Introduction
Based on World Health Organization estimates,
China's birth defect rate is 5.6% at present, which is
significantly higher than the birth defect rate of 4.72% in
developed countries (1). In
addition, birth defects have risen from fourth place in 2000 to 2nd
place in 2011 in the league table of child mortality causes in
China, accounting for 19.10% of child deaths (2). In the 21st century, the development of
genetic testing technology and the ability to reveal the causes of
genetic diseases have promoted the revolutionary transformation of
medical practice from the symptoms-based diagnostics of traditional
medicine to the cause-based diagnostics of modern medicine
(3).
With the large population in China and the
increasing number of patients carrying genetic diseases, inadequate
numbers of qualified genetic professionals and testing capabilities
represent significant challenges (1). The diagnosis and treatment of
inherited metabolic disorders has become an important task for
clinicians (4,5). In 2009, Ng et al (6) first applied next-generation sequencing
to capture the sequences of human genome exons in order to analyze
the pathogenic variations of single-gene disorders. Since then, the
technique of whole-exome sequencing (WES) has been widely used, due
to its ability to provide potential molecular genetic proof for
genetic diseases in suspected cases after clinical assessment
(7).
Guangxi, a province located in southwestern China,
has a population including several ethnic minorities that is
genetically heterogeneous (8).
Based on our experience, the application of WES in the local
population with a genetic disease presentation has proven to be
beneficial. Guangxi Maternal and Child Health Hospital (Nanning,
China) is one of the leading hospitals responsible for the
diagnosis and treatment of genetic diseases in this region
(9). In the present study, the data
of 1,360 cases of WES in a single center were reviewed and
summarized to assess the practical diagnostic value of WES and to
explore how to improve the ability of this technique to find
disease-causing genes.
Patients and methods
Basic information
A total of 1,360 cases subjected to WES at the
Guangxi Maternal and Child Health Hospital (Nanning, China) between
January 2017 and July 2019 were reviewed. Their age ranged from 1
day to 42 years (4.72±7.67) and the ratio of males to females was
1.74:1. Among them, 456 were inpatients and 904 were outpatients.
The clinical characteristics of all patients were evaluated. The
patients' ethnicities included Han, Zhuang, Yao, Uygur, Miao as
well as other ethnic minorities which is representative of the
ethnic composition of this region (10). Therefore, the present cohort may be
considered as a representative sample. The present study was a
retrospective study, and the data were collected as part of the
routine clinical procedure and no informed consent is required.
However, each patient or guardians signed the informed consent
before performing the WES. The publication of the article is
approved by the Ethics Committee of Guangxi Zhuang Autonomous
Region Maternal and Child Health Hospital and the Children's
Hospital (Reference File No.:2017, [2-11]).
WES and biometric filtering DNA
extraction
Blood samples (2 ml of peripheral blood) were
collected from the patients and their parents for WES. Genomic DNA
was isolated using the Lab-Aid DNA kit (Zeesan Biotech Co., Ltd)
and stored at -80˚C.
WES and copy number variation (CNV)
analysis
Human exome sequencing libraries were constructed
using the Agilent SureSelect Human All Exon V5 kit (Agilent
Technologies, Inc.), and amplicons were generated and sequenced
with the Illumina HiSeq 2500 system (Illumina, Inc.). All
procedures were performed according to the manufacturer's
instructions. After sequencing, reads were aligned to an indexed
human reference genome (GRCh37/hg19) with Burrows-Wheeler
transformation 0.7.15-r1140(11).
Duplicate reads were removed using Picard v.1.85 (http://picard.sourceforge.net) prior to further
processing. Base recalibration and variant calling were performed
using the Genome Analysis Toolkit v.2.3-4Lite (12). Finally, identified variants were
saved in a variant call format. In addition, in the present study,
there was an attempt to reveal CNVs with a read depth-based CNV
detection method (13-16).
The detection methods can be separated into four steps: Raw
coverage normalization, correction for sample-specific coverage
biases, CNV calling, and partially-mapped read analysis and all
copy number variants were finally confirmed by Illumina Human Cyto
SNP12 kit. Translational Genomics Expert (LifeMap Sciences, Inc.)
was used for variant prioritization. Integrative Genomics Viewer
(IGV; v.2.4.15; http://software.broadinstitute.org/software/igv/)
was used to visualize WES data and assess the coverage of the
exons.
Quality control
The capture area of WES was 60M. The average
sequencing depth of each sample was >120X and produced 12G clean
data. At the same time, the actual qualified data output of the
library was >95% with an average Q30≥80% for each sample and the
accurate coverage of the exon region (>20X) should be ≥97%; base
type distribution was uniform with no GC separation. Sanger
sequencing was used to verify the mutations and their origins.
In order to reduce the interference of noise signals
in the results, a series of measures was implemented. First, in the
experimental process, each sample was detected under the same
experimental conditions to reduce any intra-batch differences.
Furthermore, normalization of WES data was performed to avoid any
differences caused by the capture or sequencing processes. In
addition, mass correction on the exome region of the sample was
performed including a GC correction. Finally, biological
information processing on the same exome region of different
samples on the same batch was routinely performed.
Sequencing data analysis
The Genome Analysis Toolkit (GATK) was used for
variant calling (GATK HaplotypeCaller). The TGex software version
3.4.1 (LifeMap Sciences) was used to annotate the selected single
nucleotide variants (SNVs) and indels. ‘Rare deleterious’ mutations
were defined as those that met the following criteria: a) They led
to a stop-gain, stop-loss, nonsynchronous, frameshift or
splice-site mutation and (b) the reference genome GRCh37/hg19 from
the Genome Reference Consortium was used (https://www.ncbi.nlm.nih.gov/grc).
