Introduction
Multiple myeloma (MM) is a serious hematological
malignancy characterized by mass aggregation of malignant plasma
cells in the bone marrow and the existence of monoclonal protein (M
protein) in blood, urine, or both (1). To improve the life quality and prolong
the survival time of patients with MM, various available
therapeutic options including chemotherapy, autologous/allogeneic
stem cell transplantation and targeted drug therapy have been
widely applied in the clinic to treat this disease (2). In the last few decades, great
advancement in cellular and molecular mechanisms of MM has been
made, leading to the development of a wide range of new drugs, such
as adriamycin (ADR), bortezomib (BOR) and dexamethasone (DEX)
(3). Although these drugs had
significantly improved the quality of life of patients to some
extent, the clinical outcome remains unsatisfactory because of
acquired drug resistance, which has become the main obstacle of the
failure of chemotherapy in the clinical treatment of MM. Therefore,
the further exploration of the potential molecular mechanism
underlying the acquired chemotherapy resistance in MM is extremely
urgent for seeking effective treatment strategies for MM.
High-mobility group box 1 (HMGB1), an important
non-histone protein in the nucleus, functions as a key moderator in
DNA arrangement, replication, damage repair and transcription by
stabilizing nucleosome function (4). An obvious upregulation of HMGB1 has
been identified in several types of tumor tissues compared with
that of the corresponding normal tissues, which is linked with the
tumor development and progression, including inflammation and
angiogenesis, invasion, progression and metastasis (5,6).
Moreover, HMGB1 can be released from diverse tumor cells in
response to chemotherapy or radiotherapy, and thus participates in
the regulation of the chemoresistance and sensitivity of cancer
cells (7,8). The study of Liu et al (9) found that the release of HMGB1 from
leukemia cell lines increased under the stimulation of
chemotherapy, and treatment with HMGB1-neutralizing antibodies
rendered these cells more sensitive to chemotherapy. One in
vivo experiment revealed that HMGB1 can promote tumor growth by
enhancing the drug resistance of lung cancer cells (7). In human lung adenocarcinoma,
HMGB1-regulated autophagy is also considered as a critical
contributor to docetaxel resistance (10). In MM cells, HMGB1 was upregulated
and its expression was negatively associated with the survival of
patients with MM; meanwhile, bortezomib-resistant MM cells also
exhibited a higher HMGB1 expression and HMGB1 knockdown can enhance
the sensitivity of MM cells to chemotherapy in vivo
(11,12). However, the association between
HMGB1 and MM chemoresistance, and the potential molecular mechanism
has not been clarified thoroughly.
Nuclear factor-κB (NF-κB), a tightly regulated
transcription factor, functions as a moderator in almost all types
of cancer development (13). NF-κB
signaling was found to be constitutively activated in some tumor
cells, including leukemia (14),
lung (15), lymphoma (16) and breast (17) cancer cell lines. Besides, the
increased NF-κB levels indicate a poor prognosis in glioblastoma
(18) and ovarian cancer (19), and the blockage of NF-κB signaling
has been demonstrated to exhibit an anti-tumor response (19). Except for the role in cancer
development, activated NF-κB has also been recognized as a key
regulator in chemoresistance. Increasing studies have supported
that the chemotherapy-resistant tumor cells showed significantly
higher expression of NF-κB compared with the matched parental
cells. In carcinoma cell lines, NF-κB activity was found to be
negatively associated with cellular sensitivity to chemotherapy
(20); in several lung cell lines,
long term treatment with increasing doses of cisplatin would render
them becoming resistant to cisplatin (21). The aforementioned data revealed that
NF-κB signaling acts as a potential mediator in the development of
acquired cisplatin resistance.
A gene expression profiling based on 320 diagnosed
patients with MM identified the engagement and significance of
NF-κB signaling in MM cells (22),
and the gene mutations involving this pathway occurred on at least
40% of MM cell lines (MMCLs) and 17% of MM tumors (23,24).
NF-κB signaling has been identified to function as a facilitator in
proliferation and drug-resistance of myeloma cells, which played an
important role in the interactions of the myeloma cells with the
bone marrow microenvironment (25).
However, the exact molecular mechanism of NF-κB in the development
by which the MM cell develops acquired resistance remains to be
explored.
A previous study identified the important role of
HMGB1 in the occurrence and development of diverse tumors through
the NF-κB signaling pathway (26).
Thus, it is postulated that HMGB1 is most likely to be involved in
the regulation of MM chemoresistance via NF-κB signaling. To verify
this, the present study established adriamycin-, bortezomib- and
dexamethasone-resistant MM cells, and then a series of experiments
was performed to confirm the hypothesis. The present study may help
to better understand the molecular mechanisms of MM
chemoresistance, and further develop novel therapeutic strategies
in the treatment of MM.
Material and methods
Cell culture
The MMCL RPMI8266 was purchased from the American
Type Culture Collection. All cells were maintained in RPMI1640
containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.) at 37˚C in a humidified atmosphere with 5%
CO2.
Cell transfection
Short hairpin RNA (shRNA) against human HMGB1
(Sigma-Aldrich; Merck KGaA) and control shRNA (Sigma-Aldrich; Merck
KGaA) were transfected into cells using the
Lipofectamine® 2000 reagent (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Briefly, a
mixture of 1 ml Lipofectamine 2000 and 50 ml Opti-MEM I (Gibco;
Thermo Fisher Scientific, Inc.) was added to the serum-free medium
when the cell density reached ~50%. After 10 min, serum-free medium
was mixed with 1 mg FAM-shRNA and 50 ml Opti-MEM I. After
incubating at room temperature for 5 min, diluted FAM-shRNA and
Lipofectamine 2000 were mixed and incubated for 20 min. The
successfully transfected cells were screened with 0.3 µg/ml
puromycin. The interference sequence against HMGB1 was
5'-GGACAAGGCCCGTTATGAA-3', and the control sequence was
5'-TTCTCCGAACGTGTACGT-3'. At 48 h post transfection, transfected
cells were collected for next analysis.
Establishment of drug-resistance cell
lines
The parental RPMI8266 cells (5x105) were
seeded into 6-well plates containing 10 nM bortezomib
(Sigma-Aldrich; Merck KGaA) when they reached the exponential
growth phase, and were incubated in 37˚C with 5% CO2
condition. The cell culture medium was replaced every 2-3 days
until the cells return to the normal growth state. After 1-2 weeks,
bortezomib (10 nM) was added to the culture medium again; when this
process has been repeated for three times, the final concentration
of bortezomib was increased to 20 nM. The aforementioned steps were
repeated until the bortezomib concentration was increased to 200
nM. After 8-10 months, the bortezomib-resistant RPMI8266 cells were
obtained. A similar approach was also used to establish adriamycin-
and dexamethasone-resistant cell lines. Finally, ADR-resistant
RPMI8226 cells (160 nM of ADR), BOR-resistant RPMI8226 cells (200
nM of BOR), and DEX-resistant RPMI8226 cells (180 nM of DEX) were
successfully established.
Cell cycle analysis
Cells (2x105 cells/well) were cultured in
six-well plates for 48 h. Then, the cells were washed with PBS and
permeabilized with precooled 75% ethanol at 4˚C overnight. After
washing, the cells were incubated with 500 µl propidium iodide (PI;
5 mg/ml; Sigma-Aldrich; Merck KGaA) for 30 min at room temperature,
according to the manufacturer's instructions. The alterations in
cell cycle were determined by using the program M software (System
II Software; version 3.0) on an Epics flow cytometer (Coulter
Immunology).
Cell apoptosis analysis
RPMI8226, RPMI8226/ADR, RPMI8226/BOR and
RPMI8226/DEX cells were respectively treated with a specific
concentration of ADR (160 nM), BOR (200 nM) and DEX (180 nM) for 48
h. Subsequently, the cells (1x105) were incubated with
Annexin V-FITC and propidium iodide (PI; Beyotime Institute of
Biotechnology). Finally, the degree of cell apoptosis was
quantified by flow cytometry (BD Biosciences, Inc.) analysis.
