Introduction
Non-alcoholic fatty liver disease (NAFLD) is a
multisystem disease that increases the risk of type 2 diabetes,
cardiovascular disease and heart disease (1). NAFLD can range from simple steatosis
to steatohepatitis and can develop into cirrhosis and subsequently
liver cancer in certain cases. NAFLD is endemic worldwide, with a
prevalence of 25% in the Asian population. In 2016, the number of
NAFLD cases in the United States was 85.3 million (2,3). A
previous study has suggested that maternal obesity is closely
related to the occurrence of NAFLD in the offspring. Ayonrinde
et al (4) conducted a cohort
study on 1,170 17-year-old adolescent mothers and found that
maternal obesity and weight gain in early to mid-term pregnancy
increased the incidence of NAFLD in their babies. A prospective
study by Patel et al (5) of
14,541 pregnant women revealed that the higher the body mass index
of the mother prior to pregnancy, the greater the probability of
NAFLD in their offspring. Hence, it is very important to understand
how maternal obesity promotes the formation of NAFLD in
offspring.
Some studies have suggested that maternal obesity is
related to the onset of NAFLD in the offspring and the oxidation of
their lipids (6,7). Previous articles have commonly used
male rats that were fed a high-fat diet to establish NAFLD models
(8,9). Borengasser et al (10) induced obesity in female Spring
Dawley rats (SD) via a high fat diet. Tests on their male offspring
demonstrated that the liver energy of the male offspring was
reduced and mitochondrial function was damaged; additionally,
maternal obesity also reduced the level of fatty acid oxidation in
the offspring. Additionally the physical state of the mother,has
also been observed to induce NAFLD changes in offspring. Wankhade
et al (11) fed C57BL/6J
female mice a high-fat diet and extracted DNA from their liver
tissue for quantitative analysis and found that high-fat diet
resulted in DNA methylation in the offspring leading to NAFLD.
Either insulin resistance and changes in intestinal flora may be
responsible for maternal obesity and the occurrence of NAFLD in
offspring (12,13). The mechanism behind maternal obesity
and the development of NAFLD in offspring remains unknown.
Autophagy is a catabolic process inherent to
eukaryotic cells, via which cells carry damaged organelles,
misfolded proteins or pathogens into the lysosome for degradation
in order to maintain cell homeostasis (14). Autophagy serves a vital role in
maintaining a balance in cell energy metabolism, controlling
organelle quality and the cellular stress response (15). Some studies have shown that liver
transcription factor EB can induce liver cell autophagy by
regulating the expression of lysosome and autophagy-related genes,
such as LC3-II, hence improving liver steatosis (16,17).
It has also been shown that the upregulation of sirtuin-3 may lead
to the deacetylation and activation of manganese superoxide
dismutase, which may inhibit autophagy after depletion of cellular
superoxide and promote the formation of fat (18).
Recent NAFLD studies have focused on comparing a
before and after scenario regarding NAFLD occurrence, but to the
best of our knowledge there is a lack of studies investigating the
relationship between maternal obesity and the susceptibility of
their offspring to NAFLD and autophagy (19,20).
Hence, the present study generated obese maternal mice via a
high-fat diet to create NAFLD models in their offspring to explore
the influence of maternal obesity on the susceptibility of NAFLD in
the offspring and its underlying molecular mechanisms. The results
may provide a new direction for the treatment of NAFLD in offspring
caused by maternal obesity.
Materials and methods
Ethics approval
Animal experiments performed in the present study
comply with the relevant provisions of the Regulations of the
People's Republic of China on the Administration of Experimental
Animals and have been reviewed and approved by the Animal
Experiments and Animal Experiment Ethics Committee of The Second
Affiliated Hospital of Jiaxing University, Jiaxing Second Hospital
(Jiaxing, China; approval no. jxey2017002).
Animals
The obesity model of maternal mice was established
for 4 weeks and the gestation period was 3 weeks. Offspring were
weaned for 17 weeks after 4 weeks of lactation. A total of 20,
4-week-old C57BL/6J female mice were obtained from the Experimental
Animal Center of Jiaxing University (Jiaxing, China), where they
were housed in specific pathogen free grade conditions. The
temperature was kept at 24±2˚C, a relative humidity of 50±10% and
12/12 h light and dark cycle lighting was maintained. Free access
to water and food was provided. The mice were randomly divided into
2 groups. One group was fed a high-fat diet [obese mother (OM)
group, n=10] containing 60.0% fat, 19.4% protein, and 20.6%
carbohydrates (TROPHIC Animal Feed High-Tech Co., Ltd.); the other
group was fed a normal diet [chow mother (CM), n=10] containing 4%
fat, 18% protein, 53% carbohydrates and 0.07% cholesterol
(SHOOBREE, Synergetic Medical Bioengineering Co., Ltd.). Common
male mice (A total of 10 8-week-old mice from Jiaxing University)
were mated with the 2 groups of female mice (1 male:2 female) and
their offspring were defined as offspring of obese mother (OM-O)
and offspring of obese mother (CM-O). The offspring of the 2 groups
were weaned at 4 weeks old. Subsequently, 10 male offspring from
the OM-O and CM-O groups were continuously fed a high-fat diet
until they were sacrificed at 17 weeks old. Briefly, a sterile
transparent glass container with a gauze pad at the bottom was
prepared and 2-4 ml ether (Huadong Medicine Co., Ltd.) was added to
the gauze pad, the container was sealed for 10-20 sec to prevent
volatilization, a mouse was added to it, the container was resealed
and inhalation anesthesia was performed, since mice breathe fast
and tetraplegia is thought to be a sign of successful anesthesia.
After anesthesia,1 ml venous blood was collected. Subsequently, all
the mice were euthanized by neck dislocation. Finally, blood and
liver tissue obtained from the mice were stored at -80˚C for
further analysis.
Pathological liver examination
At 17 weeks old, the OM-O and CM-O mice were
sacrificed, the liver was quickly resected, a small piece of fresh
liver tissue was taken, and the liver specimens were fixed with 4%
paraformaldehyde at 4°C for 24 h. They were then
prepared and embedded in a paraffin block and 4-µm thick sections
were cut .and used for regular hematoxylin-eosin (each 10 min at
room temperature), oil red O staining (8 min at room temperature)
and Masson staining (ponceau staining for 8 min and aniline blue
for 15 min at room temperature) to analyze liver tissue changes
under confocal microscopy (magnification, x200).
Immunohistochemistry
The liver was harvested, and representative sections
were fixed in 4% paraformaldehyde (Huadong Medicine Co., Ltd.) at
room temperature for 24 h. Liver sections from OM-O and CM-O mice
were then embedded in paraffin and cut into 4 µm sections. The
paraffin sections were then dewaxed and hydrated at room
temperature by successively adding xylene Ⅰ (15 min), xylene II (15
min), anhydrous ethanol Ⅰ (5 min) and anhydrous ethanol II (5 min).
The slices were subsequently removed and placed in a ventilated
laboratory to dry and then washed in distilled water. At
100°C, EDTA antigen repair buffer (PH8.0; cat. no.