Interpretation of variations
Pathogenicity assessment was based on the American
College of Medical Genetics and Genomics (ACMG)/Association for
Molecular Pathology (AMP) 2015 guidelines and the variants were
classified as either ‘benign’, ‘likely benign’, ‘uncertain
significance’, ‘likely pathogenic’ or ‘pathogenic’ based on
InterVar (http://wintervar.wglab.org/)
(17).
Statistical analysis
Data analysis was conducted by χ2 test
using the IBM SPSS version 19.0 statistical analysis software.
P<0.05 was considered significant. χ2 test was used
to compare the frequencies of positive (disease-related genes
found)/negative (No association was found between disease and gene)
detection among medical staff.
Results
Baseline characteristics
In the present study information and WES data was
collected on 1,360 patients from the Pediatric Intensive Care Unit
and the Pediatrics, Eugenics, Child Rehabilitation, Neonatal,
Otolaryngology and General Pediatric Inpatient Departments as well
as the Surgery Clinic of Guangxi Maternal and Child Health Hospital
(Nanning, China) between January 2017 and July 2019. Of the 1,360
patients, 456 were hospitalized and 904 were out-patients. The
positive rates (detection rate of disease-associated genes) of
hospitalized and out-patients were 43.20% (198/456) and 45.02%
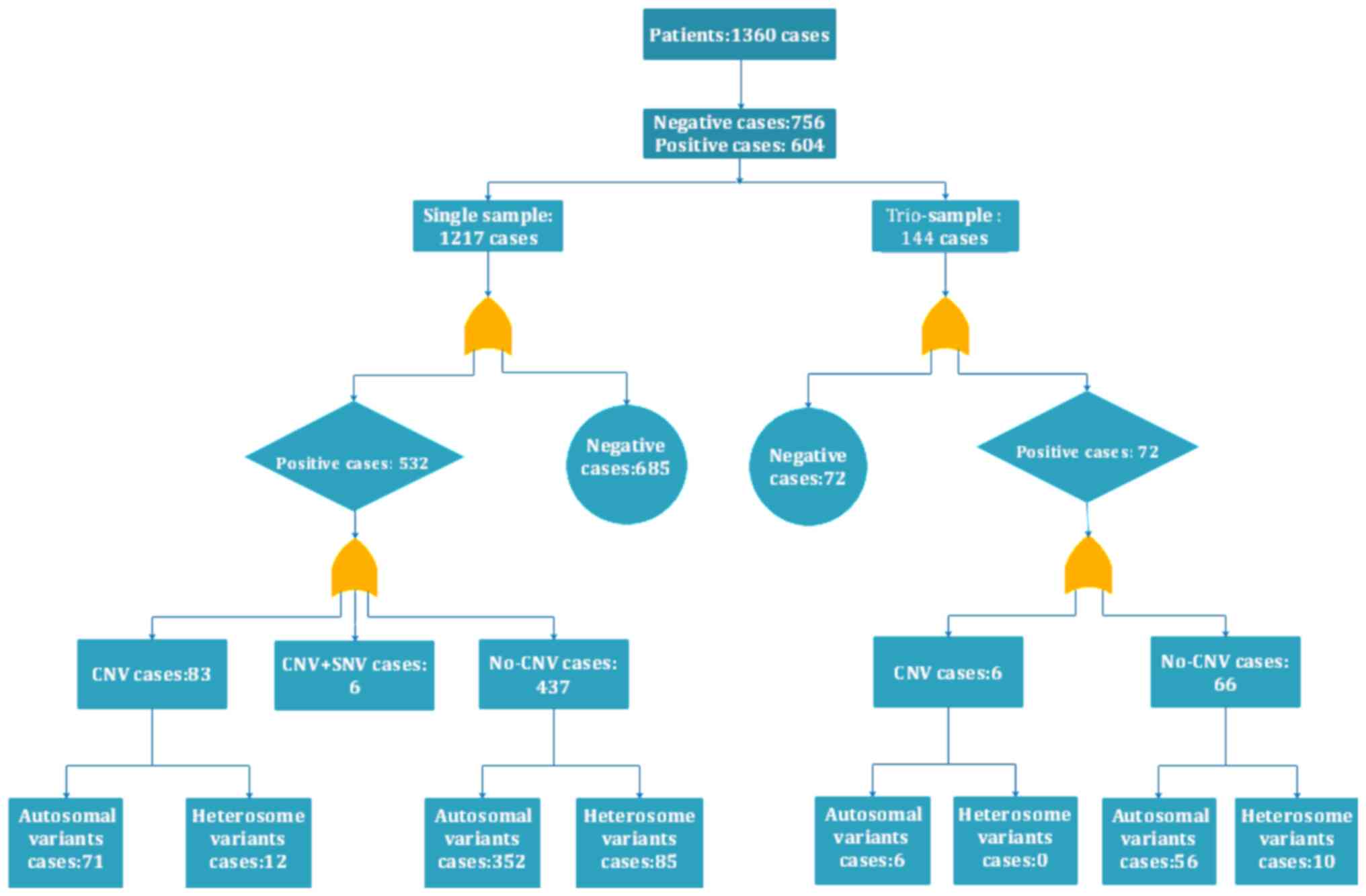
(407/904), respectively. Among all of the cases, 1,217 were
single-sample patients (where only the proband was tested) and 144
were trio-samples (the proband and their parents). Of the 1,217
single-sample patients, 532 were positive (43.71%), while 72
positive samples were detected within 144 family samples (50.00%).