Cell viability assay
Cell viability was detected by the Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.). Briefly, the
parental cells or drug-resistant cells (2x105 cells/ml)
were plated into 96-well plates. After adherence, the cells were
treated with ADR/BOR/DEX at the increasing concentrations of 0, 1,
5, 10, 20, 40 and 80 nM for 48 h at 37˚C. At 2 h before test, 10 µl
CCK-8 reagent and 150 µl DMSO were added into the medium. For the
drug sensitivity analysis, the cells were transfected with HMGB1
shRNA and control shRNA. After 48 h, the cells were treated with
ADR/BOR/DEX at the increasing concentrations of 0, 20, 40, 80, 160,
320 and 640 nM for 48 h at 37˚C. Absorbance was measured at 490 nm
using a microplate reader (Bio-Rad Laboratories, Inc.; Model 680),
and the following formula was used to calculate cell viability:
Cell viability (%) = OD value of test sample/OD value of control
sample x 100%. The inhibition rates of chemotherapy drugs on
RPMI8226/ADR, RPMI8226/BOR, RPMI8226/DEX cells were performed by
CCK-8 assay and IC50 values were calculated using the
GraphPad 5.0 software (GraphPad Software, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from different cell samples
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions and was quantified
with a nanophotometer. The obtained RNA samples were
reverse-transcribed into single stranded DNA with MLV-reverse
transcriptase at 42˚C for 1 h (Invitrogen; Thermo Fisher
Scientific, Inc.). The Applied Biosystems 7500 Real-time PCR System
was used for real-time PCR amplifications using the SYBR Green PCR
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The PCR thermal cycle conditions were 95˚C for 1 min, 55˚C for 1
min, 72˚C for 1 min for 25-30 cycles, and 72˚C for 10 min. The
relative expression levels of genes was calculated using the
2-∆∆Cq method (27). The
primers used were as follows: HMGB1 forward,
5'-ATATGGCAAAAGCGGACAAG-3'; HMGB1 reverse,
5'-GCAACATCACCAATGGACAG-3'; NF-κB forward,
5'-AGGGCCAGAGACGGATATGT-3'; NF-κB reverse,
5-AGATGTTAGTTGGGCGGTGG'-3'; β-actin forward,
5'-ACTCGTCATACTCCTGCT-3' and β-actin reverse,
5'-GAAACTACCTTCAACTCC-3'.
Western blot analysis
Cell lysates were prepared using RIPA buffer [Thermo
Fisher Scientific, Inc.; 20 mmol/l Tris-HCl, pH 7.5; 150 mmol/l
NaCl; 1 mmol/l Na2EDTA; 1 mmol/l EGTA; 1% Triton; 2.5 mmol/l sodium
pyrophosphate; 1 mmol/l b-glycerophosphate; 1 mmol/l Na3VO4; 1
mg/ml leupeptin; 1 mmol/l phenylmethylsulfonylfluoride (PMSF); and
1 mmol/l PMSF] in the presence of a protease inhibitor and PhosStop
(Roche Diagnostics). Cytoplasmic and nuclear proteins were
respectively isolated using a ProteoJET Cytoplasmic and Nuclear
Protein Extraction kit (Thermo Fisher Scientific, Inc.). Protein
from cultured medium was extracted by evaporating the medium.
Concentration of the extracted protein was detected by a
bicinchoninic acid kit (Beyotime Institute of Biotechnology). Then,
proteins (20-40 µg) were separated on 8-12% pre-made protein
electrophoresis gels and transferred to polyvinylidene difluoride
membranes (Merck KGaA). The membranes were blocked with 5% nonfat
milk for 1-2 h at room temperature. Then, the membranes were
incubated with primary antibodies (all purchased from Abcam),
including anti-HMGB1 (cat. no. ab18256; 1:1,000), anti-Cylin D1
(cat. no. ab226977; 1:5,000), anti-PCNA (cat. no. ab18197; 1:500),
anti-cleaved PARP (cat. no. ab32064; 1:1,000), anti-cleaved
caspase-3 (cat. no. ab2302; 1:1,000), anti-TLR4 (cat. no. ab13556;
1:500), anti-p-IKKα/β (cat. no. ab194528; 1:500), anti-IKKα/β (cat.
no. ab178870; 1:1,000), anti-p-IKBα (cat. no. ab133462; 1:1,000),
anti-IKBα (cat. no. ab32518; 1:1,000), anti-p-p65 (cat. no.
ab86299; 1:20), anti-p65 (cat. no. ab32536; 1:1,000), or
anti-β-actin primary antibodies (cat. no. ab8227; 1:1,000)
overnight at 4˚C. After washes with TBST, the membranes were
incubated with secondary antibodies, goat anti-mouse HRP
secondary-antibody (cat. no. ab6808; 1:1,000; Abcam) at room
temperature for 1 h. The bands were detected using an exposure
meter (Bio-Rad Laboratories, Inc.) with an enhanced
chemiluminescence detection kit for HRP (Sangon Biotech Co., Ltd.).
ImageJ densitometry software (version 1.6, National Institutes of
Health) was used to quantitatively analyze western blotting
results. β-actin (cat. no. ab8226; 1:1,000; Abcam) and Lamin A
protein (cat. no. MA3-1000; Invitrogen; Thermo Fisher Scientific,
Inc.; 1:200) were used as a loading control for cytoplasmic and
nuclear proteins.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD) of three replicates per test in a single experiment.
A two-tailed Student's t-test was used to determine significant
differences between two groups. Tukey's multiple comparison tests
was used to analyze the significance of the difference between
means. P<0.05 was considered to indicate a statistically
significant difference. GraphPad Prism V5.0 was used to perform all
statistical analyses.
Results
Establishment of MMCLs resistant to
adriamycin, bortezomib and dexamethasone
To verify the successful establishment of three
chemotherapy-resistant RPMI8266 cell lines, CCK-8 assay was
conducted to determine the cell viability of parental RPMI8266 and
drug-treated RPMI8266 after incubation with the increasing doses
(0, 1, 5, 10, 20, 40 and 80 nM) of ADR, BOR, and DEX. As shown in
Fig. 1A, the cells viability of
chemotherapy-resistant RPMI8266 cells was gradually significantly
higher compared with that of the parental cells as the drug
concentration increased (PRMI8226/ADR, P<0.5 and P<0.001;
PRMI8226/BOR, P<0.001; PRMI8226/DEX, P<0.05 and P<0.01),
which was most notable in RPMI8266/ADR and RPMI8266/BOR; thus, the
drug sensitivity of the three chemotherapy-resistant cells is
decreased. Meanwhile, it was found that the drug-resistant cells
exhibited a significantly decreased cell apoptosis compared with
the parental RPMI8266, in the case of drug treatment (P<0.001;
Fig. 1B). Besides, there was no
significant difference in terms of cell cycle between the three
chemotherapy-resistant cell lines and the parental RPMI8266 based
on the results of flow cytometry analysis (Fig. 1C). In summary, the aforementioned
results indicated the successful establishment of ADR-resistant,
BOR-resistant and DEX-resistant MMCLs.
