Introduction
Prostate cancer (PCa) is one of the most common
types of malignant cancer affecting the urinary system in men, with
>1 million new cases diagnosed every year (1). It is a major leading cause of
cancer-related mortality, a serious threat to men's health and
imposes a heavy economic burden worldwide. PCa generally develops
in men aged ≥50 years, with an average age of ~66 at the time of
diagnosis (2). The 5-year survival
rate for most patients with local or regional PCa is 90% (3). Although significant progress in early
diagnosis and clinical therapy of PCa have been achieved in the
last several decades, the overall 5-year survival rate remains ~30%
for men diagnosed with advanced PCa, and metastasis and recurrence
are the leading causes of death in patients with PCa (4). The high mortality rate is primarily
due to the metastatic spread of tumor cells to lymph nodes, the
bladder, bone, spinal cord and some other distant organs (5-7).
Therefore, it is necessary to understand the underlying pathogenic
mechanisms and to identify novel factors involved in the
progression and metastasis of PCa, to assist in identifying novel
effective methods for the prevention, diagnosis and therapeutic
intervention of this disease.
Cancer metastasis is a multistage process that
includes cell invasion, cell migration, colonization, and adaption
to and growth in foreign tissues (8,9). At
each step of this process, cells undergo morphological changes
driven by dynamic remodeling of the actin cytoskeleton (10). Ezrin-radixin-moesin (ERM) proteins
are membrane-cytoskeleton linkers that serve critical roles in the
regulation of cytoskeleton reorganization and further cell
morphological changes, migration and adhesion (11,12).
ERM proteins exist in two conformational states, an inactive
conformation with the N-terminal FERM domain and C-terminal tail
domain (C-ERMAD) forming an intramolecular interaction, and an
active conformation where these domains are dissociated. The active
conformation of ERM functions as linkers between the cytoskeleton
and transmembrane proteins (13,14).
Several lines of evidence have demonstrated that aberrant
localization, expression or activation of ERM proteins are involved
in the progression of several types of cancer (15).
The phosphorylation of the FERM domain (T567, T564
and T558, for ezrin, radixin and moesin, respectively) has been
identified as the critical step in the activation of ERM proteins
(12,16). Serine threonine kinase 10 (STK10),
also called lymphocyte-oriented kinase, is a serine/threonine
kinase predominantly expressed in lymphoid organs (17). As one of the kinases responsible for
the phosphorylation and activation of ERM proteins, STK10 has been
demonstrated to be associated with migration and Lymphocyte
function-associated antigen-1 (LFA-1)-mediated adhesion of
lymphocytes (18,19). Several studies also found that
mutations of STK10 are associated with testicular germ cell tumors
and aggressive lymphoma. According to the data from The Human
Protein Atlas (proteinatlas.org/ENSG00000072786-STK10), STK10 is
expressed in ~17 types of cancer, including PCa; however, the
biological functions of STK10 in the pathogenesis of PCa remains
undetermined.
In the present study, to investigate the function of
STK10 in the pathology of PCa, an STK10-knockout (KO) DU145
prostate cell line was generated using the CRISPR-Cas9 gene editing
system, and the effects of STK10 KO on the tumor biological
behaviors, including cell proliferation, apoptosis and migration,
were further analyzed. The present data indicated that STK10 KO
resulted in decreased migration of PCa cells via regulation of ERM
and p38 MAPK activity. These findings uncovered an important role
of STK10 in the tumorigenesis of PCa, particularly in cancer
metastasis.
Materials and methods
Cell culture
The PCa cell line (DU145) was purchased from the
American Type Culture Collection and grown in RPMI-1640 medium
(Hyclone; Cytiva) with 10% (vol/vol) FBS (Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C with 5% CO2 in a humidified
incubator.
STK10 KO using the CRISPR Cas9 gene
editing system
The guide-RNA (5'-GGCGGACGTGCTCATATTCG-3') was
designed using the online CRISPR design tool (zlab.bio/guide-design-resources) to
target exon 1 of the STK10 gene. Two partially complementary
oligonucleotides (5'-CACCGG CGGACGTGCTCATATTCG-3' and
5'-AAACCGAATATG AGCACGTCCGCC-3') were annealed and cloned into the
PX459 plasmid (cat. no. 62988; Addgene, Inc.), which was digested
using the BbsI restriction enzyme. This plasmid was termed
the STK10-KO plasmid, which could guide hSpCas9 to the
genomic target site in the STK10 gene. DU145 cells were
seeded into a 6-cm dish (8x105 cells) and transfected
with 4 µg STK10-KO plasmids or PX459 plasmids (Ctrl) using
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.).