Among the 604 positive patients, including 532 single and 72 family
samples, 89 cases had CNVs with an associated positive rate of
14.74%, 83.28% were the non-CNVs and 1.98% (114 cases) carried
compound heterozygous mutations. The deletions of one or more exons
with merge point mutations accounted for 10 cases and CNV merged
with SNV accounted for 6 cases. Among them, 478 mutations had been
reported previously and 240 mutations were novel (unpublished). A
total of 198 mutations were de novo variations. A flow chart
of the analysis of all of the cases studied is presented in
Fig. 1.
Genetic variants and pathogenicity
assessment
A total of 150 genetic syndromes, involving 510
genes (with respect to abnormal CNVs and point mutations) and 718
variations were detected in the 604 positive cases, of which 89
were CNVs, including variations associated with deletions and
duplications. Based on the ACMG/AMP 2015 guidelines, 718 mutations
were classified as being pathogenic (65.13%), likely pathogenic
(25.05%) and as having uncertain significance (9.82%). Even if
certain genes may be consistent with clinical manifestations, they
may not have been extensively studied and may only be identified as
variants of uncertain significance. In such instances, the progress
of research requires them to be tracked on a regular basis to
determine whether the rating may be updated in order to provide
patients with treatment and genetic counseling. At present, the
‘consensus recommendations for the clinical application of genetic
testing for children's genetic diseases’ recommends that only
pathogenic and likely pathogenic mutations may be used for prenatal
or preimplantation diagnosis (18).
Classification of clinical symptoms
and statistics
Among the patients, certain probands had a single
symptom while others had multiple systemic phenotypes. The patients
were divided into 20 categories according to their major clinical
symptoms. The positive detection rate for each clinical
manifestation is presented in Table
I. The major clinical manifestations included short stature,
motor deterioration, language development retardation, mental
retardation, skeletal deformity, seizures and gonadal dysgenesis or