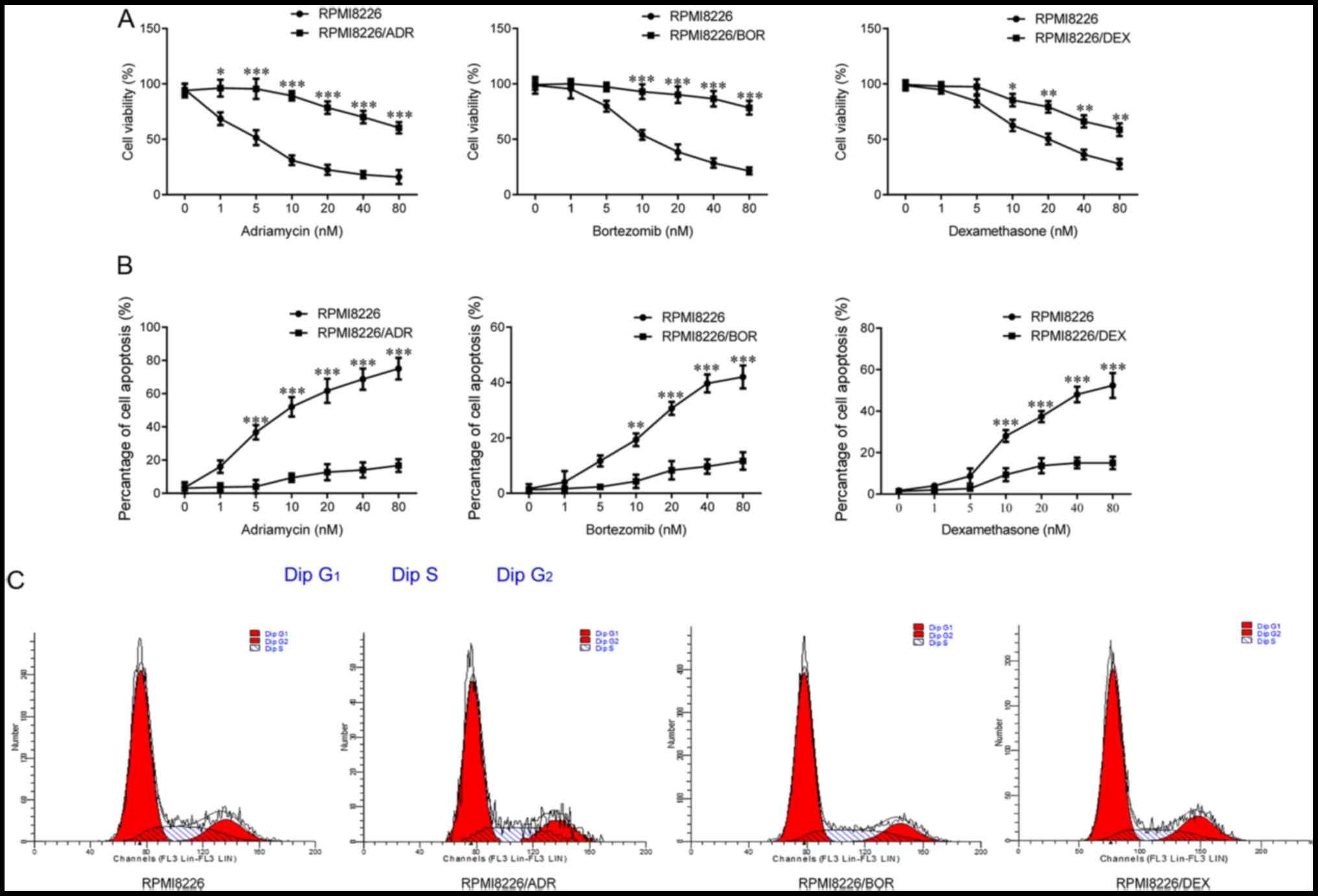 | Figure 1Establishment of multiple myeloma
cell lines resistant to adriamycin, bortezomib and dexamethasone.
Chemotherapy-sensitive cells, RPMI8226, and three
chemotherapy-resistant cells, RPMI8226/ADR, RPMI8226/BOR and
RPMI8226/DEX cells, were treated with increasing concentration of
adriamycin, bortezomib and dexamethasone for 48 h. (A) The cell
viability was determined using the Cell Counting Kit-8 assay. (B)
The extent of cell apoptosis was analyzed by measuring Annexin
V-positive cells by flow cytometry. (C) The cell cycle was measured
by flow cytometric analysis. Data are presented as mean ± SD from
three independent experiments. *P<0.05,
**P<0.01, ***P<0.001, vs RPMI8226. ADR,
adriamycin; BOR, bortezomib; DEX, dexamethasone. |
Release of HMGB1 is increased from
chemotherapy-resistant MMCLs
Next, the expression levels of HMGB1 was detected in
the drug-sensitive and drug-resistant MM cells. As shown in
Fig. 2A, the protein expression of
HMGB1 in the lysate of all three chemotherapy-resistant MMCLs was
higher compared with that of the chemotherapy-sensitive MM cells.
In addition, it was found that the amount of HMGB1 in the
drug-resistant RPMI8226 cells were much higher compared with that
in PRMI8226 cells, in both the cell nucleus and culture media;
moreover, the increase in HMGB1 in the culture media was higher
compared with that in the nucleus (Fig.
2B). These findings revealed that chemotherapy-resistant MM
cells released higher level of HMGB1 compared with the parental
cells, and the increased release of HMGB1 is likely to cause the
chemotherapy drug resistance of MM cells; to further verify this, a
target-specific shRNA was designed to silence HMGB1 expression.
Both RT-qPCR and western blotting results revealed that HMGB1 shRNA
transfection contributed to a notable decrease in HMGB1 expression
(P<0.05, P<0.01, P<0.001; Fig.
2C and D), suggesting the
successful knockdown of HMGB1 in MM cells.
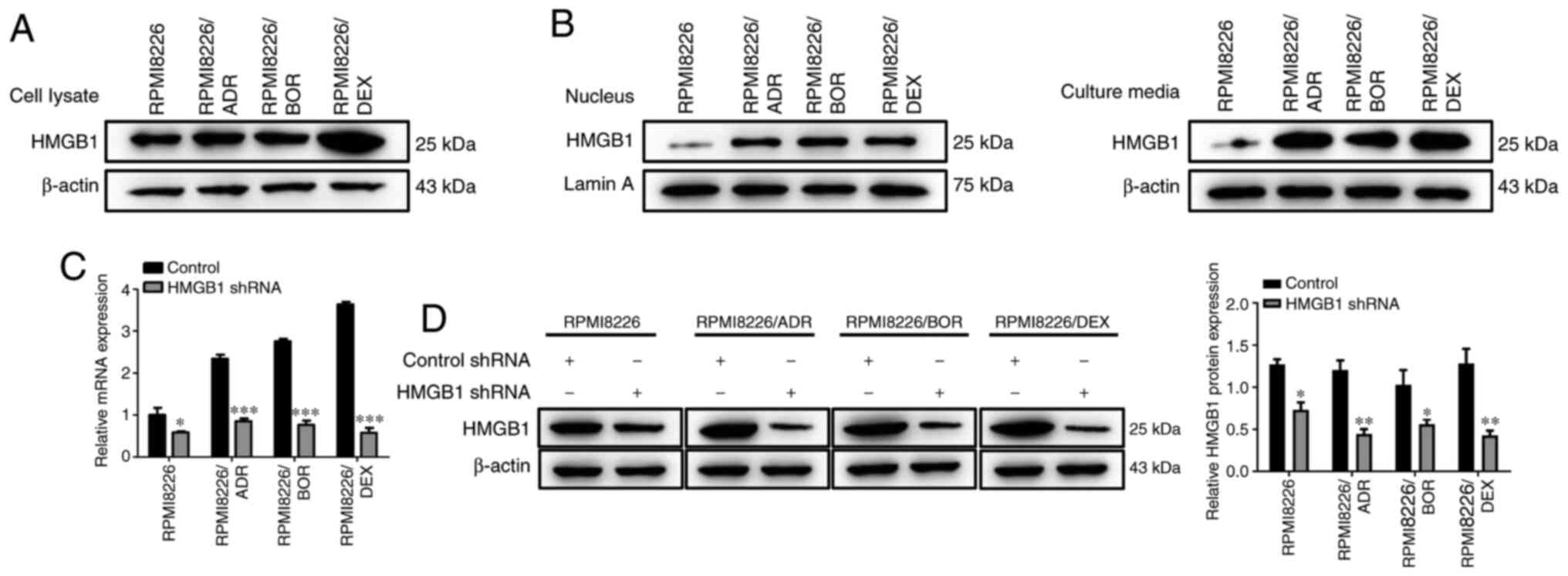 | Figure 2Expression level of HMGB1 is
increased in chemotherapy-resistant multiple myeloma cell lines.