The supernatant was exchanged for fresh medium 12 h later. After 2
days, the cells were incubated in fresh medium with 1 µg/ml
puromycin for 1 week. Subsequently, the concentration of cells were
adjusted to 10 cells/ml by limiting dilution method and the cells
were sub-cultured into 96-well plates (100 µl/well) for single
clone selection. Single STK10-KO and control cell clones
were identified by western blot analysis and Sanger sequencing
using specific primers (forward, 5'-GCCTCGATATTCCCACA GCA-3' and
reverse, 5'-CAGGGCACACTTGACCGAG-3'; sequencing primer,
5'-CGGGTCTGGGGAGAACCCCG-3').
STK10 expression vector
construction
Expression constructs of STK10 were cloned from
cells using the PrimeSTAR® HS PCR kit (cat. no. R040A;
Takara Bio, Inc.) with the following primers: forward,
5'-TGCTGGATATCTGCAGAATTCACGCGGC GTCCTCCAACTC-3' and reverse,
5'-TAGTCCAGTGTGG TGGAATTCAGAAGCATCCGCAGAACTGTAGGG-3'. The following
temperature protocol was used: 98˚C for 10 min, followed by 35
cycles of 98˚C for 10 sec, 60˚C for 10 sec and 72˚C for 3 min; then
72˚C for 10 min. STK10 fragment was cloned into the pcDNA3.1
myc-His(-) B plasmid (cat. no. V855-20; Thermo Fisher Scientific,
Inc.). Final constructs contained the affinity tags Myc and His,
and were transcribed under the control of the CMV promoter.
Western blot analysis
DU145 cells were grown in 60-mm dishes and then
cultured in serum-free RPMI-1640 medium for 6 h, following which,
they were stimulated with 50 ng/ml human recombinant epidermal
growth factor (EGF; R&D Systems, Inc.) for the appropriate
times (5, 15 and 30 min) at 37˚C with 5% CO2 in a
humidified incubator. DU145 cells were grown in a six-well plate
and then transfected with the indicated STK10 expression vectors or
empty vectors using Lipofectamine® 3000, according to
the manufacturer's instructions. Cells were collected 48 h after
transfection. STK10-KO DU145 and control cell lysates were
prepared using RIPA lysis buffer [1% Nonidet P-40, 0.5% sodium
deoxycholate, 0.1% SDS in phosphate-buffered saline (PBS)] with
freshly supplemented protease inhibitors cocktail (Roche
Diagnostics). The concentration of proteins was detected using a
bicinchoninic acid assay, and 40 µg protein was separated using 10%
SDS-PAGE. Standard protocols were used for electrophoresis and
immunoblotting analyses (20).
Membranes were blocked with 5% milk in PBS for 1 h at room
temperature. An anti-STK10 antibody (dilution 1:1,000; cat. no.
ab70484; Abcam), anti-GAPDH rabbit polyclonal antibody (dilution
1:2,000; cat. no. D110016; BBI Solutions), recombinant anti-Ezrin
antibody (dilution 1:500; cat. no. ab40839; Abcam), recombinant
anti-Moesin antibody (dilution 1:500; cat. no. ab52490; Abcam),
recombinant anti-Radixin antibody (dilution 1:1,000; cat. no.