abnormal genital organs. In order to investigate whether the
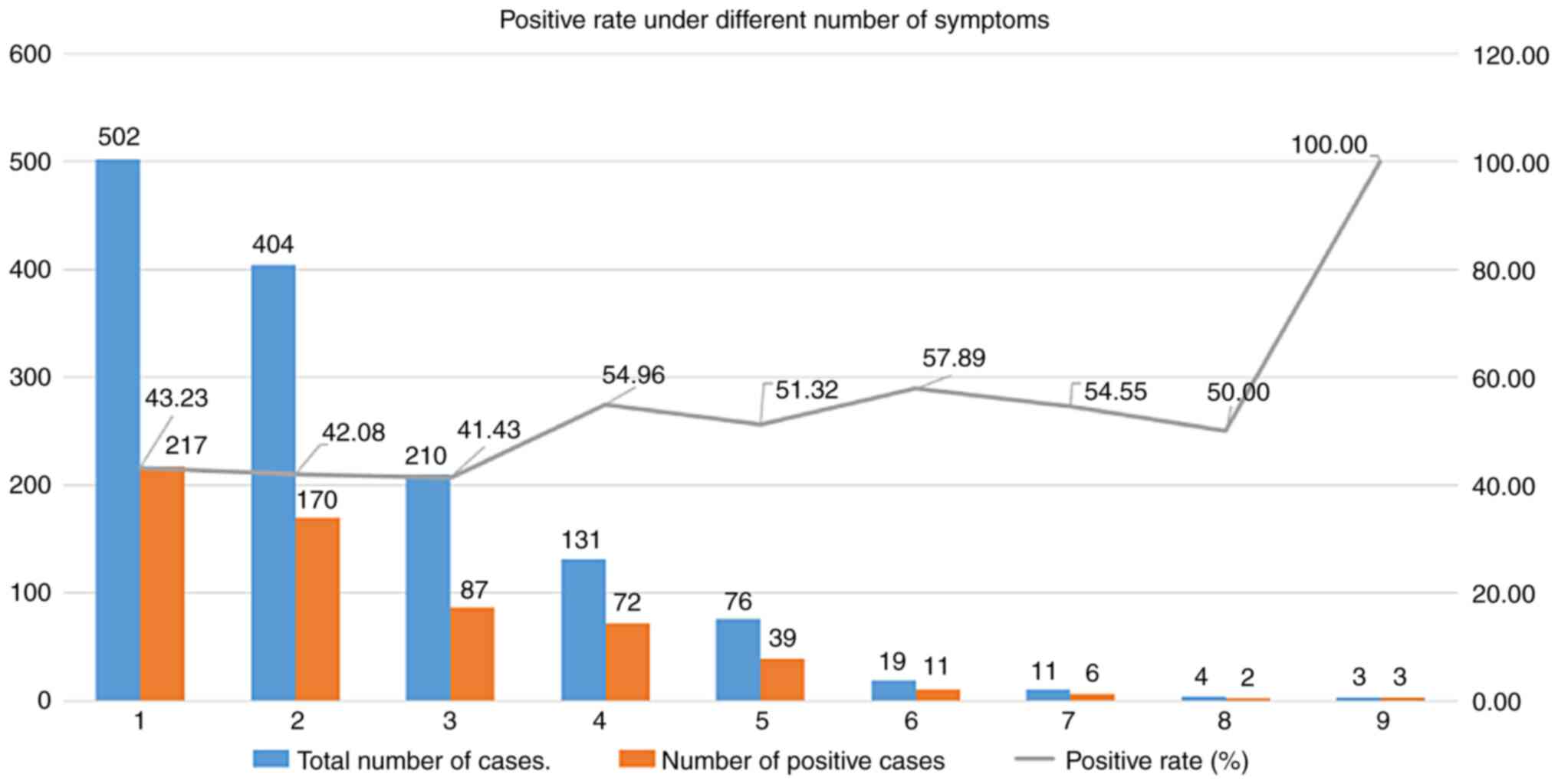
positive rate of WES increased with the number of clinical
symptoms, the number of clinical symptoms exhibited by each of the
subjects was determined, and according to those numbers, the
patients were assigned to categories 1-9 and the positive rates in
those groups were then compared in a graph. Through analysis and
comparison, it was determined that the positive rate of WES
increased with the number of clinical symptoms in the range of 1 to
8. The positive rate was ~40% when the number of symptoms was 1-3,
but when the number of symptoms was 4-8, the positive rate was ~50%
(Fig. 2). It is recommended,
wherever possible, that the interpretation of WES data should be
accompanied by clinical information (19). In the positive cases, the incidence
of autosomal variation was 4.59 times [491 (autosomal variation
cases): 107 (sex chromosomes variation cases)] that of sex
chromosome abnormality. The patients with sex chromosome
abnormalities were mainly male children (<14 years old) and the
male-to-female ratio was 15.75:1. Clinical manifestations of sex
chromosome abnormalities included cryptorchidism, micro-penis and
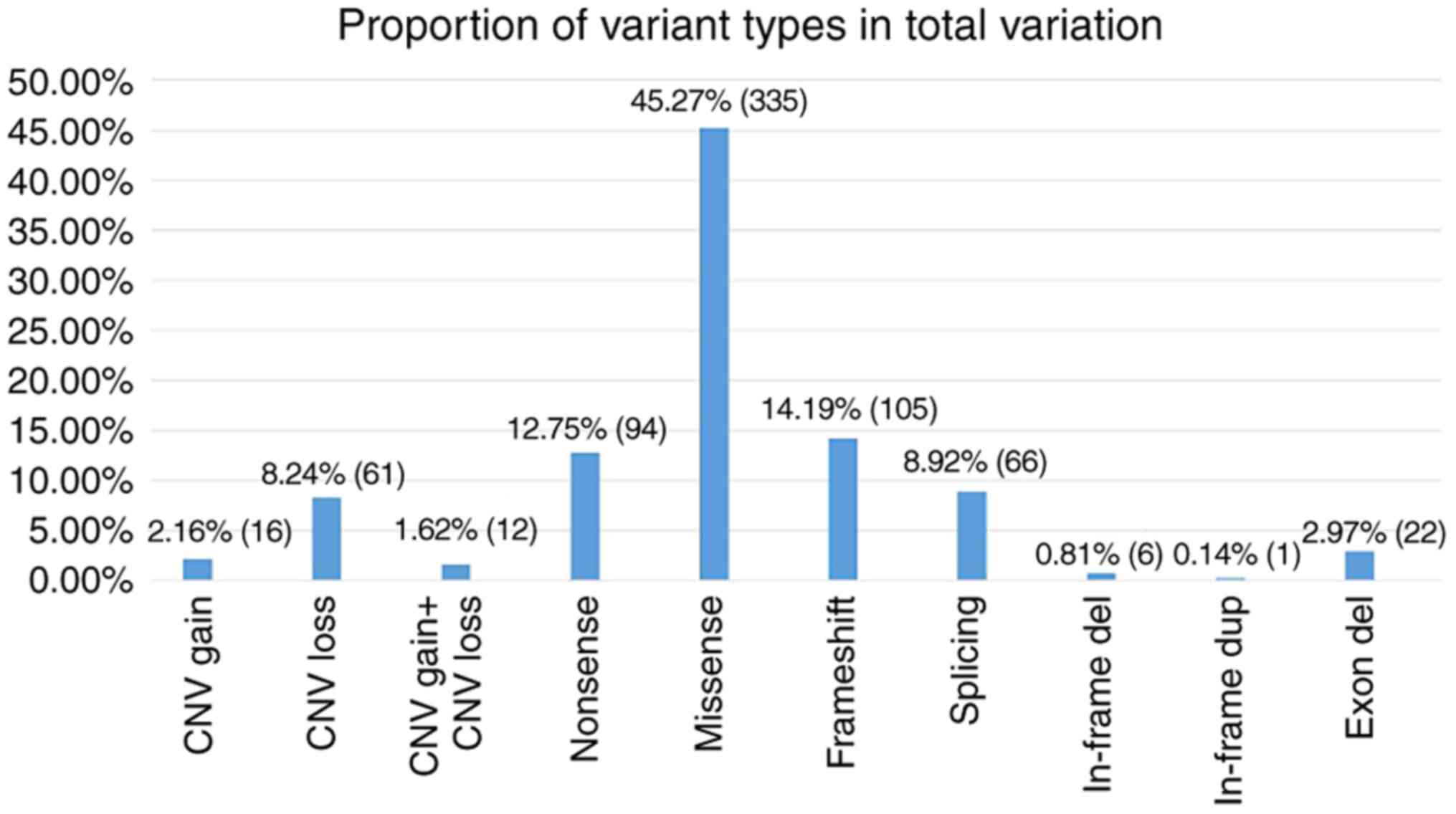
hypospadias. There were 629 non-CNVs, namely nonsense, missense,
frameshift, splicing, in-frame deletions, in-frame duplications and
exon-deletions. The proportion of each variant is presented in
Fig. 3. In order to determine
whether the capabilities of clinicians and data analysts affect the
positivity rate of WES, a pediatric outpatient department, which
has the largest number of test samples, was also selected to
observe whether there were any differences in the positive status
of the samples submitted by individual doctors. The positive
detection rates among the samples analyzed by the data analysts
were also determined. As presented in Table II, different clinicians had
different professional levels of expertise and experience to
evaluate patients and this directly affected the WES-positive rate,
but there was no significant difference between different data
analysts.