P0086; Beyotime Institute of Biotechnology) was used for antigen
repair. Hydrogen peroxide (3%) was added and incubated at room
temperature for 10 min to inactivate the activity of endogenous
enzymes. Finally, 2% bovine serum albumin (cat. no. 37520 Thermo
Fisher Scientific, Inc.) was added and incubated for 10 min at room
temperature. Primary antibodies (all ABclonal Biotech Co., Ltd.)
against alpha-smooth muscle actin (α-sma) (cat. no. A7240),
transforming growth factor beta1 (TGF-β1) (cat. no. A2124), IL-6
(cat. no. A0286) and their corresponding biotinylated secondary
antibodies (HRP Goat Anti-Rabbit IgG; cat. no. AS014; ABclonal)
were added to the sections. All antibodies were diluted at 1:500.
DAB (cat. no. RC062; Shanghai Recordbio Biological Technology Co.,
Ltd.) color development was performed and the slides coverslipped.
Sections were assessed and imaged under confocal microscopy
(magnification, x200). Quantitative analysis was then performed
using Image J software (1.52V; National Institutes of Health).
Biochemical index detection
Blood samples (1 ml) were collected from the
eyeballs of 10 mice from the OM-O and CM-O groups at 17 weeks of
age. Whole blood was placed at 4˚C, centrifuged at 2,200 x g for 10
min at 4˚C and the supernatant was collected. Total cholesterol
(TC) assay kit (cat. no. A111-1-1; Nanjing Jiancheng Biological
Co., Ltd.), triglycerides (TG) assay kit (cat. no. A110-1-1;
Nanjing Jiancheng Biological Co., Ltd.), high- density lipoprotein
cholesterol (HDL) assay kit (cat. no. A112-1-1; Nanjing Jiancheng
Biological Co., Ltd.), low-density lipoprotein cholesterol (LDL)
assay kit (cat. no. A113-1-1; Nanjing Jiancheng Biological Co.,
Ltd.) and interleukin-6 (IL-6) Elisa kit (cat. no. A007-1-2;
Nanjing Jiancheng Biological Co., Ltd.) were detected according to
the manufacturer's instructions.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from the liver with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific
Inc.) and reverse transcribed to cDNA using the HiFiScript gDNA
Removal cDNA Synthesis kit (CoWin Biosciences) according to the
manufacturer's protocol. The expression of Beclin-1, ATG3, ATG5,
ATG12, P62 and LC3B were detected with SYBR Green mix (BioTek
China) using the Thermo Fisher-QS3 (Thermo Fisher Scientific Inc.).
β-actin was used as an internal control. The thermocycling
conditions were as follows: Pre-denaturation at 94°C for
5 min; followed by 40 cycles of denaturation at 94°C for
30 sec, annealing at 60°C for 30 sec and extension at
72°C for 30 sec. The primer sequences used in this study
are summarized in Table I. The
relative mRNA expression was measured using the 2-ΔΔCq
method (21).
 | Table ISequences of primers used for
RT-qPCR. |
Table I
Sequences of primers used for
RT-qPCR.
| Gene | Forward
(5'-3') | Reverse
(5'-3') |
|---|
| ATG5 |
GCAGAATGACAGATTTGACCAGTTT |
GGTTTCCAGCATTGGCTCTATC |
| ATG12 |
TCGGTGCTGTGGGAAGAG |
GTGCCAACCAAGTAAATGC |
| ATG3 |
GAAGAAGATGATGGTGATGGGGG |
TTCCTCGTCTTCTTCATCACACA |
| Beclin-1 |
GAGTGGAATGAAATCAATGCTGC |
TTTCCACCTCTTCTTTGAACTGC |
| LC3 |
CCACACCCATCGCTGAC |
AAGGTTTCTTGGGAGGCGTAG |
| P62 |
TGCTCCACCAGAAGATCCC |
CGGCTTCTCTTCCCTCCATGT |
| β-actin |
CTAGGCACCAGGGTGTGATG |
CTCATTGTAGAAGGTGTGGTGC |
Western blotting
Total protein was extracted from the liver tissue of
OM-O and CM-O mice for western blotting. A protease phosphate
inhibitor (cat. no. P1045; Beyotime Institute of Biotechnology) and
Phenylmethanesulfonyl fluoride (cat. no. ST506; Beyotime Institute
of Biotechnology) was added to RIPA lysis buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology) to extract total proteins from
the liver tissues of the OM-O and CM-O groups. The bicinchoninic
acid (cat. no. P0010S; Beyotime Institute of Biotechnology) protein
determination kit was used to measure total protein concentration.
The protein samples were adjusted to the same concentration and
then mixed with 1/4 volume of protein loading buffer, boiled and
denatured. The total protein content (2 µg) was then separated by
10% SDS polyacrylamide gel electrophoresis and transferred to a
PVDF membrane. The membrane was blocked using 5% skimmed milk
powder at room temperature for 2 h and the corresponding antibody
stock solution (1:1,000) was diluted. Primary antibodies (all
ABclonal Biotech Co., Ltd.) against Beclin-1 (cat. no. A10101),
ATG3 (cat. no. A5809), ATG5 (cat. no. A0203), ATG12 (cat. no.
A19610), p62 (cat. no. A7758), LC3B (cat. no. A5618), adenosine
5'-monophosphate (AMP)-activated protein kinase (AMPK) (cat. no.
A1229), phosphorylated (p)-AMPK (cat. no. A1229), mammalian target
of rapamycin (mTOR) (cat. no. A2445), p-mTOR (cat. no. AP0115) and
α-sma (cat. no. A7240), TGF-β1 (cat. no. A2124), IL-6 (cat. no.
A0286) were added and the sections incubated overnight at 4˚C. The
following day, the membrane was washed three times in TBST
(TBS/0.1% Tween x10; cat. no. PS103, Epizyme) for 10 min each time
and the horseradish peroxidase-labeled secondary antibody (Goat
Anti-Rabbit IgG; cat. no. AS029; ABclonal, 1:1,000) was diluted
with 5% skimmed milk powder and incubated on a shaker for 90 min at
room temperature. After washing the membrane three times in TBST, a
luminescent liquid (ECL: cat.no. P0018S, Beyotime) was added and
exposed with the Bio-Rad imaging system. ImageJ software (National
Institutes of Health; 1.52 V) was used to determine the gray value.
β-actin (cat. no. AC038; ABclonal; 1:1,000) or tubulin (cat. no.
AC008; ABclonal; 1:1,000) were used as the loading controls and the
gray value of the target protein was compared with the gray value
of the internal control to determine relative expression
levels.
Statistical analysis
Data were presented as means ± standard error of the
mean (SEM). All the experiments were repeated 3 times. An unpaired
Student's t-test was used for comparisons between 2 groups.
Statistical analyses were performed using IBM SPSS Statistics 22
software (IBM Corp.) *P<0.05 were considered to
indicate a statistically significant difference.
Results
Maternal obesity promotes NAFLD in
mouse offspring
In the present study, 4-week-old female C57BL/6J
mice (n=20) were randomly selected and 1 group was fed a high-fat
diet (OM group, n=10), and the other group was fed a normal diet
(CM group n=10). After 3 weeks of feeding, the weight began to show
differences between the 2 groups and feeding was continued for 1
week. After 4 weeks of high-fat feeding, the body weight of the OM
group was significantly higher compared with that of the CM group
and the average weight of the OM group was greater compared with
the CM group (P<0.05; Fig. 1A).