(A) The HMGB1 protein expression level in the cell lysate of
chemotherapy-sensitive cells, RPMI8226, and three
chemotherapy-resistant cells, RPMI8226/ADR, RPMI8226/BOR and
RPMI8226/DEX cells, were detected by western blotting. β-actin was
used as a loading control. (B) The HMGB1 protein expression level
in the nucleus and the culture media of chemotherapy-sensitive
cells, RPMI8226, and three chemotherapy-resistant cells,
RPMI8226/ADR, RPMI8226/BOR, and RPMI8226/DEX cells was detected by
western blotting. (C) The endogenous HMGB1 expression was
interfered with shRNA specific to HMGB1. RPMI8226, RPMI8226/ADR,
RPMI8226/BOR and RPMI8226/DEX cells were plated into 48-well plate.
The cells were cultured overnight and transfected with HMGB1 shRNA
and control shRNA for 48 h. Then, the lysates were prepared for
detecting HMGB1 expression by reverse transcription-quantitative
PCR. (D) The HMGB1 protein expression was detected by western
blotting and quantified. β-actin was used as a loading control.
Data are presented as mean ± SD of three independent experiments.
*P<0.05, **P<0.01,
***P<0.001, vs. RPMI8226. HMGB1, high-mobility group
box 1; ADR, adriamycin; BOR, bortezomib; DEX, dexamethasone; shRNA,
short hairpin RNA. |
Interference with endogenous HMGB1
increases drug sensitivity in chemotherapy-resistant MM cells
Next, drug sensitivity analysis was performed by
treating the three HMGB1 shRNA-transfected chemotherapy-resistant
MM cells with increasing concentration of ADR, BOR and DEX. Based
on the result of the CCK-8 assay, the IC50 values of
HMGB1 shRNA-transfected PRMI8226/ADR, PRMI8226/BOR and PRMI8226/DEX
cells were calculated as 34.34, 25.7 and 53.35 nM, respectively,
which were significantly lower compared with that of control
shRNA-transfected chemotherapy-resistant cells. Thus, interference
with endogenous HMGB1 in chemotherapy-resistant MM cells enhances
drug sensitivity and decreases resistance (Fig. 3A). Cell apoptosis of RPMI8226,
RPMI8226/ADR, RPMI8226/BOR and RPMI8226/DEX cells treated with a
specific concentration of ADR (160 nM), BOR (200 nM) and DEX (180
nM) was analyzed by flow cytometry, which found that silencing
HMGB1 in drug-resistant MM cells rendered them significantly more
sensitive to ADR-, BOR- and DEX-induced cell apoptosis (Fig. 3B). The aforementioned finding was
also supported by the decrease in cyclin D1 and PCNA protein
expression, and the activation of the pro-apoptotic protein cleaved
PARP and cleaved caspase-3 (Fig.
3C). Overall, the data suggest that interference of HMGB1 in
chemotherapy-resistant MM cells significantly improved their drug
sensitivity.
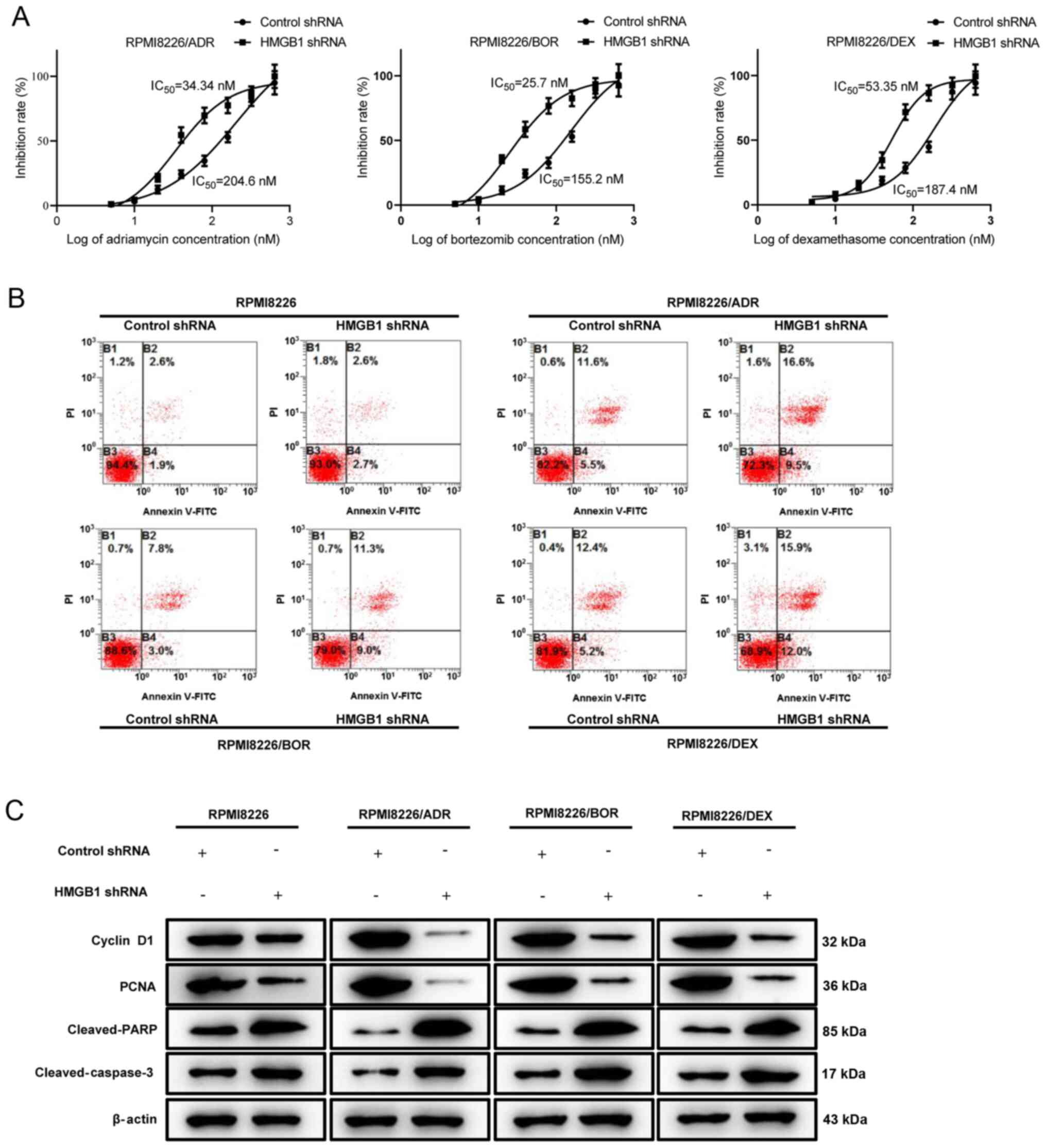 | Figure 3Interference with endogenous HMGB1
increases drug sensitivity in chemotherapy-resistant multiple
myeloma cells. Chemotherapy-sensitive cells, RPMI8226, and three
chemotherapy-resistant cells, RPMI8226/ADR, RPMI8226/BOR and
RPMI8226/DEX cells, were transfected with HMGB1 shRNA and control
shRNA. (A) RPMI8226/ADR, RPMI8226/BOR and RPMI8226/DEX were treated
with increasing concentrations of ADR, BOR, and DEX, respectively,
for 48 h. The inhibition rate was determined by the Cell Cycle
Kit-8 assay. The IC50 value was calculated. (B)
RPMI8226, RPMI8226/ADR, RPMI8226/BOR and RPMI8226/DEX cells were
respectively treated with a specific concentration of ADR (160 nM),
BOR (200 nM) and DEX (180 nM) for 48 h. Cell apoptosis was analyzed
by measuring Annexin V-positive cells by flow cytometry. (C)
RPMI8226, RPMI8226/ADR, RPMI8226/BOR and RPMI8226/DEX cells were
respectively treated with a specific concentration of ADR (200 nM),
BOR (160 nM), and DEX (180 nM) for 48 h. The protein expression
levels of cyclin D1, PCNA, cleaved-PARP, cleaved-caspase-3 were
detected by western blotting. β-actin was used as a loading
control. Data are presented as mean ± SD from three independent
experiments. HMGB1, high-mobility group box 1; ADR, adriamycin;
BOR, bortezomib; DEX, dexamethasone; shRNA, short hairpin RNA. |
Interference with endogenous HMGB1
inhibits NF-κB signaling activity in chemotherapy-resistant MM
cells
The important role of NF-κB signaling in regulating
drug sensitivity has been identified in various tumors.