 | Table IPositive rate under different clinical
symptoms. |
Table I
Positive rate under different clinical
symptoms.
| Phenotypic
abnormality | HPO | Number of subjects
tested | Positive cases, n
(%) |
|---|
| Growth
abnormality | HP:0000002 | 239 | 118 (49.37) |
| Abnormality of the
nervous system | | | |
|
Seizures | HP:0001250 | 229 | 102 (44.54) |
|
Intellectual
disability | HP:0001249 | 104 | 38 (36.54) |
|
Delayed
speech and language | HP:0000750 | | |
| Development or
autistic behavior | HP:0000729 | 116 | 38 (32.76) |
|
Global
developmental delay | HP:0001263 | 93 | 51 (54.84) |
|
Cerebral
palsy | HP:0100021 | 143 | 59 (41.26) |
|
Motor
deterioration | HP:0002333 | 206 | 99 (48.06) |
| Abnormality of the
respiratory system | HP:0002086 | 165 | 67 (40.61) |
| Abnormality of the
genitourinary system | | | |
|
Abnormality
of the genital system | HP:0000078 | 182 | 66 (36.26) |
|
Abnormality
of the urinary system | HP:0000079 | 195 | 66 (33.85) |
| Abnormality of the
skeletal system | HP:0000924 | 215 | 116 (53.95) |
| Abnormality of head
or neck | HP:0000152 | 290 | 161 (55.52) |
| Abnormality of the
skin | HP:0000951 | 108 | 58 (53.70) |
| Abnormality of the
endocrine system | HP:0000818 | 225 | 112 (49.78) |
| Hearing or vision
abnormality | HP:0000364 | 94 | 55 (58.51) |
| | HP:0000504 | | |
| Abnormality of
blood and blood-forming tissues | HP:0001871 | 75 | 27 (36.00) |
| Abnormality of the
digestive system | HP:0025031 | 52 | 14 (26.92) |
| Abnormality of the
cardiovascular system | HP:0001626 | 125 | 54 (43.20) |
| Table IIClinicians and data analysts' ability
to influence the impact of whole-exome sequencing results. |
Table II
Clinicians and data analysts' ability
to influence the impact of whole-exome sequencing results.
| Staff group/ID | Positive cases | Negative cases | Positive rate
(%) | Chi-square | P-value |
|---|
| Clinicians | | | | 14.43 | 0.025 |
|
A | 140 | 194 | 0.42 | | |
|
B | 32 | 17 | 0.65 | | |
|
C | 21 | 34 | 0.38 | | |
|
D | 12 | 19 | 0.39 | | |
|
E | 4 | 8 | 0.33 | | |
|
F | 8 | 12 | 0.40 | | |
|
G | 1 | 7 | 0.13 | | |
| Data analysts | | | | 3.46 | 0.75 |
|
A1 | 127 | 150 | 0.46 | | |
|
B1 | 74 | 82 | 0.47 | | |
|
C1 | 23 | 33 | 0.41 | | |
|
D1 | 18 | 23 | 0.44 | | |
|
E1 | 89 | 94 | 0.49 | | |
|
F1 | 14 | 24 | 0.37 | | |
|
G1 | 70 | 69 | 0.50 | | |
Discussion
In general, Mendelian (single-gene) diseases are
considered to be rare, occurring at a rate of 40-82 cases per 1,000
live births, with an estimated 7.9 million infants being born
annually with serious birth defects of genetic or partially genetic
origin (20,21). The US Food and Drug Administration
has approved ~400 types of orphan drugs, but these drugs have
limited efficacy and are suitable for treatment of only ~5.00% of
rare diseases. Most rare diseases may only rely on symptomatic or
placebo treatments, as the causes of most of these genetic diseases
still remain elusive (22-24).
In recent years, with the advancement of precision medicine and the
rapid development of large-scale parallel sequencing technology,
the application of WES has increased, not only for research
purposes but also for clinical diagnosis. The effectiveness of WES
has been well documented for certain diseases, such as those
relating to the nervous system, dermatology and seizures (25-27).
This may help to elucidate the pathogenesis of these diseases.
In general, genetic variations may affect biological
function by enhancing (e.g., dose effects and alterations of
transcriptional activity) or decreasing (e.g., haplo-insufficiency
and dominant-negative mutation) the function of key genes (28).