At 8 weeks old, the female mice were mated with ordinary male mice
and the offspring named OM-O and CM-O, respectively. The results
demonstrated no difference in the body weight between these 2
groups at 4 weeks of weaning. After high-fat feeding for 12 weeks,
the body weight of the OM-O group was high, and after 17 weeks, the
weight of the OM-O group was significantly higher compared with
that of the CM-O group (P<0.05; Fig.
1B). In addition, serum TC and TG in the OM-O group were found
to be significantly higher compared with the CM-O group
(P<0.05), while HDL was significantly lower compared with the
CM-O group (P<0.05), while LDL had an upward trend in the OM-O
group compared with the CM-O group (Fig. 1C-F). Following HE staining of
offspring liver sections, it was identified that when compared with
the liver tissue of the CM-O group, the OM-O group exhibited
markedly higher fatty vacuolization (Fig. 1G). Observation of the fat content,
according to the degree of redness of the oil red O-stained tissue
sections, revealed that the fat content of the OM-O group was
significantly higher compared with the CM-O group (Fig. 1H).
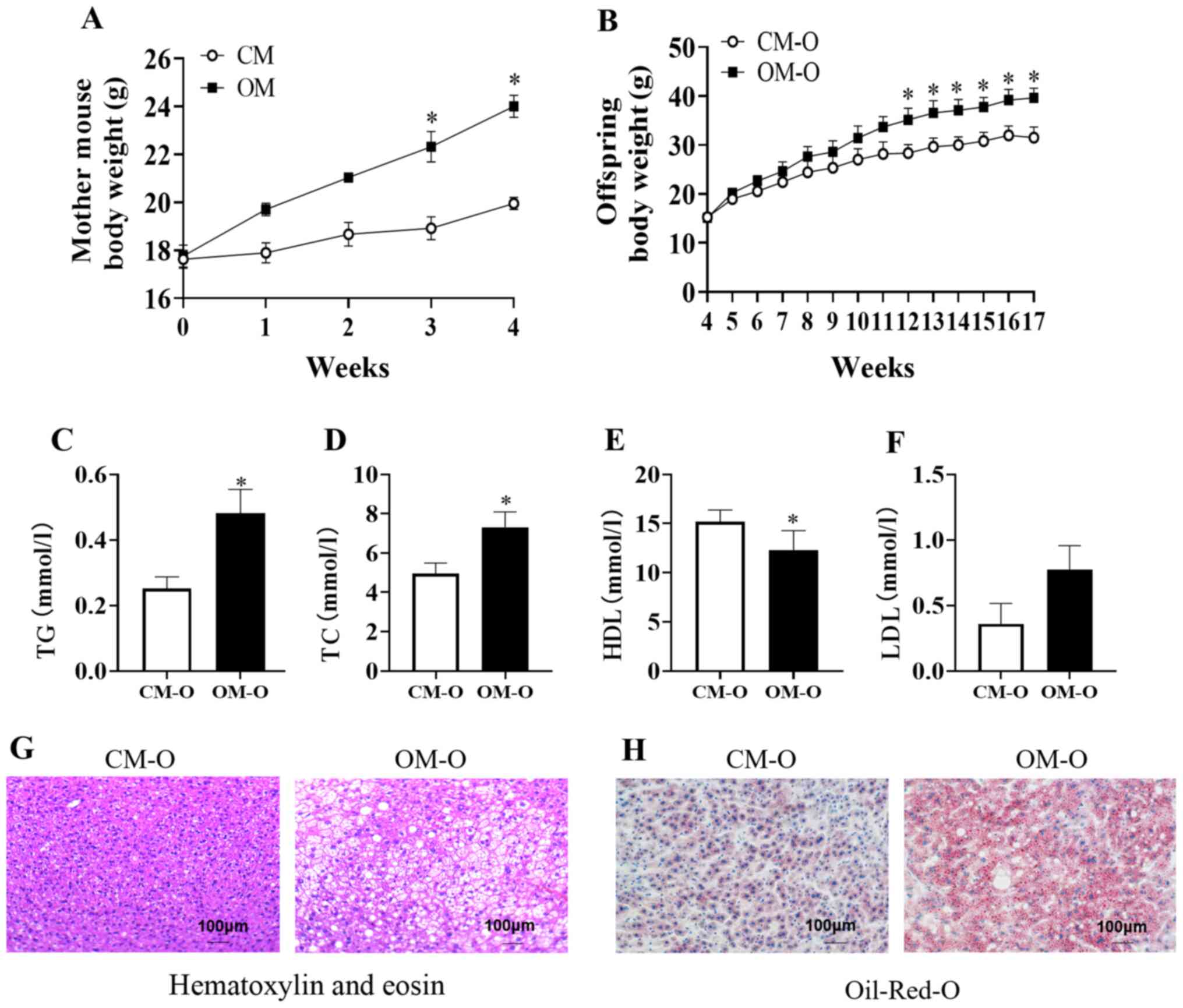 | Figure 1Body weight of mother and NAFLD in
mice offspring. (A) Body weight of mouse groups, CM and OM on
different diets (n=10/group). (B) Body weight of offspring of CM
and OM, CM-O and OM-O groups, respectively from 4-17 weeks
(n=10/group). (C-F) Serum TG, TC, LDL, HDL at 17 weeks of offspring
mice (n=6/group). (G and H) Hematoxylin-eosin and Oil-Red-O
staining of liver sections from CM-O and OM-O groups at 17 weeks
(n=3/group; magnification, x100). Data are shown as mean ± SEM.