ab52495; Abcam), anti-Ezrin (pThr567)/Radixin (pThr564)/Moesin
(pThr558) antibody (dilution 1:500; cat. no. ab76247; Abcam),
anti-p38 MAPK antibody (dilution 1:1,000; cat. no. 9212; Cell
Signaling Technology, Inc.), anti-phospho-p38 MAPK antibody
(dilution 1:1,000; cat. no. 9211; Cell Signaling Technology, Inc.),
anti-ERK1/2 antibody (dilution 1:1,000; cat. no. 9102; Cell
Signaling Technology, Inc.), anti-phospho-ERK1/2 antibody (dilution
1:1,000; cat. no. 4370; Cell Signaling Technology, Inc.),
anti-phospho-PI3K p85 antibody (dilution 1:1,000; cat. no. 4228;
Cell Signaling Technology, Inc.), anti-PI3K p85 antibody (dilution
1:1,000; cat. no. 4292; Cell Signaling Technology, Inc.),
anti-PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase,
catalytic subunit (p110)α (dilution 1:1,000; cat. no. 4249; Cell
Signaling Technology, Inc.), anti-PI3K p110γ (dilution 1:1,000;
cat. no. 5405; Cell Signaling Technology, Inc.), anti-Cyclin D1
(dilution 1:500; cat. no. 2978; Cell Signaling Technology, Inc.),
anti-Cyclin D3 (dilution 1:1,000; cat. no. 2936; Cell Signaling
Technology, Inc.), anti-zinc finger E-box binding homeobox 1
(dilution 1:500; ZEB1; cat. no. 3396; Cell Signaling Technology,
Inc.) and anti-E-Cadherin (dilution 1:1,000; cat. no. 3195; Cell
Signaling Technology, Inc.) were used as the primary antibodies and
incubated overnight at 4˚C. Subsequently, the membranes were
incubated with IRDyeCW800-conjugated anti-rabbit immunoglobulin
(dilution 1:10,000; cat. no. 926-32213; LI-COR Biosciences, Inc.).
The images were captured using the LI-COR Odyssey imaging system
(LI-COR Odyssey 9120 imaging system; LI-COR Biosciences, Inc.) and
adjusted to grayscale images according to the fluorescence
intensity of the bands.
Immunofluorescence staining
DU145 cells grown on coverslips (8x104
cells per well in a 12-well plate) for 48 h were fixed with 4%
paraformaldehyde in PBS for 10 min at room temperature and washed
with PBS three times. To permeabilize the cells, 0.5% Triton X-100
in PBS-Tween (PBST) was used to permeabilize cells for 10 min at
room temperature. Coverslips were incubated with 10% goat serum
(Gibco; Thermo Fisher Scientific, Inc.) in PBST for 30 min to block
non-specific antibody binding. An anti-STK10 antibody (1:500; cat.
no. ab70484; Abcam) was used as the primary antibody and incubated
overnight at 4˚C. Then, the coverslips were washed three times with
PBST. Cells were incubated with the goat anti-rabbit IgG (H+L)
Highly Cross-Adsorbed secondary antibody-Alexa Fluor Plus 594
(1:500; cat. no. A32740; Thermo Fisher Scientific, Inc.) for 2 h at
room temperature. Coverslips were washed three times with PBST, and
incubated with DAPI for 10 min at room temperature. Coverslips were
mounted using Fluorescence Mounting Medium (Dako; Agilent
Technologies, Inc.) and imaged by fluorescence microscopy
(magnification, x400; Nikon Corporation).
Cell Counting Kit-8 (CCK-8) cell
viability assay
Cell viability was detected using a CCK-8 assay
(Dojindo Molecular Technologies, Inc.), according to the
manufacturer's instructions. DU145 cells were seeded into a 96-well
plate with 1x103 cells/well. Viability was detected
following incubation of the cells with 10% CCK-8 solution in
complete medium for 24 h. The absorbance was detected at 450 nm
using a microplate reader (BioTek Instruments, Inc.).
BrdU incorporation and apoptosis
detection
Cell proliferation and the cell cycle were analyzed
using a Phase-Flow™ FITC BrdU kit (BioLegend, Inc.) following the
manufacturer's instructions. First, BrdU solution (5 µg/ml) was
added to the cell suspension and cultured for 1 h. Then, the cells
were dyed with an anti-BrdU antibody and 7-AAD, and analyzed by
flow cytometry (BD FACSVerse™; BD Biosciences, Inc.). To detect
cell apoptosis, an Annexin V-FITC Apoptosis Detection kit
(eBioscience; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The results were analyzed using software
FlowJo version 10 (FlowJo LLC).