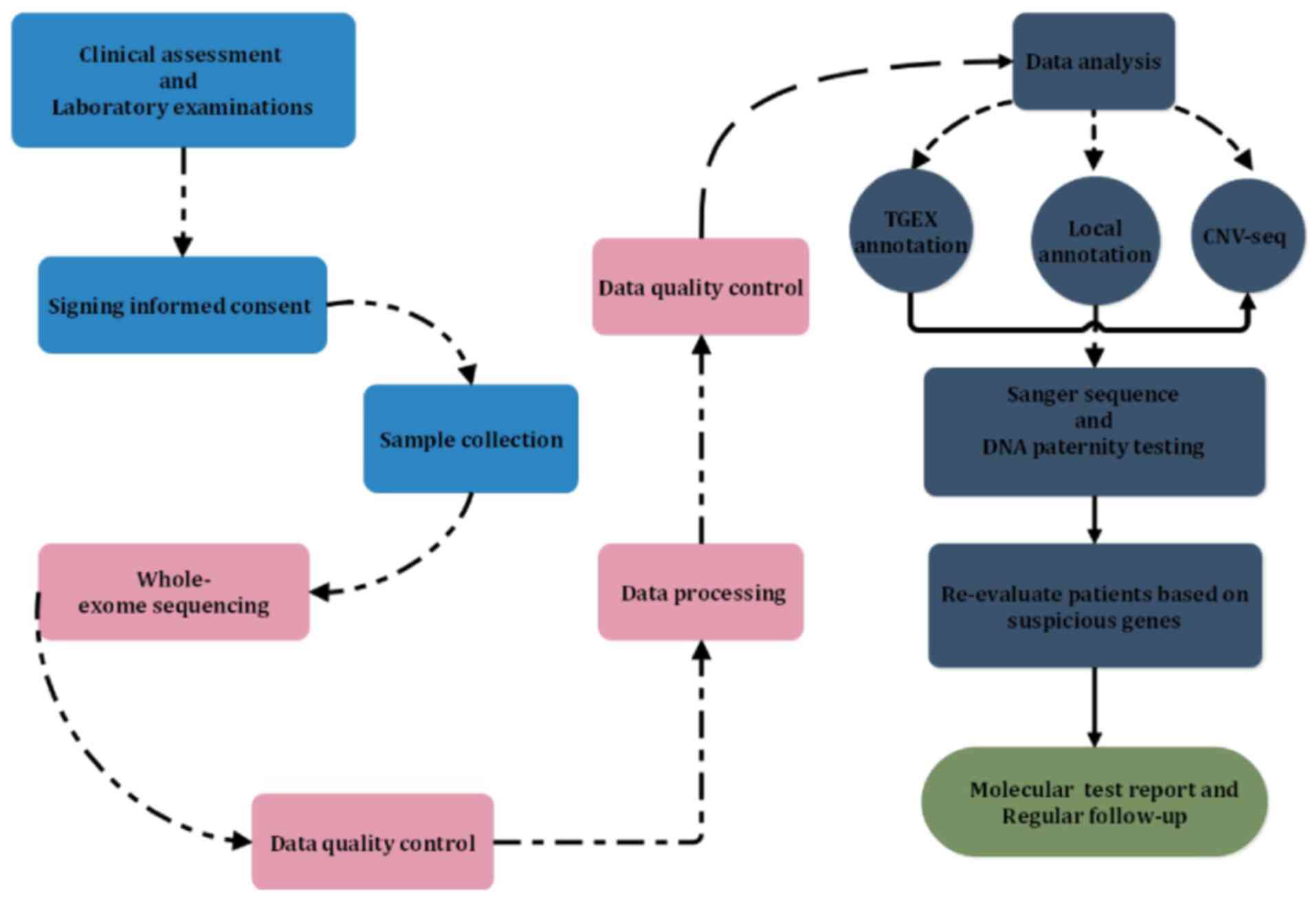
WES was performed on all 1,360 patients of the
present study. The examination and analytical processes are
depicted in a schematic in Fig. 4,
including those performed prior to, during and after WES. The
overall positive rate obtained with this protocol was 47.05%, which
was slightly higher than that for a previous case series, in which
yields of 25.00-26.00% were reported (29) and it was higher than the positive
rate of other genetic testing methods, such as whole-genome
sequencing analysis (34.00-42.00%), karyotype analysis
(4.20-10.00%), chromosome microarray analysis (6.50-20.00%) and
fluorescence in situ hybridization analysis (3.00%)
(30-36).
Among them, the trio-samples positive rate was 6.29% higher than
the rate obtained for single samples, and this was similar to the
results obtained from a previous study (37).
Analysis of trio-samples may not only identify the
mutations associated with the disease detected based on the
clinical manifestations but may also, in turn, be compared and
evaluated from the severity of the mutations to the clinical
performance of the patient. At the same time, it is possible to
rapidly eliminate irrelevant variations by using the genetic model,
allowing for the identification of suspected disease-causing genes,
ultimately increasing the positive detection rate and efficiency.
In addition, trios facilitate the detection of de novo
variants and allow for appraisal of compound heterozygous variants
during interpretation (rather than during the confirmatory testing
process). When WES was performed, factors to be considered by
clinicians included cost, time-to-result and the presence of
consanguinity or family history. For certain diseases that are
rapidly progressing and have serious clinical symptoms, this study
would suggest that priority should be given to performing WES where
trios are available.
In the present study, the major clinical
manifestations of 1,360 patients were classified into 20 main
categories in order to determine which symptoms contributed the
most to the positive rate of WES. Among the 20 classifications, the
positive rate of audio or visual abnormalities was the highest
(58.51%), followed by abnormality of the head or neck (55.52%) and
global developmental delay (54.84%). The manifestations with the
lowest positive rate were abnormalities of the digestive system
(26.92%). A total of 52 patients with abnormality of the digestive
system were analyzed, revealing that the major symptoms were
diarrhea, constipation, vomiting and gastro-esophageal reflux. The
age of the affected children ranged from several days to several
months. At this stage, the appearance of such symptoms was mainly
related to the developmental characteristics of the infants with an
immature digestive system and minimal establishment of microflora.
Therefore, it would be difficult to adapt to changes in the quality
and quantity of food. In addition, those subjects are likely to
have low body immunity and incorrect feeding regimens employed by
certain parents as well as other factors may lead to these
symptoms. However, such symptoms should not be ignored during the
analysis, as they may also be characteristic of certain hereditary
diseases, such as Hirschsprung disease 2 [online mendelian
inheritance in man (OMIM) ID, 600155].