*P<0.05. NAFLD, non-alcoholic fatty liver disease;
OM, obese mother; CM, chow mother; OM-O, offspring of obese mother;
CM-O, offspring of chow mother; TC, total cholesterol; TG,
triglycerides; HDL, high-density lipoprotein cholesterol; LDL,
low-density lipoprotein cholesterol. |
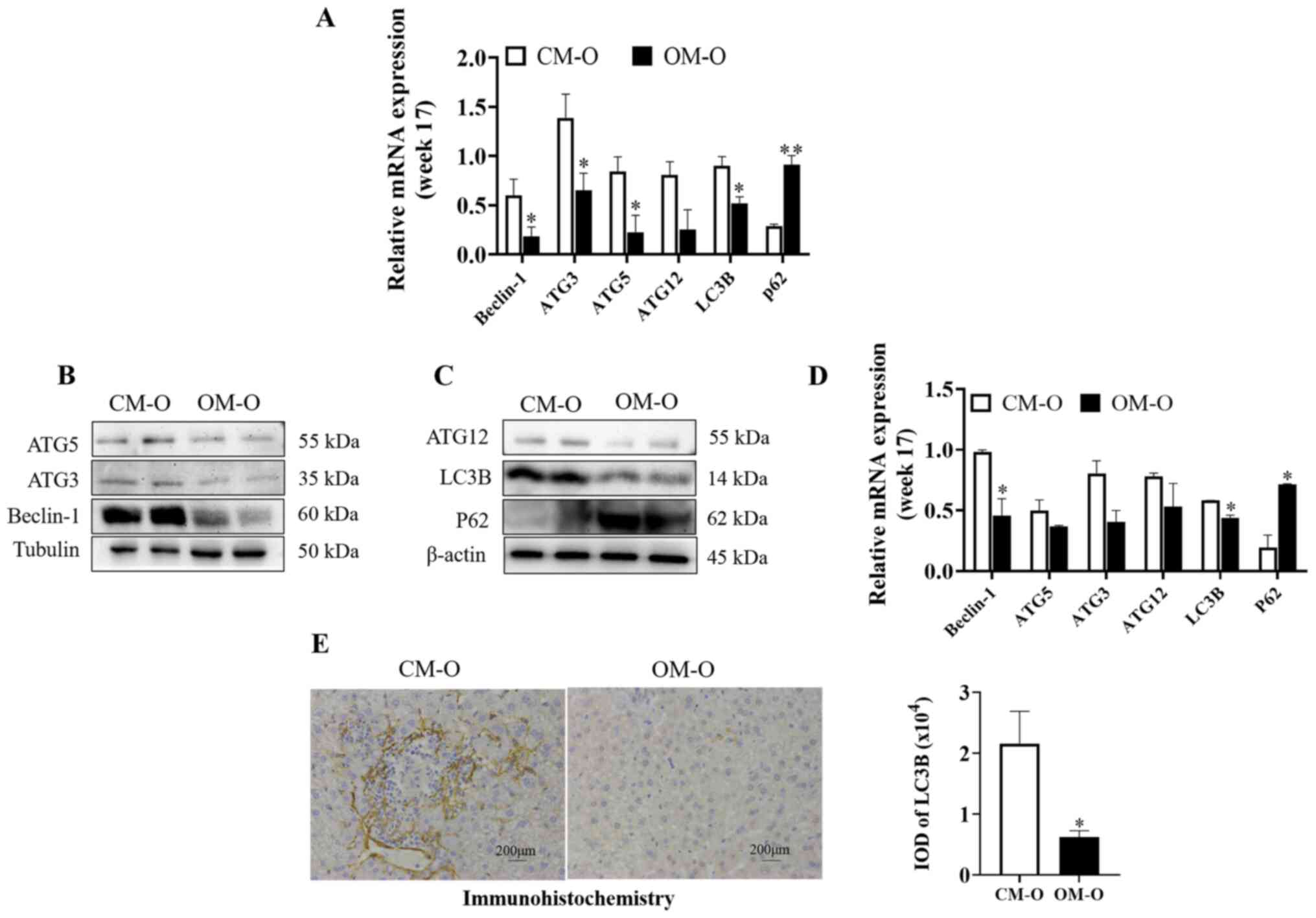
Maternal obesity promotes the decrease
of the liver autophagy level in offspring
It was hypothesized that maternal obesity may
promote NAFLD in offspring by autophagy regulation. Previous
studies have suggested that Beclin-1, Atg3, Atg5 and Atg12 are
components of autophagy mechanism, and LC3b is a marker gene of
autophagy (22,23). Hence, the present study investigated
autophagy related genes by RT-qPCR. At 17 weeks, Beclin-1, ATG3,
ATG5, ATG12, and LC3B levels were significantly lower in the OM-O
group compared with the CM-O group, while P62 showed the opposite
trend (P<0.05) (Fig. 2A). It was
also revealed that there were significant differences in Beclin-1,
LC3B and P62 between the two groups (P<0.05) (Fig. 2B-D). Furthermore, ATG3, ATG5 and
ATG12 levels were decreased. By immunohistochemical detection of
the expression level of LC3B, the same result was observed
(P<0.05) (Fig. 2E). These
results indicated that the level of autophagy in the liver of the
offspring of the obese mice was decreased.
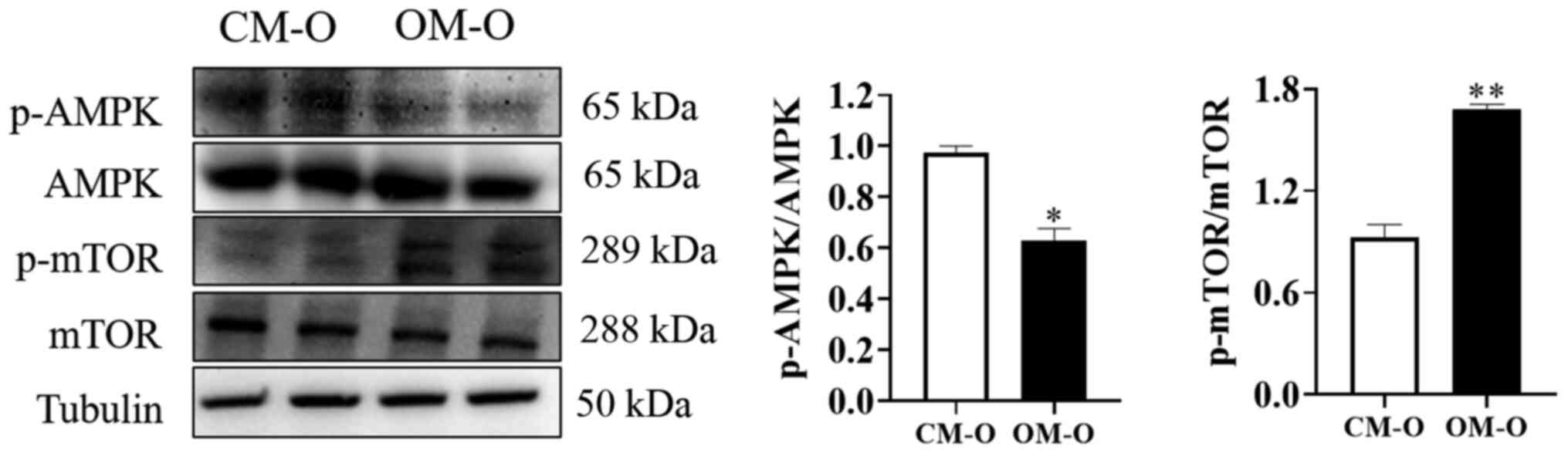
Maternal obesity promotes the decrease
of AMPK phosphorylation levels and the increase of mTOR
phosphorylation levels in offspring livers
AMPK is a key energy sensor that regulates cellular
metabolism to maintain energy homeostasis. Additionally, it is
known that autophagy is inhibited by mTOR (24,25).
AMPK and its downstream site changes in mTOR phosphorylation were
therefore investigated after feeding mice for 17 weeks. The results
of showed that the total AMPK and mTOR protein levels remained
unchanged in both CM-O and OM-O groups. However, compared with
CM-O, AMPK phosphorylation levels were decreased and mTOR
phosphorylation levels were increased in OM-O (P<0.05; Fig. 3). These results indicated that the
formation of NAFLD in the offspring of obese female mice may be
related to changes in AMPK/mTOR phosphorylation.
Maternal obesity promotes liver
fibrosis in offspring mice
Masson staining revealed that the level of fibrosis
in the OM-O group was significantly higher compared with CM-O group
(P<0.05; Fig. 4A). In order to
verify the association between the occurrence of NAFLD in the
offspring and any changes in liver fibrosis, an ELISA kit was used
to determine the serum IL-6 level in the mice offspring at 17
weeks. α-sma and TGF-β1 lead to liver fibrosis, which further leads
to changes in NAFLD. α-sma and TGF-β1 were chosen as liver fibrosis
markers in the present study (26)
and western blotting and immunohistochemistry to investigate their
expression was performed. The results demonstrated that α-sma and
TGF-β1 expression in the OM-O group at 17 weeks were
significantly higher compared with the CM-O group (P<0.05;
(Fig. 4B and C). A significant increase in IL-6 levels
at 17 weeks was also observed in the OM-O group when compared with
the CM-O group (P<0.05; Fig.