Unsurprisingly, a significant increase in TLR4 and NF-κB mRNA
expression was found in all chemotherapy-resistant MM cells
(P<0.05, P<0.01; Fig. 4A).
Given the high expression of TLR4 and HMGB1 in the MM cells with
acquired drug resistance, it was speculated that the enhanced drug
sensitivity induced by silencing HMGB1 may be associated with NF-κB
signaling pathway. To further verify this, the effect of HMGB1 on
NF-kB activity was next examined. Western blotting showed that
HMGB1 shRNA significantly attenuated the TLR4 level, the
phosphorylation of IKKα/β, IκBα,and p65 in the cell lysate of the
three drug-resistant cells compared with the control shRNA cells
(Fig. 4B). Moreover, interference
with HMGB1 also drastically attenuated the level of phosphorylated
p65 in the nucleus (P<0.05; P<0.001; Fig. 4C). These data revealed an apparent
suppression of shRNA HMGB1 on NF-κB signaling activity,
demonstrating that the effect of interference with endogenous HMGB1
on drug sensitivity is most likely because of NF-κB signaling
activity inhibition.
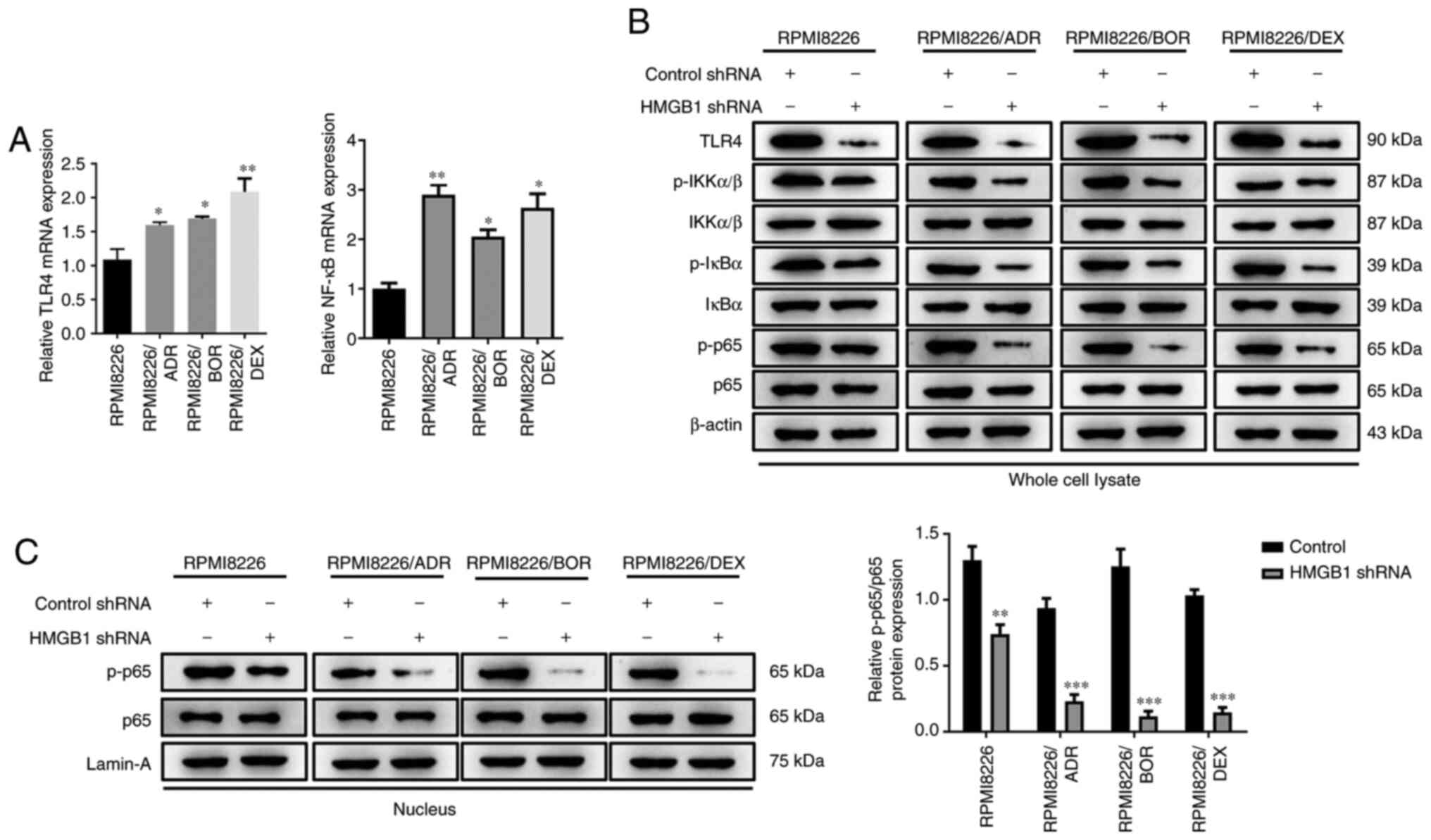 | Figure 4Interference with endogenous HMGB1
inhibits NF-κB signaling activity in chemotherapy-resistant
multiple myeloma cells. Chemotherapy-sensitive cells, RPMI8226, and
three chemotherapy-resistant cells, RPMI8226/ADR, RPMI8226/BOR and
RPMI8226/DEX cells, were transfected with HMGB1 shRNA or control
shRNA. (A) The relative mRNA expression of TLR4 and NF-κB mRNA were
detected by reverse transcription-quantitative PCR. (B) The protein
expression of TLR4, p-IKKα/β, total IKKα/β, p-IκBα, total IκBα,
p-p65 and total p65 in whole cell lysate of RPMI8226, RPMI8226/ADR,
RPMI8226/BOR and RPMI8226/DEX cells were detected by western
blotting. β-actin was used as a loading control. (C) The protein
expression of p-p65 and total p-65 in the nucleus of RPMI8226,
RPMI8226/ADR, RPMI8226/BOR and RPMI8226/DEX cells were detected by
western blotting and quantified. Lamin-A was used as a loading
control. Data are presented as mean ± SD from three independent
experiments. *P<0.05, **P<0.01,
***P<0.001 vs. RPMI8226. HMGB1, high-mobility group
box 1; ADR, adriamycin; BOR, bortezomib; DEX, dexamethasone; p-,
phosphorylated; shRNA, short hairpin RNA; TLR4, Toll-like receptor
4; IKKα/β, I-κ-B kinase α/β. |
Activation of NF-κB signaling reverses
HMGB1 silencing-induced enhancement of chemotherapy sensitivity in
MM cells
To further identify the regulatory function of NF-κB
signaling in the process of HMGB1 silencing-mediated drug
sensitivity of MM cells, HY-N6826 (an agonist of NF-κB signaling
pathway) was used to treat the MM cells transfected with HMGB1
shRNA. The results showed that the IKKα/β, IκBα and p65 in the cell
lysate were more phosphorylated in HY-N6826-treated cells in
contrast to the cells transfected with HMGB1 shRNA alone (Fig. 5A). Meanwhile, the nuclear
translocation of p65 was also significantly increased, as confirmed
by the elevation of the nuclear level of phosphorylated p65
(Fig. 5B). These data suggested the
successful activation of NF-κB by HY-N6826. Fig. 5C displayed that the treatment of
NF-κB agonist significantly reversed the inhibition of HMGB1
knockdown on MM cell viability (P<0.05, P<0.01). Moreover,
HMGB1 silencing-induced cell apoptosis was also dramatically
decreased when NF-κB signaling was activated (Fig. 5D; P<0.05). Similar outcome was
also confirmed by the protein expression of cyclin D1, PCNA,
cleaved-PARP and cleaved-caspase 3 (Fig. 5E). The aforementioned data manifest
that the activation of NF-κB signaling could effectively reverse
the impact of HMGB1 silencing on the drug sensitivity of MM
cells.