Cell migration assay
Cells were starved in serum-free medium for 24 h,
Transwell chambers with an 8 µm pore size (EMD Millipore) were
inserted into a 24-well culture plate. DU145 cells
(1x105) were resuspended in 400 µl serum-free medium and
added to the upper chamber. Complete medium (600 µl) was added to
the outside of the inserts and incubated at 37˚C in a cell
incubator for 48 h. Cells were fixed with 4% paraformaldehyde for
10 min at room temperature and stained with 0.5% crystal violet for
5 min at room temperature. Cells which had not migrated were
removed using a cotton swab. Cells which had migrated were imaged
using a light microscope (magnification, x100). The number of cells
in five random fields of view were counted, and the mean was
calculated.
In vivo mice study
Male NPSG immune deficient mice
(NOD-PrkdcscidIl2rgnull, weight:
19.46-22.23 g; 4-weeks old) were injected with DU145 cells
(1x106) in 100 µl medium mixed with Matrigel (ratio 1:1;
Invitrogen; Thermo Fisher Scientific, Inc.) in the flank regions.
All mice were housed with a 12-h light/dark cycle, with ad
libitum access to food and water. Adequate humanitarian care
was provided. For this study, the endpoints were a tumor burden
>10% of body weight, any tumor >20 mm in diameter,
ulceration, necrosis or infection, and the presence of a tumor that
interfered with eating or walking in an adult mouse. A total of 8
mice were used in this experiment, and none of them reached the
endpoint of the study. After 29 days, all mice were euthanized with
CO2, before the longest dimension of the tumors reached
20 mm. Initial CO2 delivery to the micro-isolator was
accomplished by opening the CO2 cylinder valve such that
animals were slowly exposed to increasing levels of CO2
(displacing ~40% of the chamber volume per min). Cardiac arrest for
5 min was used to confirm death. Tumor tissues were collected and
weighed. Tumors were measured using a digital caliper, and the
tumor volume was calculated as follows: (width)2 x
length/2. All experimental manipulations were approved by the
Animal Ethics Committee of Ruijin Hospital Affiliated to Shanghai
Jiao Tong University School of Medicine (Shanghai, China).