4D). In addition, the results of western blotting demonstrated
that there was significantly higher IL-6 and TGF-β1 in the liver in
OM-O group compared with the CM-O group and that α-sma expression
significantly increased in the OM-O group compared with the CM-O
group (P<0.05) (Fig. 4E and
G). These results indicated that
while maternal obesity led to the development of NAFLD in the
offspring, it was accompanied by an increase in liver fibrosis.
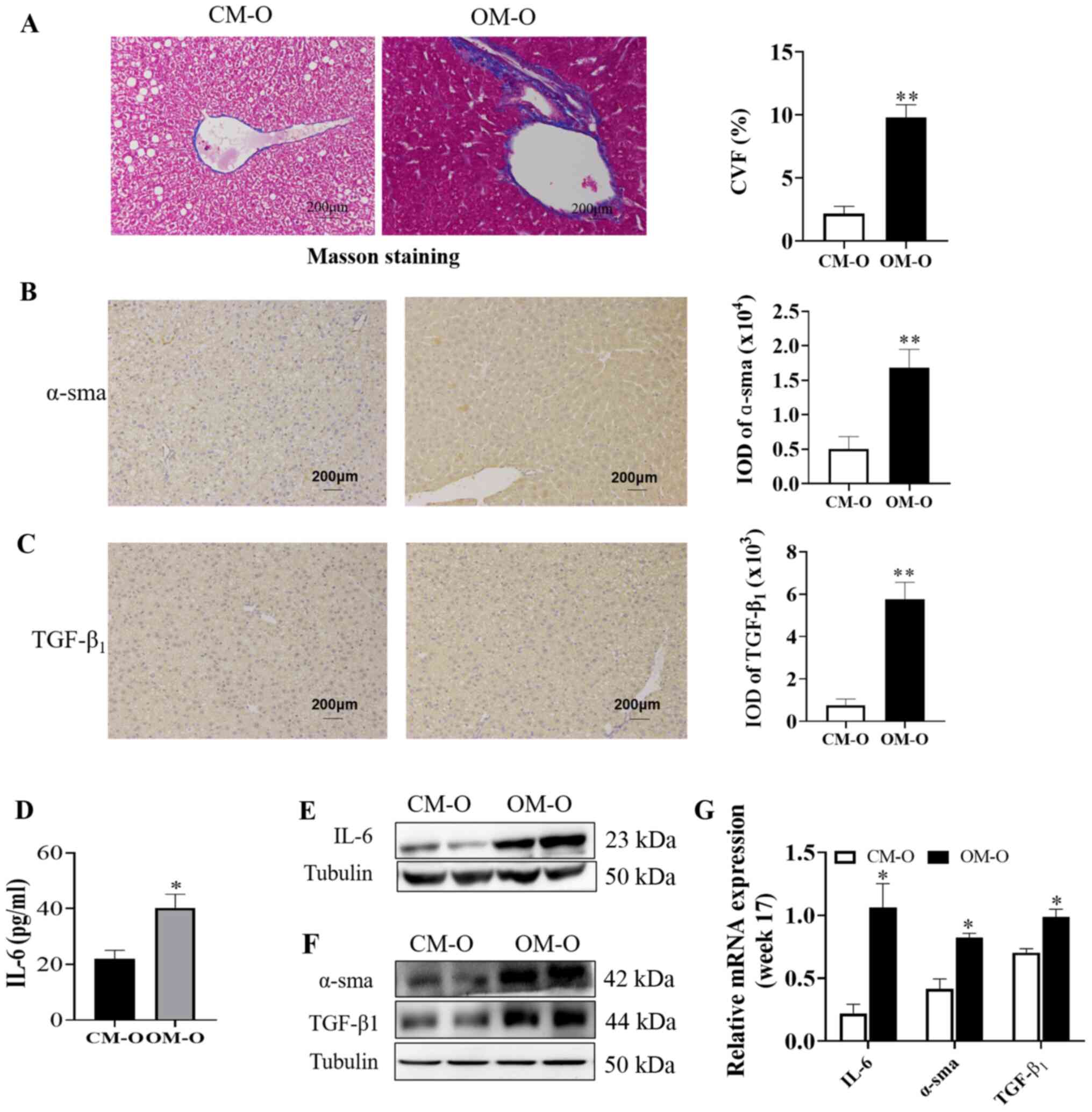 | Figure 4NAFLD is associated with liver
fibrosis. (A) Representative image of Masson staining in liver
sections (n=3/group; magnification, x200). (B and C) Representative
pictures of α-sma and TGF-β1 immunohistochemical staining,
respectively in liver are shown (n=3/group; magnification, x200).
(D) Serum IL-6 was detected in offspring at 17 weeks (n=6/group).
(E and F) Representative blots of IL-6, α-sma and TGF-β1 expression
in offspring mice liver are shown (n=3/group). (G) Expression of
IL-6, α-sma and TGF-β1 in 17 weeks old offspring by WB (n=3/group).
*P<0.05, **P<0.01. NAFLD, non-alcoholic
fatty liver disease; CM-O, offspring of chow mother; OM-O,
offspring of obese mother; IL-6, interleukin-6; α-sma, alpha-smooth
muscle actin; TGF-β1, transforming growth factor beta 1; IOD,
integral optical density; CVF, Collagenvolume fraction. |
Discussion
NAFLD global prevalence is increasing year on year
and it is widely known that maternal obesity is closely related to
the occurrence of NAFLD in the offspring (27). Cantoral et al (28) found that maternal overweight before
pregnancy was closely related to liver fat content of offspring in
a cohort study. Another study demonstrated that intervention of
maternal rats with high-fat diet during pregnancy or early
pregnancy can lead to severe liver steatosis and NAFLD risk in
offspring (29). In the present
study, an obese female mouse model was established via a high-fat
diet and it was found that the mice of the OM-O group were more
likely to form NAFLD than the CM-O group. By investigation of
biochemical indicators and tissue sections of the offspring in the
present study, it was revealed that TC, TG and LDL of the OM-O
group increased, while the HDL levels showed a downward trend
compared with the CM-O group. Notably, the present study by
performing HE and oil red staining demonstrated that the OM-O group
had more lipid droplets than the CM-O group. Previous
epidemiological studies have reported that maternal obesity is an
independent risk factor for the development of NAFLD in offspring
(27,30,31).
However, the present study did not provide sufficient data to prove
that the degree of maternal obesity was positively associated with
the severity of NAFLD in the offspring. It is likely that this may
be caused by some uncontrollable factors regarding the maternal
generation in epidemiological studies, such as dietary structure,
circadian rhythm etc. The findings of the present study
demonstrated that maternal obesity is one of the susceptibility
factors for NAFLD in the offspring, but it cannot yet be stated
definitively that the higher the obesity of the maternal
generation, the more severe the occurrence of NAFLD in the
offspring. However, the results of the present study are sufficient
to provide a warning to mothers that obesity serves an important
role in the occurrence and exacerbation of NAFLD in the
offspring.
A study demonstrated that autophagy may serve a role
in NAFLD formation in offspring (32). Lipophagy (a type of selective
autophagy) mainly involves the selective degradation of cytoplasmic
lipid droplets, so hepatocyte autophagy is currently considered a
defense mechanism in the prevention of NAFLD (33). On the contrary, a study has
demonstrated that excessive triglycerides and free fatty acids in
NAFLD caused lipid effects, such as insulin resistance and
oxidative stress to inhibit autophagy activity and then a decrease
in autophagy activity further aggravates the production of NAFLD
(34). Yet other studies have
reported that using rapamycin and carbamazepine promotes autophagy
to reduce NAFLD occurrence (35,36).