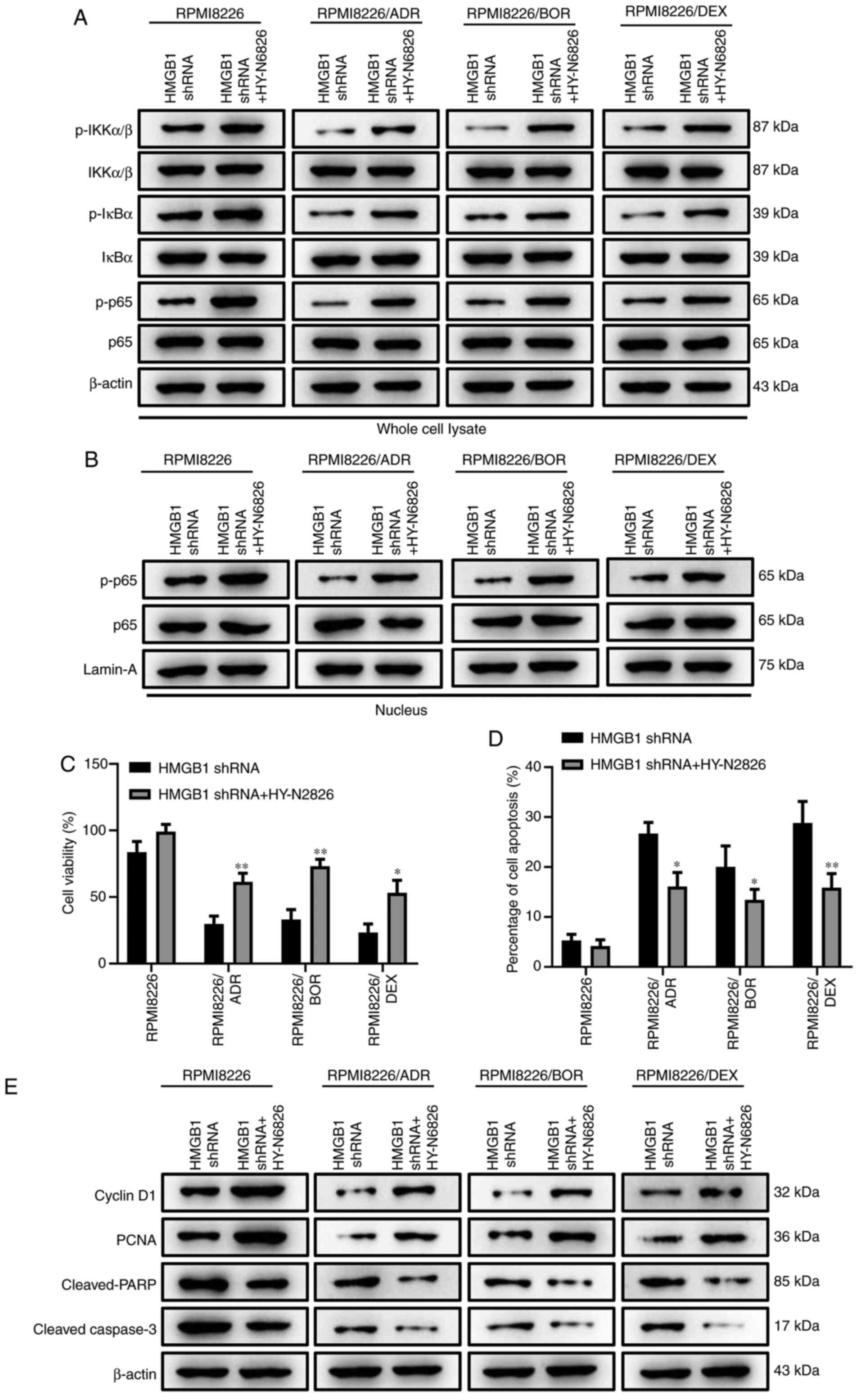 | Figure 5Activation of NF-κB signaling
reverses HMGB1 silencing-induced enhancement of chemotherapy
sensitivity in multiple myeloma cells. The MM cells transfected
with HMGB1 shRNA were treated with an agonist of nuclear factor-κB
signaling pathway, HY-N6826 (asatone). (A) The protein expression
of phosphorylated p-IKKα/β, total IKKα/β, p-IκBα, total IκBα, p-p65
and total p65 in whole cell lysate of RPMI8226, RPMI8226/ADR,
RPMI8226/BOR and RPMI8226/DEX cells were detected by western
blotting. β-actin was used as a loading control. (B) The protein
expression of p-p65 and total p-65 in the nucleus of RPMI8226,
RPMI8226/ADR, RPMI8226/BOR and RPMI8226/DEX cells were detected by
western blotting. Lamin-A was used as a loading control. (C) The
cell viability was determined using the Cell Counting Kit-8 assay.
(D) The quantification of Annexin-positive cells based on the
result of flow cytometry assay. (E) The protein expression of
cyclin D1, PCNA, cleaved-PARP, cleaved-caspase-3 was detected by
western blotting. β-actin was used as a loading control. Data are
presented as mean ± SD from three independent experiments.
*P<0.05, **P<0.01 vs. RPMI8226.HMGB1,
high-mobility group box 1; ADR, adriamycin; BOR, bortezomib; DEX,
dexamethasone; p-, phosphorylated; shRNA, short hairpin RNA;
IKKα/β, I-κ-B kinase α/β. |
Discussion
As a malignant hematological tumor, MM is
characterized by the abnormal proliferation of plasma cells in the
bone marrow, which has been seriously threatened human health and
survival (28). Currently, diverse
clinical therapies including chemotherapy, targeted therapy and
immunotherapy have been used for the treatment of MM, which are all
in the face of a challenging problem of drug resistance that has
become a major obstacle in successfully treating MM. Although the
common anticancer chemotherapy drugs, adriamycin, bortezomib and
dexamethasone have exhibited a satisfactory therapeutic outcome in
the treatment of MM, the widespread emergence of acquired
resistance to chemotherapy greatly limits the use of these drugs
(29). Therefore, it is urgent to
define the exact mechanism of chemotherapy resistance that is
clinically relevant in MM, which may be helpful to predict and
overcome drug resistance, thereby improving chemotherapy and
ultimately the outcome of patients with cancer.
In order to explore the exact mechanism of drug
resistance in MM, three drug-resistant MMCLs were first
constructed. Based on the result by which the increased cell
viability and the decreased cell apoptosis of drug-resistant MM
cells compared with the parental cells, as well as the similar cell
cycle between the three chemotherapy-resistant cell lines and the
parental cells, the successful establishment of ADR-resistant,
BOR-resistant, and DEX-resistant MMCLs was confirmed.