Statistical analysis
Each experiment was performed at least three times
independently, and results are presented as the mean ± standard
deviation. All statistical analyses were performed using GraphPad
Prism version 7 (GraphPad Software, Inc.). Comparisons between two
groups were performed using an unpaired Student's t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Generation of STK10-KO cell lines
using the CRISPR-Cas9 gene editing system
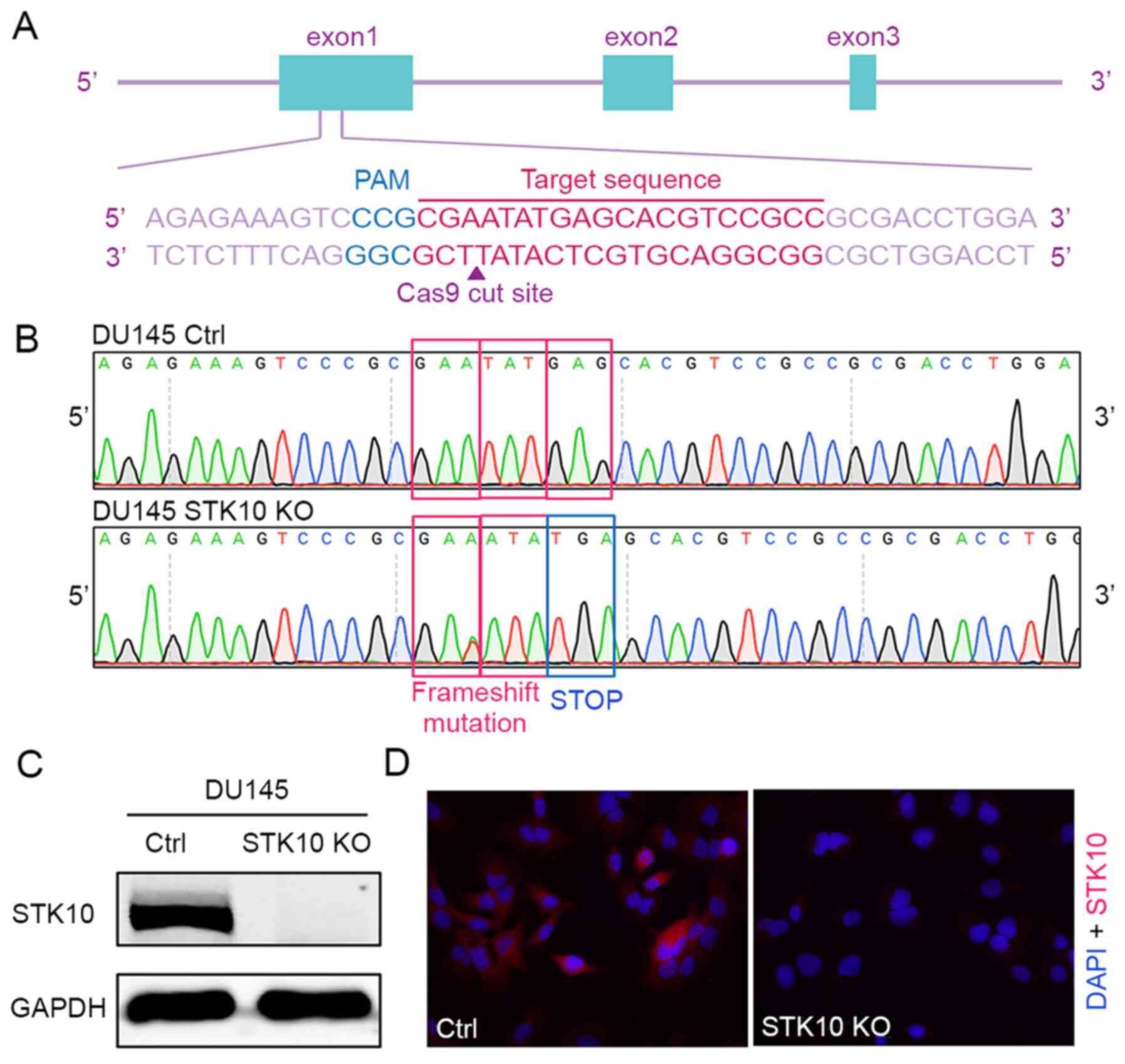
The CRISPR-Cas9 gene editing system was used to
generate a STK10-KO PCa cell line. The oligonucleotides for
the guide RNA were designed, synthesized and cloned into the pX459
vector (Fig. 1A). Then, the vectors
were transfected into DU145 cells. The indel mutations in these
cell lines were confirmed by DNA sequencing of the PCR products of
target DNA. The insertion of one base (c.66_67insA) causes
frameshift mutation, resulting in the formation of a stop codon and
deficiency of the STK10 protein (Fig.
1B). Finally, western blotting and immunofluorescence staining
with an anti-STK10 antibody showed that the expression of STK10
protein was abolished in the STK10-KO cell line (Fig. 1C and D).
Target deletion of STK10 in PCa cells
promotes proliferation but suppresses migration
The effects of STK10 deficiency on the spontaneous
proliferation of PCa cells was first evaluated using a BrdU
incorporation assay and cell cycle distribution was assessed using
fluorescence activated cell sorting (FACS). The results indicated
that target deletion of STK10 markedly decreased the percentage of
cells in the G0/G1 phase (46.23±1.77 vs. 62.77±2.01%; P<0.001),
but increased the proportion of cells in the S phase (19.03±0.61
vs. 11.40±0.60%; P<0.001) and G2 phase (20.07±1.78 vs.
15.37±1.40%; P<0.01) in DU145 PCa cells (Fig. 2A and D). Furthermore, a CCK-8 assay was
performed for 1-4 days to evaluate the proliferation of DU145
control and STK10-KO cells. As shown in Fig. 2C, the cell viability and
proliferative capacity of STK10-KO DU145 cells was
significantly increased when compared with the control cells. These
data suggest that the KO of STK10 promoted cell cycle progression
of PCa cells.