The present study demonstrated that beclin-1, ATG3, ATG5, ATG12 and
LC3B expression in OM-O livers was lower than mice of the CM-O
group, while the expression of P62 was increased, which indicated
that OM-O livers were inhibited during this period. In the present
study, the livers of the OM-O group had severe lipid droplet
deposition and a large increase in serum IL-6, fully indicating
that the decrease in autophagy was aggravated to some extent, which
was confirmed by previous studies (37-39).
Liu et al (40) found that
in SD rats fed a high-fat diet, the early stage of steatosis
revealed higher levels of LC3 II/I, lower p62 and higher fat
cholesterol in the late stage, suggesting that autophagy is usually
activated in the early stages of steatosis, but then blocked in at
a late stage. The aforementioned results indicated that autophagy
is more likely to be blocked in the late obese generation, and the
decrease of autophagy level is more likely to increase the
susceptibility to NAFLD in the offspring of an obese mother.
The present study also demonstrated that the liver
NAFLD formation induced by maternal obesity may be related to the
changes of autophagy upstream signal AMPK and mTOR. Recently, a
liver-specific genetic study confirmed the important role of AMPK
in HFD induced hepatic steatosis in NAFLD mice (41). While, a large number of studies have
also shown that the activation of AMPK signal pathway can reduce
the deposition of lipid droplets and the occurrence of NAFLD. Zhou
et al (42) found that a low
dose of aspirin may reduce lipid droplet deposition via AMPK
activation in the offspring of obese mothers. Devarajan et
al (32) also reported that the
occurrence of NAFLD in offspring with energy limitation during the
perinatal period was related to AMPK changes. AMPK is the main
positive regulator of autophagy and mTOR is the key negative
regulator of autophagy (43).
Soto-Avellaneda et al (44)
demonstrated that statins can alter the synthesis of cholesterol
free fatty acids by inhibiting the activation of mTOR site and
autophagy, hence reducing the deposition of lipids in non-adipose
tissues, such as the liver. The aforementioned results were
consistent with the findings of the present study and that the
change of AMPK-mTOR pathway may promote the formation of NAFLD in
obese children.
NAFLD is a type of systemic liver disease, which
ranges from simple steatosis to steatohepatitis, and can progress
to fibrosis, cirrhosis, and finally liver cancer in some cases
(45). Previous studies have
revealed that the inflammatory factor, IL-6 and fibrosis
indicators, TGF-β1 and α-sma, are highly expressed in NAFLD
(46-48).
In the process of liver fibrosis, apart from TGF-β1 and α-sma, as
an important indicator of liver fibrosis, serum IL-6 also has been
widely reported (49). Wei et
al (50) established a rat
model of liver fiber by carbon tetrachloride. Through the detection
of serum IL-6, it was found that the content of serum IL-6 in the
CCL4 group was significantly higher compared with that in the wild
group, but following the addition of plumbagin, the hepatic
fibrosis induced by CCL4 was significantly improved and serum IL-6
was significantly decreased (50).
A study by Al-Hashem et al (51) treated mice with hepatic fibrosis
with metformin and it was revealed that the liver fibrosis of mice
treated with metformin was significantly improved and the level of
serum IL-6 was significantly decreased. A large number of clinical
studies have found that compared with healthy subjects, the content
of serum IL-6 in patients with NAFLD is significantly increased
(52,53). Hence, it was hypothesized that the
level of IL-6 may be used as an important biological indicator of
liver fibrosis. In liver fibrosis, IL-6 is not studied as a single
indicator (54), hence in the
present study liver α-SMA and TGF-β1 were studied as well. In the
present study, in order to verify whether maternal obesity is
responsible for fibrosis change in the offspring, ELISA and western
blotting was used to measure IL-6, α-SMA and TGF-β1 in the
offspring and it was demonstrated that with the continuous
development of NAFLD at 17 weeks, the expression levels of IL-6,
TGF-β1, and α-sma in the livers of the OM-O group was significantly
higher than CM-O mice. Mouralidarane et al (55) published similar findings regarding
liver fibrosis in the offspring of obese female mice at different
time points.
In addition, there is growing evidence that innate
immunity is the driving force behind the progression of NAFLD
(56,57). As an important immune organ, the
liver contains a coordinated network of innate immune cells, such
as Kupffer cell (KCs), dendritic cell (DC), lymphocytes,
neutrophils and mast cells. Mouralidarane et al established
an obese maternal mouse model to induce hyperlipidemia in their
offspring during pregnancy and found that the liver phenotype of
their offspring deteriorated significantly in adulthood. In
addition, it was also found the number of KCs in the innate immune
system of the liver increased but its cell phagocytosis decreased,
is not conducive to liver lipid clearance (55). KCs can release a large number of
cytokines and chemokines, such as tumor necrosis factor (TNF)-α,
IL-6 etc. further promoting hepatocyte production and complement
protein release and a large number of complement and inflammatory
factors lead to liver fat deformation and inflammatory changes
(58). The expression of
inflammatory cytokines IL-6, TNF-α and TGF-β1 has also been
demonstrated to change with the regulation of the immune system
(59-61).
Based on the finding of previous studies, it was hypothesized that
innate immunity serves an important role in the formation and
development of NAFLD, while inflammatory factors may be regulated
by the innate immune response, hence playing an important role in
the development of disease. The findings of previous studies
indicated that the occurrence of NAFLD susceptibility in the
offspring due to maternal obesity may be related to innate
immunity. Future studies need to study this in more detail.
The study has certain limitations. Only the changes
of the OM-O and CM-O groups were observed at 17 weeks, meaning that
the changes of offspring mice beyond this time was not assessed. At
the same time, with regard to the changes of liver inflammation in
mice, only the changes of IL-6 were assessed, despite there being
other inflammatory factors that may serve a role. Future studies
should assess the relationship between autophagy and NAFLD in obese
offspring at different times,and elucidate the role of different
inflammatory factors in NAFLD obese offspring.
In conclusion, the present study revealed the
phenomenon of altered autophagy in a NAFLD mouse model of offspring
caused by maternal obesity and in addition, demonstrated the role
of AMPK/mTOR signal in this process. Changes in autophagy may serve
a key role in the development of maternal obesity and NAFLD in
offspring, which may lead to the early prevention of NAFLD.
Autophagy may be used as a new target to prevent maternal obesity
causing NAFLD in the offspring.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by Natural Science Foundation
of Zhejiang Province (grant no. LY18H040014) and Natural Science
Foundation of Anhui Province (grant no. KJ2019A0353).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YT and GJ designed the study. SH, FZ, XH, PY, FS and
KX performed the experiments. SH, FZ, JS and ZY analyzed the data.
SH and FZ wrote the manuscript. SH and FZ confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal
Experiments and Animal Experiment Ethics Committee of The Second
Affiliated Hospital of Jiaxing University, Jiaxing Second Hospital
(Jiaxing, China; approval no. jxey2017002).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Byrne CD and Targher G: NAFLD: A
multisystem disease. J Hepatol. 62 (Suppl 1):S47–S64.