HMGB1 has been found to be overexpressed in a
variety of hematological malignancies, such as leukemia and
lymphoma (30), and its high
expression has been shown to be a strong predictor of poor survival
in diverse malignancies, including colorectal cancer, gastric
cancer, nasopharyngeal carcinoma and squamous-cell carcinoma of the
head and neck (31). In MM, a study
based on GEP analysis revealed a higher expression level of HMGB1
in the bone marrow plasma cells of patients with MM compared with
that in healthy donors, and the high level of HMGB1 also indicated
a poor survival in these patients; meanwhile, a significant
upregulation of HMGB1 has been observed in bortezomib resistant-MM
cells (12). Besides, silencing
HMGB1 in MM cells could enhance the inhibitory effect of
chemotherapy with dexamethasone via regulating the mTOR pathway
(11). In the present study, all
three drug-resistant MMCLs expressed an increased expression level
of HMGB1 compared with the parental cells; this observation was
confirmed by the prior finding of the higher HMGB1 expression in
bortezomib-resistant MM cells (11). These findings confirmed a close
association between HMGB1 and drug resistance in MM. Actually,
several studies had been carried out to explore the association
between HMGB1 expression and chemotherapy resistance in various
tumor cells. For example, a significantly enhanced expression of
HMGB1 was found in cisplatin- and methotrexate-treated human
osteosarcoma cells (32); PTX has
been shown to induce HMGB1 release from human pancreatic cancer
cells and colon cancer cells (33);
Liu et al (9) found that
chemotherapy treatment significantly improved the level of HMGB1 in
the supernatants of leukemia cell cultures (9); the study of Zheng et al
(7) revealed that HMGB1 levels were
much higher in the cisplatin-resistant cell line compared with the
cisplatin-sensitive cell line, and the levels of HMGB1 level in
cytoplasm gradually elevated with the increasing concentrations of
cisplatin.
To further explore the regulatory role of HMGB1 in
drug resistance in MM cells, an shRNA was designed to interfere
with the expression of HMGB1. The results of CCK-8 and flow
cytometry assays manifested that HMGB1 knockdown significantly
decreased the IC50 value and promoted cell apoptosis of
drug-resistant MM cells; this finding suggests that HMGB1 might be
a driver to promote MM cells to develop chemotherapy drug
resistance. Based on a previous study (34), it is postulated that the mechanisms
of drug-resistance causing an increase in HMGB1 in myeloma cells
may be due to that the chemotherapeutic drugs-induced autophagy in
MM cells, and the translocation of HMGB1 from the nucleus to the
cytoplasm is a key molecular event in this process. Previous
studies provided empirical evidence that extracellular HMGB1 is
associated with both sensitivity and resistance to anticancer
therapy. The present finding was consistent with the findings by
Zheng et al (7) that
knockdown of HMGB1 enhanced the chemosensitivity of human lung
cancer cells to chemotherapeutic drugs, including 5-FU, DDP and
OXA. Similarly, an associated study showed that the blockage of
HMGB1 release could effectively inhibit cell viability and
strengthen the sensitivity of DU145R cells to PTX, indicating a
promotion of PTX resistance induced by the released HMGB1 in
prostate cancer cells (35).
Meanwhile, HMGB1 overexpression can attenuate the leukemia cell
sensitivity to adriamycin, vincristine and cytarabine, while HMGB1
knockdown enhanced this effect (36). Of note, if the drug sensitivity
analysis was also performed in HMGB1-transfected cells, the
confidence of the present results may be higher; this can be
considered as a minor limitation of the present study, and would be
served as the research focus in the future study.
HMGB1 a DNA binding protein located in nucleus, has
been identified to participate in the regulation of drug resistance
and sensibility in various tumors via the mediation of various
signaling pathways. For example, HMGB1 can promote the formation
and fusion of the autophagosome with the lysosome by activating the
PI3K-MEK-ERK pathway, thereby enhancing the resistance of leukemia
cells to anticancer therapies (9).
Moreover, overexpression HMGB1 induces drug resistance of the
leukemia cells by inducing autophagy via the PI3K/Akt/mTORC1
pathway, while knockdown HMGB1 inhibiting leukemia cell autophagy
and increasing drug sensitivity through increasing the
phosphorylation of Akt and p70S6k (9,36). As
a novel proinflammatory cytokine, HMGB1 has been demonstrated to
interact with binding to Toll-like receptor 4 (TLR4) to activate
NF-κB (37). In the present study,
the interference with endogenous HMGB1 was found to remarkably
decrease TLR4 expression and NF-κB signaling activity, which is
consistent with the previous study (38). Activated NF-κB has been considered
as an important role of cisplatin resistance, which could
negatively regulate the cellular sensitivity of carcinoma cell
lines to chemotherapy (38).
Furthermore, the cisplatin-resistant lung cancer cells displayed an
elevated NF-κB expression in contrast to their parental cells,
supporting a potential regulatory role of NF-κB in acquired
cisplatin resistance (21). Based
on the aforementioned finding, it was hypothesized that there is a
potential association between HMGB1-mediated drug resistance and
NF-κB signaling pathway.
To confirm the hypothesis, further rescue
experiments were performed and the results showed that the
activation of NF-κB signaling reverses HMGB1 silencing-induced
enhancement of drug sensitivity in MM cells. Numerous studies have
explored the regulatory association between HMGB1 and NF-κB
signaling. van Beijnum et al (39) found that HMGB1 administration can
activate NF-κB pathway via TLR4, thereby involving the regulation
of inflammation and activation of immune cells. The study by Huang
et al (40) indicated that
HMGB1 silencing significantly inhibited the activation of NF-κB by
suppressing the nuclear translocation and DNA-binding activity of
NF-κB/p65. In murine models, the treatment of cells with an
AKT-NF-kB inhibitor (genistein) rendered them sensitive to
cisplatin-induced apoptosis by increasing NF-κB activity (41).
In conclusion, based on the establishment of three
drug-resistant MMCLs, high expression of HMGB1 was observed in the
chemotherapy-resistant MM cells, and the interference of endogenous
HMGB1 enhanced the drug sensibility and inhibited NF-κB signaling
activity in chemotherapy-resistant MM cells. The activation of
NF-κB signaling restored HMGB1 silencing-induced enhancement of
drug sensitivity, manifesting a significant role of NF-κB signaling
pathway in HMGB1-mediated drug resistance. These results provided a
greater understanding of the potential molecular mechanism of HMGB1
in regulating drug resistance of MM cells, which may be helpful for
the development of novel therapeutic strategies and drugs to treat
MM.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by Ningxia Hui Autonomous
Region Key Research and Development Project (grant no.
2019BEG03053).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JN, RY, HW and LC initiated the work and designed
the experiments. JN performed the majority of the experiments. JN
and RY wrote the manuscript. HW and LC provided samples and
critical suggestions. All authors read and approved the final
manuscript. JN and LC confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sparano F, Cavo M, Niscola P, Caravita T
and Efficace F: Patient-reported outcomes in relapsed/refractory
multiple myeloma: A systematic review. Support Care Cancer.
26:2075–2090. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
McCullough KB, Hobbs MA, Abeykoon JP and
Kapoor P: Common adverse effects of novel therapies for multiple
myeloma (MM) and Their management strategies. Curr Hematol Malig
Rep. 13:114–124. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Abdi J, Chen G and Chang H: Drug
resistance in multiple myeloma: Latest findings and new concepts on
molecular mechanisms. Oncotarget. 4:2186–2207. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Raucci A, Di Maggio S, Scavello F,
D'Ambrosio A, Bianchi ME and Capogrossi MC: The Janus face of HMGB1
in heart disease: A necessary update. Cell Mol Life Sci.
76:211–229. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Venereau E, De Leo F, Mezzapelle R,
Careccia G, Musco G and Bianchi ME: HMGB1 as biomarker and drug
target. Pharmacol Res. 111:534–544. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang H, Antoine DJ, Andersson U and Tracey
KJ: The many faces of HMGB1: Molecular structure-functional
activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol.
93:865–873. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zheng H, Chen JN, Yu X, Jiang P, Yuan L,
Shen HS, Zhao LH, Chen PF and Yang M: HMGB1 enhances drug
resistance and promotes in vivo tumor growth of lung cancer cells.
DNA Cell Biol. 35:622–627. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang YX, Yuan YQ, Zhang XQ, Huang DL, Wei
YY and Yang JG: HMGB1-mediated autophagy confers resistance to
gemcitabine in hormone-independent prostate cancer cells. Oncol
Lett. 14:6285–6290. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pan B, Chen D, Huang J, Wang R, Feng B,
Song H and Chen L: HMGB1-mediated autophagy promotes docetaxel
resistance in human lung adenocarcinoma. Mol Cancer.