The three most common variations were missense,
frameshift and nonsense mutations and the least common ones were
in-frame duplications. It is worth noting that of the 604 positive
cases, 22 had exon deletions, such as ASCC1 (activating signal
cointegrator 1 complex subunit 1) c.932C>G(p.Ser311Ter) or exon5
deletion. These small deletions should always be noted, as they may
be missed during routine and low-depth CNV analyses. During the
analysis of an autosomal recessive disease, when one mutation is
found in a certain gene, which is relevant to the clinical
symptoms, we recommended that the exons of this particular gene
should be carefully viewed using IGV software.
WES may help clinicians to determine the cause of
the disease and can also influence the treatment strategy, such as
pyridoxine-dependent epilepsy caused by the ALDH7A1 gene (aldehyde
dehydrogenase 7 family member A1; OMIM ID, 266100). Patients with
this type of epilepsy are generally insensitive to treatment with
anticonvulsants but may be treated effectively with large doses of
pyridoxine (vitamin B6). WES, which Raffan and Semple (38) called ‘game-changing technology’ in
2011, is becoming an essential tool for clinicians dealing with
rare and common genetic disorders.
It may be assumed that due to the maturity of WES
technology and the standardization of the process, the data
analysis performed by different analysts according to standardized
procedures does not exhibit any obvious differences in the results
obtained. In order to avoid the influence of subjective factors,
indicators of different diseases were quantified in the present
study, so as to minimize variations caused by those subjective
factors. For instance, short stature was evaluated according to
items including the standard deviation, the annual increase in
height and growth hormone stimulation test results.
There are numerous challenges associated with WES.
First of all, there is an increasing number of non-paternity
infants (from donor gametes), which makes it difficult to identify
and interpret suspected pathogenic variants. Furthermore, there are
complex issues surrounding genetic counseling associated with WES
(e.g., variants of unknown significance). Finally, establishing a
close cooperation network between analysts and clinicians in the
clinical environment may be challenging.
In summary, there are numerous difficulties and
challenges that require to be overcome to successfully apply WES
sequencing technology in the fields of healthcare and disease
prevention. Fortunately, the situation is constantly improving. In
the foreseeable future, it is anticipated that WES will become a
valuable and possibly even a first-line diagnostic tool in the
clinical setting for the diagnosis of complex genetic diseases.
Acknowledgements
The authors would like to thank Dr Dev Sooranna
(Division of Cancer, Academic Department of Obstetrics &
Gynaecology, Imperial College, London, SW10 9NH, UK) for editing
the manuscript.
Funding
Funding: This study was supported by the Guangxi Zhuang Region
Health Department (grant no. Z20190311).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QZ and ZQ designed the study and analyzed the
relevant literature. ZQ wrote the manuscript and prepared the
figures. SY and XZ processed the data, HW and JS collected the
blood samples and performed the DNA extraction experiments. QZ, ZQ,
XZ and JS were involved in performing the WES and the patient
examinations. QZ and SY confirm the authenticity of all the raw
data. All the authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Review
Committee of the Maternal and Child Health Hospital of Guangxi
(Nanning, Guangxi) and was performed in accordance with the
approved guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yu M, Ping Z, Zhang S, He Y, Dong R and
Guo X: The survey of birth defects rate based on birth registration
system. Chin Med J (Engl). 128:7–14. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Cai L, Zheng LA and He L: The forty years
of medical genetics in China. J Genet Genomics. 45:569–582.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhang Q, He L and Shen YP: The arrival of
the clinical whole genome era. Zhonghua Er Ke Za Zhi. 57:401–404.
2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
4
|
Cram DS and Zhou D: Next generation
sequencing: Coping with rare genetic diseases in China. Intractable
Rare Dis Res. 5:140–144. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhao X, Wang P, Tao X and Zhong N: Genetic
services and testing in China. J Community Genet. 4:379–390.
2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ng SB, Turner EH, Robertson PD, Flygare
SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler
EE, et al: Targeted capture and massively parallel sequencing of 12
human exomes. Nature. 461:272–276. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dillon OJ, Lunke S, Stark Z, Yeung A and
Thorne N: Melbourne Genomics Health Alliance. Gaff C, White SM and
Tan TY: Exome sequencing has higher diagnostic yield compared to
simulated disease-specific panels in children with suspected
monogenic disorders. Eur J Hum Genet. 26:644–651. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen P, He G, Zou X, Zhang X, Li J, Wang
Z, Gao H, Luo L, Zhang Z, Yu J and Han Y: Genetic diversities and
phylogenetic analyses of three Chinese main ethnic groups in
southwest China: A Y-Chromosomal STR study. Sci Rep.
8(15339)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Business Wire: The Maternal and Children
Health Hospital of Guangxi Zhuang Autonomous Region and LifeMap
Sciences Join Hands to Improve Diagnosis for Rare Diseases.
Business Wire, Inc., 2018. https://www.businesswire.com/news/home/20180417005741/en/Maternal-Children-Health-Hospital-Guangxi-Zhuang-Autonomous.%20Journal.
Accessed April 17, 2018.
|
|
10
|
Hu P, Qin YH, Jing CX, Lu L, Hu B and Du
PF: Does the geographical gradient of ApoE4 allele exist in China?
A systemic comparison among multiple Chinese populations. Mol Biol
Rep. 38:489–494. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Angel GD, Levy-Moonshine A, Jordan T, Shakir K, Roazen D,
Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.11–11.10.33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Abel HJ, Duncavage EJ, Becker N, Armstrong
JR, Magrini VJ and Pfeifer JD: SLOPE: A quick and accurate method
for locating non-SNP structural variation from targeted
next-generation sequence data. Bioinformatics. 26:2684–2688.