2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fan JG, Kim SU and Wong VJJ: New trends on
obesity and NAFLD in asia. J Hepatol. 67:862–873. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Estes C, Anstee QM, Arias-Loste MT, Bantel
H, Bellentani S, Caballeria J, Colombo M, Craxi A, Crespo J, Day
CP, et al: Modeling NAFLD disease burden in China, France, Germany,
Italy, Japan, Spain, United Kingdom, and United States for the
period 2016-2030. J Hepatol. 69:896–904. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ayonrinde OT, Adams LA, Mori TA, Beilin
LJ, de Klerk N, Pennell CE, White S and Olynyk JK: Sex differences
between parental pregnancy characteristics and nonalcoholic fatty
liver disease in adolescents. Hepatology. 67:108–122.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Patel S, Lawlor DA, Callaway M,
Macdonald-Wallis C, Sattar N and Fraser A: Association of maternal
diabetes/glycosuria and pre-pregnancy body mass index with
offspring indicators of non-alcoholic fatty liver disease. BMC
Pediatr. 16(47)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Pereira TJ, Fonseca MA, Campbell KE, Moyce
BL, Cole LK, Hatch GM, Doucette CA, Klein J, Aliani M and Dolinsky
VW: Maternal obesity characterized by gestational diabetes
increases the susceptibility of rat offspring to hepatic steatosis
via a disrupted liver metabolome. J Physiol. 15:3181–3197.
2015.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Bouanane S, Merzouk H, Benkalfat NB,
Soulimane N, Merzouk SA, Gresti J, Tessier C and Narce M: Hepatic
and very low-density lipoprotein fatty acids in obese offspring of
overfed dams. Metabolism. 59:1701–1709. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Liou CJ, Wu SJ, Shen SC, Chen LC, Chen YL
and Huang WC: Phloretin ameliorates hepatic steatosis through
regulation of lipogenesis and Sirt1/AMPK signaling in obese mice.
Cell Biosci. 10(114)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wilson RB, Chen YJ, Sutherland BG, Sawyez
CG, Zhang R, Woolnough T, Hetherington AM, Peters KM, Patel K,
Kennelly JP, et al: The marine compound and elongation factor 1A1
inhibitor, didemnin B, provides benefit in western diet-induced
non-alcoholic fatty liver disease. Pharmacol Res.
161(105208)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Borengasser SJ, Lau F, Kang P, Blackburn
ML, Ronis MJ, Badger TM and Shankar K: Maternal obesity during
gestation impairs fatty acid oxidation and mitochondrial SIRT3
expression in rat offspring at weaning. PLoS One.
6(e24068)2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wankhade UD, Zhong Y, Kang P, Alfaro M,
Chintapalli SV, Thakali KM and Shankar K: Enhanced offspring
predisposition to steatohepatitis with maternal high-fat diet is
associated with epigenetic and microbiome alterations. PLoS One.
12(e0175675)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yamaguchi R, Nakagawa Y, Liu YJ, Fujisawa
Y, Sai S, Nagata E, Sano S, Satake E, Matsushita R, Nakanishi T, et
al: Effects of maternal high-fat diet on serum lipid concentration
and expression of peroxisomal proliferator-activated receptors in
the early life of rat offspring. Horm Metab Res. 42:821–825.
2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Paul HA, Bomhof MR, Vogel HJ and Reimer
RA: Diet-induced changes in maternal gut microbiota and metabolomic
profiles influence programming of offspring obesity risk in rats.
Sci Rep. 6(20683)2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Settembre C, De Cegli R, Mansueto G, Saha
PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch
TJ, et al: TFEB controls cellular lipid metabolism through a
starvation-induced autoregulatory loop. Nat Cell Biol. 15:647–658.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Zhang H, Yan S, Khambu B, Ma F, Li Y, Chen
X, Martina JA, Puertollano R, Li Y, Chalasani N and Yin XM: Dynamic
MTORC1-TFEB feedback signaling regulates hepatic autophagy,
steatosis and liver injury in long-term nutrient oversupply.
Autophagy. 14:1779–1795. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li S, Dou X, Ning H, Song Q, Wei W, Zhang
X, Shen C, Li J, Sun C and Song Z: Sirtuin 3 acts as a negative
regulator of autophagy dictating hepatocyte susceptibility to
lipotoxicity. Hepatology. 66:936–952. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chao X, Wang H, Jaeschke H and Ding WX:
Role and mechanisms of autophagy in acetaminophen-induced liver
injury. Liver Int. 38:1363–1374. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chu Q, Zhang S, Chen M, Han W, Jia R, Chen
W and Zheng X: Cherry anthocyanins regulate NAFLD by promoting
autophagy pathway. Oxid Med Cell Longev.
2019(4825949)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Warnes G: Flow cytometric assays for the
study of autophagy. Methods. 82:21–28. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
Jia J, Abudu YP, Claude-Taupin A, Gu Y,
Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al:
Galectins control MTOR and AMPK in response to lysosomal damage to
induce autophagy. Autophagy. 15:169–171. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhou Y, Wu R, Cai FF, Zhou WJ, Lu YY,
Zhang H, Chen QL and Su SB: Xiaoyaosan decoction alleviated rat
liver fibrosis via the TGFβ/Smad and Akt/FoxO3 signaling pathways
based on network pharmacology analysis. J Ethnopharmacol.
264(113021)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ayonrinde OT, Oddy WH, Adams LA, Mori TA,
Beilin LJ, de Klerk N and Olynyk JK: Infant nutrition and maternal
obesity influence the risk of non-alcoholic fatty liver disease in
adolescents. J Hepatol. 67:568–576. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Cantoral A, Montoya A, Luna-Villa L,
Roldán-Valadez EA, Hernández-Ávila M, Kershenobich D, Perng W,
Peterson KE, Hu H, Rivera JA and Téllez-Rojo MM: Overweight and
obesity status from the prenatal period to adolescence and its
association with non-alcoholic fatty liver disease in young adults:
Cohort study. BJOG. 127:1200–1209. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhou Y, Peng H, Xu H, Li J, Golovko M,
Cheng H, Lynch EC, Liu L, McCauley N, Kennedy L, et al: Maternal
diet intervention before pregnancy primes offspring lipid
metabolism in liver. Lab Invest. 100:553–569. 2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Alisi A and Vajro P: Pre-natal and
post-natal environment monitoring to prevent non-alcoholic fatty
liver disease development. J Hepatol. 67:451–453. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Simpson J, Smith AD, Fraser A, Sattar N,
Callaway M, Lindsay RS, Lawlor DA and Nelson SM: Cord blood
adipokines and lipids and adolescent nonalcoholic fatty liver
disease. J Clin Endocrinol Metab. 101:4661–4668. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Devarajan A, Rajasekaran NS, Valburg C,
Ganapathy E, Bindra S and Freije WA: Maternal perinatal calorie
restriction temporally regulates the hepatic autophagy and redox
status in male rat. Free Radic Biol Med. 130:592–600.
2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Martinez-Lopez N and Singh R: Autophagy
and lipid droplets in the liver. Annual Rev Nutr. 35:215–237.