13(165)2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Guo X, He D, Zhang E, Chen J, Chen Q, Li
Y, Yang L, Yang Y, Zhao Y, Wang G, et al: HMGB1 knockdown increases
MM cell vulnerability by regulating autophagy and DNA damage
repair. J Exp Clin Cancer Res. 37(205)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Roy M, Liang L, Xiao X, Peng Y, Luo Y,
Zhou W, Zhang J, Qiu L, Zhang S, Liu F, et al: Lycorine
downregulates HMGB1 to inhibit autophagy and enhances bortezomib
activity in multiple myeloma. Theranostics. 6:2209–2224.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mansouri L, Papakonstantinou N, Ntoufa S,
Stamatopoulos K and Rosenquist R: NF-κB activation in chronic
lymphocytic leukemia: A point of convergence of external triggers
and intrinsic lesions. Semin Cancer Biol. 39:40–48. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tew GW, Lorimer EL, Berg TJ, Zhi H, Li R
and Williams CL: SmgGDS regulates cell proliferation, migration,
and NF-kappaB transcriptional activity in non-small cell lung
carcinoma. J Biol Chem. 283:963–976. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Boudesco C, Verhoeyen E, Martin L,
Chassagne-Clement C, Salmi L, Mhaidly R, Pangault C, Fest T, Ramla
S, Jardin F, et al: HSP110 sustains chronic NF-κB signaling in
activated B-cell diffuse large B-cell lymphoma through MyD88
stabilization. Blood. 132:510–520. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chua HL, Bhat-Nakshatri P, Clare SE,
Morimiya A, Badve S and Nakshatri H: NF-kappaB represses E-cadherin
expression and enhances epithelial to mesenchymal transition of
mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2.
Oncogene. 26:711–724. 2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kına I, Sultuybek GK, Soydas T, Yenmis G,
Biceroglu H, Dirican A, Uzan M and Ulutin T: Variations in
Toll-like receptor and nuclear factor-kappa B genes and the risk of
glioma. Br J Neurosurg. 33:165–170. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rada M, Nallanthighal S, Cha J, Ryan K,
Sage J, Eldred C, Ullo M, Orsulic S and Cheon DJ: Inhibitor of
apoptosis proteins (IAPs) mediate collagen type XI alpha 1-driven
cisplatin resistance in ovarian cancer. Oncogene. 37:4809–4820.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Seubwai W, Vaeteewoottacharn K, Kraiklang
R, Umezawa K, Okada S and Wongkham S: Inhibition of NF-κB activity
enhances sensitivity to anticancer drugs in cholangiocarcinoma
cells. Oncol Res. 23:21–28. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Barr MP, Gray SG, Hoffmann AC, Hilger RA,
Thomale J, O'Flaherty JD, Fennell DA, Richard D, O'Leary JJ and
O'Byrne KJ: Generation and characterisation of cisplatin-resistant
non-small cell lung cancer cell lines displaying a stem-like
signature. PLoS One. 8(e54193)2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Broyl A, Hose D, Lokhorst H, de Knegt Y,
Peeters J, Jauch A, Bertsch U, Buijs A, Stevens-Kroef M, Beverloo
HB, et al: Gene expression profiling for molecular classification
of multiple myeloma in newly diagnosed patients. Blood.
116:2543–2553. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Peng H, Peng T, Wen J, Engler DA,
Matsunami RK, Su J, Zhang L, Chang CC and Zhou X: Characterization
of p38 MAPK isoforms for drug resistance study using systems
biology approach. Bioinformatics. 30:1899–1907. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Demchenko YN, Glebov OK, Zingone A, Keats
JJ, Bergsagel PL and Kuehl WM: Classical and/or alternative
NF-kappaB pathway activation in multiple myeloma. Blood.
115:3541–3552. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Keats JJ, Fonseca R, Chesi M, Schop R,
Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al:
Promiscuous mutations activate the noncanonical NF-kappaB pathway
in multiple myeloma. Cancer Cell. 12:131–144. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang J, Shao S, Han D, Xu Y, Jiao D, Wu
J, Yang F, Ge Y, Shi S, Li Y, et al: High mobility group box 1
promotes the epithelial-to-mesenchymal transition in prostate
cancer PC3 cells via the RAGE/NF-κB signaling pathway. Int J Oncol.
53:659–671. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Choi YW, Park JS, Han JH, Kim JH, Ahn MS,
Lee HW, Kang SY, Choi JH and Jeong SH: Strong immunoexpression of
dickkopf-1 is associated with response to bortezomib in multiple
myeloma. Leuk Lymphoma. 59:2670–2678. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang Y, Liu H, Chen X, Bai Q, Liang R,
Shi B, Liu L, Tian D and Liu M: Modified bortezomib, adriamycin and
dexamethasone (PAD) regimen in advanced multiple myeloma. Pathol
Oncol Res. 20:987–995. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sohun M and Shen H: The implication and
potential applications of high-mobility group box 1 protein in
breast cancer. Ann Transl Med. 4(217)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Murakami T, Matsuyama R, Ueda M, Mochizuki
Y, Homma Y, Kameda K, Yazawa K, Izumisawa Y, Fukushima T, Kamimukai
N, et al: High-Mobility Group Box 1 expression predicts survival of
patients after resection of adenocarcinoma of the ampulla of Vater.
World J Surg Oncol. 17(140)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R,
Vernon P, Cao L and Tang D: HMGB1 promotes drug resistance in
osteosarcoma. Cancer Res. 72:230–238. 2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Aldonza MB, Hong JY and Lee SK:
Paclitaxel-resistant cancer cell-derived secretomes elicit
ABCB1-associated docetaxel cross-resistance and escape from
apoptosis through FOXO3a-driven glycolytic regulation. Exp Mol Med.
49(e286)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li Y, Xie J, Li X and Fang J: Poly
(ADP-ribosylation) of HMGB1 facilitates its acetylation and
promotes HMGB1 translocation-associated chemotherapy-induced
autophagy in leukaemia cells. Oncol Lett. 19:368–378.
2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhou J, Chen X, Gilvary DL, Tejera MM,
Eksioglu EA, Wei S and Djeu JY: HMGB1 induction of clusterin
creates a chemoresistant niche in human prostate tumor cells. Sci
Rep. 5(15085)2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Yang L, Yu Y, Kang R, Yang M, Xie M, Wang
Z, Tang D, Zhao M, Liu L, Zhang H, et al: Up-regulated autophagy by
endogenous high mobility group box-1 promotes chemoresistance in
leukemia cells. Leuk Lymphoma. 53:315–322. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Shi Y, Zhang L, Teng J and Miao W: HMGB1
mediates microglia activation via the TLR4/NF-κB pathway in
coriaria lactone induced epilepsy. Mol Med Rep. 17:5125–5131.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Almeida LO, Abrahao AC, Rosselli-Murai LK,
Giudice FS, Zagni C, Leopoldino AM, Squarize CH and Castilho RM:
NFκB mediates cisplatin resistance through histone modifications in
head and neck squamous cell carcinoma (HNSCC). FEBS Open Bio.
4:96–104. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
van Beijnum JR, Buurman WA and Griffioen
AW: Convergence and amplification of toll-like receptor (TLR) and
receptor for advanced glycation end products (RAGE) signaling
pathways via high mobility group B1 (HMGB1). Angiogenesis.
11:91–99. 2008.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Huang Z, Zhong Z, Zhang L, Wang X, Xu R,
Zhu L, Wang Z, Hu S and Zhao X: Down-regulation of HMGB1 expression
by shRNA constructs inhibits the bioactivity of urothelial
carcinoma cell lines via the NF-κB pathway. Sci Rep.
5(12807)2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Li Y, Ahmed F, Ali S, Philip PA, Kucuk O
and Sarkar FH: Inactivation of nuclear factor kappaB by soy
isoflavone genistein contributes to increased apoptosis induced by
chemotherapeutic agents in human cancer cells. Cancer Res.
65:6934–6942. 2005.PubMed/NCBI View Article : Google Scholar
|