2010.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yoon S, Xuan Z, Makarov V, Ye K and Sebat
J: Sensitive and accurate detection of copy number variants using
read depth of coverage. Genome Res. 19:1586–1592. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li C and Hung Wong W: Model-based analysis
of oligonucleotide arrays: Model validation, design issues and
standard error application. Genome Biol.
2(RESEARCH0032)2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li X, Chen S, Chen F, Xie W, Wang J, Wang
J, Yang H and Zhang X: A kind of copy number mutation detection
method and system. CN Patent CN104221022B. Filed April 5, 2014;
issued November 21, 2017.
|
|
17
|
Li Q and Wang K: InterVar: Clinical
interpretation of genetic Variants by the 2015 ACMG-AMP guidelines.
Am J Hum Genet. 100:267–280. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Editorial Board, Chinese Journal of
Pediatrics. Consensus recommendations for the clinical application
of genetic testing for children's genetic diseases. Zhonghua Er Ke
Za Zhi. 57:172–176. 2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
19
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jamuar SS and Tan EC: Clinical application
of next-generation sequencing for Mendelian diseases. Hum Genomics.
9(10)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Harris J: Germline manipulation and our
future worlds. Am J Bioeth. 15:30–34. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Baird PA, Anderson TW, Newcombe HB and
Lowry RB: Genetic disorders in children and young adults: A
population study. Am J Hum Genet. 42:677–693. 1988.PubMed/NCBI
|
|
23
|
Valdez R, Grosse SD and Khoury MJ: The
need for a next-generation public health response to rare diseases.
Genet Med. 19:489–490. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhao M and Wei DQ: Rare Diseases: Drug
discovery and informatics resource. Interdiscip Sci. 10:195–204.
2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Reches A, Hiersch L, Simchoni S, Barel D,
Greenberg R, Ben Sira L, Malinger G and Yaron Y: Whole-exome
sequencing in fetuses with central nervous system abnormalities. J
Perinatol. 38:1301–1308. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Caylor RC, Grote L, Thiffault I, Farrow
EG, Willig L, Soden S, Amudhavalli SM, Nopper AJ, Horii KA, Fleming
E, et al: Incidental diagnosis of tuberous sclerosis complex by
exome sequencing in three families with subclinical findings.
Neurogenetics. 19:205–213. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kobayashi Y, Tohyama J, Kato M, Akasaka N,
Magara S, Kawashima H, Ohashi T, Shiraishi H, Nakashima M, Saitsu H
and Matsumoto N: High prevalence of genetic alterations in
early-onset epileptic encephalopathies associated with infantile
movement disorders. Brain Dev. 38:285–292. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jackson M, Marks L, May GHW and Wilson JB:
The genetic basis of disease. Essays Biochem. 62:643–723.
2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Retterer K, Juusola J, Cho MT, Vitazka P,
Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J,
Monaghan KG, et al: Clinical application of whole-exome sequencing
across clinical indications. Genet Med. 18:696–704. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Stavropoulos DJ, Merico D, Jobling R,
Bowdin S, Monfared N, Thiruvahindrapuram B, Nalpathamkalam T,
Pellecchia G, Yuen RKC, Szego MJ, et al: Whole genome sequencing
expands diagnostic utility and improves clinical management in
pediatric medicine. NPJ Genom Med. 1(15012)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gudbjartsson DF, Helgason H, Gudjonsson
SA, Zink F, Oddson A, Gylfason A, Besenbacher S, Magnusson G,
Halldorsson BV, Hjartarson E, et al: Large-scale whole-genome
sequencing of the Icelandic population. Nat Genet. 47:435–444.
2015.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Dai R, Yu Y, Xi Q, Hu X, Zhu H, Liu R and
Wang R: Prenatal diagnosis of 4953 pregnant women with indications
for genetic amniocentesis in Northeast China. Mol Cytogenet.
12(45)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shaffer LG, Bejjani BA, Torchia B,
Kirkpatrick S, Coppinger J and Ballif BC: The identification of
microdeletion syndromes and other chromosome abnormalities:
Cytogenetic methods of the past, new technologies for the future.
Am J Med Genet C Semin Med Genet. 145C:335–345. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Shaffer LG, Rosenfeld JA, Dabell MP,
Coppinger J, Bandholz AM, Ellison JW, Ravnan JB, Torchia BS, Ballif
BC and Fisher AJ: Detection rates of clinically significant genomic
alterations by microarray analysis for specific anomalies detected
by ultrasound. Prenat Diagn. 32:986–995. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
Miller DT, Adam MP, Aradhya S, Biesecker
LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE,
Epstein CJ, et al: Consensus statement: Chromosomal microarray is a
first-tier clinical diagnostic test for individuals with
developmental disabilities or congenital anomalies. Am J Hum Genet.
86:749–764. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ravnan JB, Tepperberg JH, Papenhausen P,
Lamb AN, Hedrick J, Eash D, Ledbetter DH and Martin CL: Subtelomere
FISH analysis of 11 688 cases: An evaluation of the frequency and
pattern of subtelomere rearrangements in individuals with
developmental disabilities. J Med Genet. 43:478–489.
2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yates CL, Monaghan KG, Copenheaver D,
Retterer K, Scuffins J, Kucera CR, Friedman B, Richard G and
Juusola J: Whole-exome sequencing on deceased fetuses with
ultrasound anomalies: Expanding our knowledge of genetic disease
during fetal development. Genet Med. 19:1171–1178. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Raffan E and Semple RK: Next generation
sequencing-implications for clinical practice. Br Med Bull.
99:53–71. 2011.PubMed/NCBI View Article : Google Scholar
|