism in cancer. J Cell Biol.
214:249–257. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ju HQ, Lu YX, Chen DL, Zuo ZX, Liu ZX, Wu
QN, Mo HY, Wang ZX, Wang DS, Pu HY, et al: Modulation of redox
homeostasis by inhibition of MTHFD2 in colorectal cancer:
Mechanisms and therapeutic implications. J Natl Cancer Inst.
111:584–596. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lin H, Huang B, Wang H, Liu X, Hong Y, Qiu
S and Zheng J: MTHFD2 overexpression predicts poor prognosis in
renal cell carcinoma and is associated with cell proliferation and
vimentin-modulated migration and invasion. Cell Physiol Biochem.
51:991–1000. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Pikman Y, Puissant A, Alexe G, Furman A,
Chen LM, Frumm SM, Ross L, Fenouille N, Bassil CF, Lewis CA, et al:
Targeting MTHFD2 in acute myeloid leukemia. J Exp Med.
213:1285–1306. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yu C, Yang L, Cai M, Zhou F, Xiao S, Li Y,
Wan T, Cheng D, Wang L, Zhao C and Huang X: Down-regulation of
MTHFD2 inhibits NSCLC progression by suppressing cycle-related
genes. J Cell Mol Med. 24:1568–1577. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu F, Liu Y, He C, Tao L, He X, Song H
and Zhang G: Increased MTHFD2 expression is associated with poor
prognosis in breast cancer. Tumour Biol. 35:8685–8690.
2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tedeschi PM, Vazquez A, Kerrigan JE and
Bertino JR: Mitochondrial methylenetetrahydrofolate dehydrogenase
(MTHFD2) overexpression is associated with tumor cell proliferation
and is a novel target for drug development. Mol Cancer Res.
13:1361–1366. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wei Y, Liu P, Li Q, Du J, Chen Y, Wang Y,
Shi H, Wang Y, Zhang H, Xue W, et al: The effect of MTHFD2 on the
proliferation and migration of colorectal cancer cell lines. Onco
Targets Ther. 12:6361–6370. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sheppard NG, Jarl L, Mahadessian D,
Strittmatter L, Schmidt A, Madhusudan N, Tegnér J, Lundberg EK,
Asplund A, Jain M and Nilsson R: The folate-coupled enzyme MTHFD2
is a nuclear protein and promotes cell proliferation. Sci Rep.
5(15029)2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Derunes C, Burgess R, Iraheta E, Kellerer
R, Becherer K, Gessner CR, Li S, Hewitt K, Vuori K, Pasquale EB, et
al: Molecular determinants for interaction of SHEP1 with Cas
localize to a highly solvent-protected region in the complex. FEBS
Lett. 580:175–178. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wigler M, Pellicer A, Silverstein S and
Axel R: Biochemical transfer of single-copy eucaryotic genes using
total cellular DNA as donor. Cell. 14:725–731. 1978.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liang T, Zhang Y, Yin S, Gan T, An T,
Zhang R, Wang Y, Huang Y, Zhou Q and Zhang J: Cardio-protecteffect
of qiliqiangxin capsule on left ventricular remodeling, dysfunction
and apoptosis in heart failure rats after chronic myocardial
infarction. Am J Transl Res. 8:2047–2058. 2016.PubMed/NCBI
|
|
22
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lehtinen L, Ketola K, Mäkelä R, Mpindi JP,
Viitala M, Kallioniemi O and Iljin K: High-throughput RNAi
screening for novel modulators of vimentin expression identifies
MTHFD2 as a regulator of breast cancer cell migration and invasion.
Oncotarget. 4:48–63. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Koufaris C, Gallage S, Yang T, Lau CH,
Valbuena GN and Keun HC: Suppression of MTHFD2 in MCF-7 breast
cancer cells increases glycolysis, dependency on exogenous glycine,
and sensitivity to folate depletion. J Proteome Res. 15:2618–2625.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20.
2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Campbell SL and Wellen KE: Metabolic
signaling to the nucleus in cancer. Mol Cell. 71:398–408.
2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Torti SV and Torti FM: Iron and cancer:
More ore to be mined. Nat Rev Cancer. 13:342–355. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Zhuang J, Song Y, Ye Y, He S, Ma X, Zhang
M, Ni J, Wang J and Xia W: PYCR1 interference inhibits cell growth
and survival via c-Jun N-terminal kinase/insulin receptor substrate
1 (JNK/IRS1) pathway in hepatocellular cancer. J Transl Med.
17(343)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Elstrom RL, Bauer DE, Buzzai M, Karnauskas
R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM and
Thompson CB: Akt stimulates aerobic glycolysis in cancer cells.
Cancer Res. 64:3892–3899. 2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chu N, Salguero AL, Liu AZ, Chen Z,
Dempsey DR, Ficarro SB, Alexander WM, Marto JA, Li Y, Amzel LM, et
al: Akt kinase activation mechanisms revealed using protein
semisynthesis. Cell. 174:897–907.e14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Schultze SM, Hemmings BA, Niessen M and
Tschopp O: PI3K/AKT, MAPK and AMPK signalling: Protein kinases in
glucose homeostasis. Expert Rev Mol Med. 14(e1)2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Rathmell JC, Fox CJ, Plas DR, Hammerman
PS, Cinalli RM and Thompson CB: Akt-directed glucose metabolism can
prevent Bax conformation change and promote growth
factor-independent survival. Mol Cell Biol. 23:7315–7328.
2003.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tong X, Zhao F and Thompson CB: The
molecular determinants of de novo nucleotide biosynthesis in cancer
cells. Curr Opin Genet Dev. 19:32–37. 2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wieman HL, Wofford JA and Rathmell JC:
Cytokine stimulation promotes glucose uptake via
phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and
trafficking. Mol Biol Cell. 18:1437–1446. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
36
|
Zhang X, Zhao H, Li Y, Xia D, Yang L, Ma Y
and Li H: The role of YAP/TAZ activity in cancer metabolic
reprogramming. Mol Cancer. 17(134)2018.PubMed/NCBI View Article : Google Scholar
|