2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
González-Rodríguez A, Mayoral R, Agra N,
Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J,
Lo Iacono O, Corazzari M, Fimia GM, et al: Impaired autophagic flux
is associated with increased endoplasmic reticulum stress during
the development of NAFLD. Cell Death Dis. 5(e1179)2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lin CW, Zhang H, Li M, Xiong X, Chen X,
Chen X, Dong XC and Yin XM: Pharmacological promotion of autophagy
alleviates steatosis and injury in alcoholic and non-alcoholic
fatty liver conditions in mice. J Hepatol. 58:993–999.
2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Guo R, Nair S, Zhang Y and Ren J:
Adiponectin deficiency rescues high-fat diet-induced hepatic
injury, apoptosis and autophagy loss despite persistent steatosis.
Int J Obes (Lond). 41:1403–1412. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lu W, Mei J, Yang J, Wu Z, Liu J, Miao P,
Chen Y, Wen Z, Zhao Z, Kong H, et al: ApoE deficiency promotes
non-alcoholic fatty liver disease in mice via impeding AMPK/mTOR
mediated autophagy. Life Sci. 252(117601)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lee DH, Park SH, Ahn J, Hong SP, Lee E,
Jang YJ, Ha TY, Huh YH, Ha SY, Jeon TI and Jung CH: Mir214-3p and
Hnf4a/Hnf4α reciprocally regulate Ulk1 expression and autophagy in
nonalcoholic hepatic steatosis. Autophagy, 2020 (Epub ahead of
print).
|
|
40
|
Liu C, Liu L, Zhu HD, Sheng JQ, Wu XL, He
XX, Tian DA, Liao JZ and Li PY: Celecoxib alleviates nonalcoholic
fatty liver disease by restoring autophagic flux. Sci Rep.
8(4108)2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Garcia D, Hellberg K, Chaix A, Wallace M,
Herzig S, Badur MG, Lin T, Shokhirev MN, Pinto AFM, Ross DS, et al:
Genetic liver-specific AMPK activation protects against
diet-induced obesity and NAFLD. Cell Rep. 26:192–208.e6.
2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhou Y, Peng H, Liu Z, Zhang KK, Jendrusch
C, Drake M, Hao Y and Xie L: Sex-associated preventive effects of
low-dose aspirin on obesity and non-alcoholic fatty liver disease
in mouse offspring with over-nutrition in utero. Lab Invest.
99:244–259. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Inoki K, Kim J and Guan KL: AMPK and mTOR
in cellular energy homeostasis and drug targets. Annu Rev Pharmacol
Toxicol. 52:381–400. 2012.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Soto-Avellaneda A and Morrison BE:
Signaling and other functions of lipids in autophagy: A review.
Lipids Health Dis. 19(214)2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Rinella ME and Sanyal AJ: Management of
NAFLD: A stage-based approach. Nat Rev Gastroenterol Hepatol.
13:196–205. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fang C, Cai X, Hayashi S, Hao S, Sakiyama
H, Wang X, Yang Q, Akira S, Nishiguchi S, Fujiwara N, et al:
Caffeine-stimulated muscle IL-6 mediates alleviation of
non-alcoholic fatty liver disease. Biochim Biophys Acta Mol Cell
Biol Lipids. 1864:271–280. 2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hart KM, Fabre T, Sciurba JC, Gieseck RL,
Borthwick LA, Vannella KM, Acciani TH, de Queiroz Prado R, Thompson
RW, White S, et al: Type 2 immunity is protective in metabolic
disease but exacerbates NAFLD collaboratively with TGF-β. Sci
Transl Med. 9(eaal3694)2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Chen P, Luo Q, Huang C, Gao Q, Li L, Chen
J, Chen B, Liu W, Zeng W and Chen Z: Pathogenesis of non-alcoholic
fatty liver disease mediated by YAP. Hepatol Int. 12:26–36.
2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Stojsavljević S, Gomerčić Palčić M,
Virović Jukić L, Smirčić Duvnjak L and Duvnjak M: Adipokines and
proinflammatory cytokines, the key mediators in the pathogenesis of
nonalcoholic fatty liver disease. World J Gastroenterol.
20:18070–18091. 2014.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Wei Y, Huang M, Liu X, Yuan Z, Peng Y,
Huang Z and Zhao T: Anti-fibrotic effect of plumbagin on
CCl4-lesioned rats. Cell Physiol Biochem. 35:1599–1608.
2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Al-Hashem F, Al-Humayed S, Amin SN, Kamar
SS, Mansy SS, Hassan S, Abdel-Salam LO, Ellatif MA, Alfaifi M,
Haidara MA and Al-Ani B: Metformin inhibits mTOR-HIF-1α axis and
profibrogenic and inflammatory biomarkers in thioacetamide-induced
hepatic tissue alterations. J Cell Physiol. 234:9328–9337.
2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Haukeland JW, Damås JK, Konopski Z, Løberg
EM, Haaland T, Goverud I, Torjesen PA, Birkeland K, Bjøro K and
Aukrust P: Systemic inflammation in nonalcoholic fatty liver
disease is characterized by elevated levels of CCL2. J Hepato.
44:1167–1174. 2006.PubMed/NCBI View Article : Google Scholar
|
|
53
|
García-Galiano D, Sánchez-Garrido MA,
Espejo I, Montero JL, Costán G, Marchal T, Membrives A,
Gallardo-Valverde JM, Muñoz-Castañeda JR, Arévalo E, et al: IL-6
and IGF-1 are independent prognostic factors of liver steatosis and
non-alcoholic steatohepatitis in morbidly obese patients. Obes
Surg. 17:493–503. 2007.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Mridha AR, Wree A, Robertson AAB, Yeh MM,
Johnson CD, Van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou
GN, et al: NLRP3 inflammasome blockade reduces liver inflammation
and fibrosis in experimental NASH in mice. J Hepatol. 66:1037–1046.
2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Mouralidarane A, Soeda J, Visconti-Pugmire
C, Samuelsson AM, Pombo J, Maragkoudaki X, Butt A, Saraswati R,
Novelli M, Fusai G, et al: Maternal obesity programs offspring
nonalcoholic fatty liver disease by innate immune dysfunction in
mice. Hepatology. 58:128–138. 2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Schuster S, Cabrera D, Arrese M and
Feldstein AE: Triggering and resolution of inflammation in NASH.
Nat Rev Gastroenterol Hepatol. 15:349–364. 2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Gao B, Jeong WI and Tian Z: Liver: An
organ with predominant innate immunity. Hepatology. 47:729–736.
2008.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Arrese M, Cabrera D, Kalergis AM and
Feldstein AE: Innate Immunity and Inflammation in NAFLD/NASH. Dig
Dis Sci. 61:1294–1303. 2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Yu Y, Liu Y, An W, Song J, Zhang Y and
Zhao X: STING-mediated inflammation in Kupffer cells contributes to
progression of nonalcoholic steatohepatitis. J Clin Invest.
129:546–555. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hurrell BP, Galle-Treger L, Jahani PS,
Howard E, Helou DG, Banie H, Soroosh P and Akbari O: TNFR2
Signaling enhances ILC2 survival, function, and induction of airway
hyperreactivity. Cell Rep. 29:4509–4524.e5. 2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Fang W, Deng Z, Benadjaoud F, Yang C and
Shi GP: Cathepsin B deficiency ameliorates liver lipid deposition,
inflammatory cell infiltration, and fibrosis after diet-induced
nonalcoholic steatohepatitis. Transl Res. 222:28–40.
2020.PubMed/NCBI View Article : Google Scholar
|