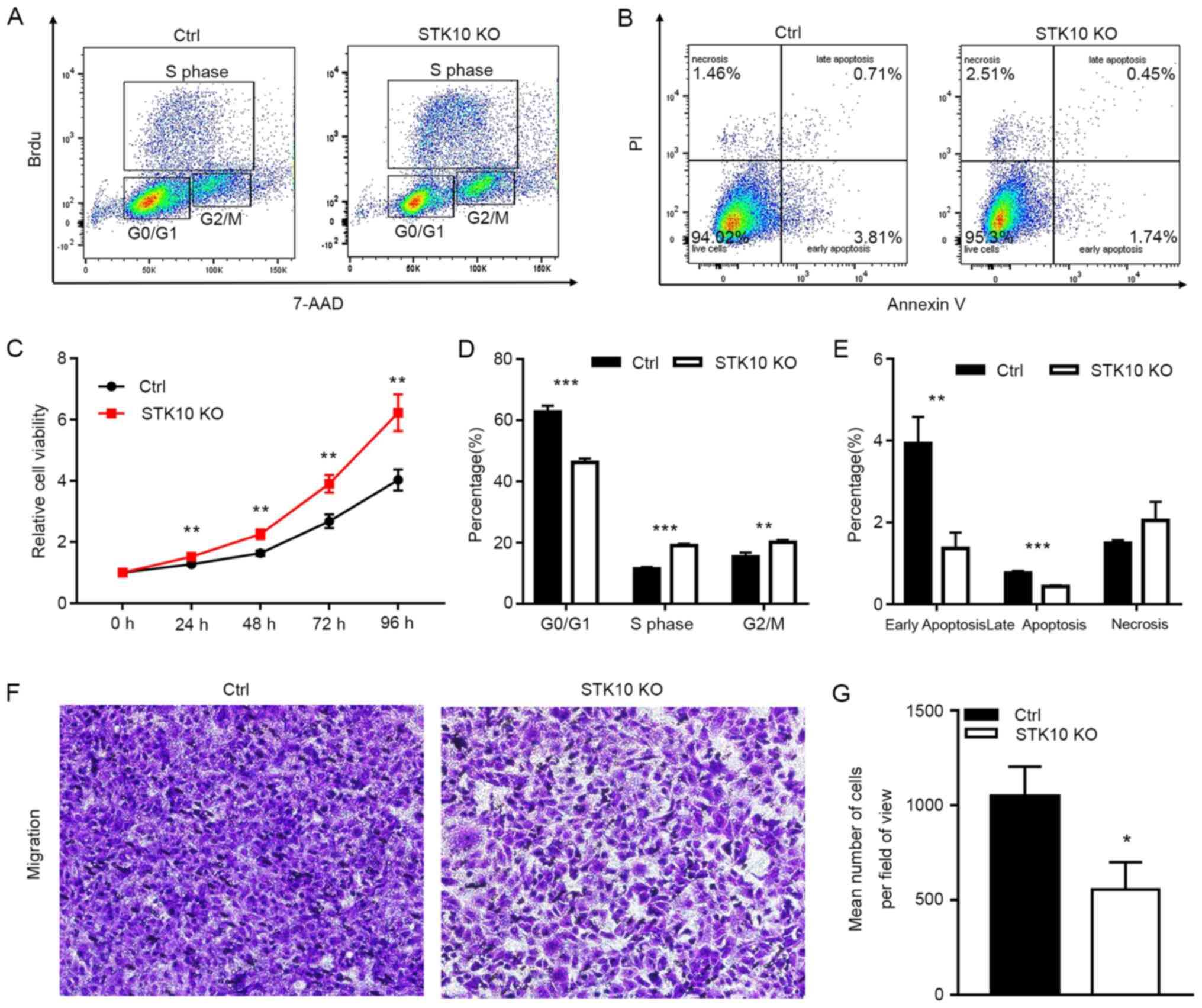 | Figure 2Effects of STK10 KO on the
proliferation, apoptosis and migration of PCa cells. (A) BrdU
incorporation and FACS analyses for control and STK10-KO
DU145 cells. (B) Effects of STK10 KO on the apoptosis of DU145
cells were examined by Annexin V-FITC/PI staining. (C)
Proliferative activity of control and STK10-KO DU145 cells
was quantified using a Cell Counting Kit-8 assay. (D and E) The
percentages of different stages of the cell cycle and apoptotic
cells were calculated. n=3 in each group. All experiments were
performed at least three times. (F and G) Representative images and
quantitative analysis of the Transwell migration assays.
Magnification, x100. Data are presented as the mean ± standard
deviation. *P<0.05, **P<0.01,
***P<0.001 vs. Ctrl. STK10, serine threonine kinase
10; KO, knockout; PCa, prostate cancer; FACS, fluorescence
activated cell sorting; AAD, 7-amino actinomyosin D; Ctrl,
control. |
Since abnormal apoptosis is one of the most
important characteristics of malignant cancer cells, Annexin V-PI
staining and flow cytometry analysis was performed to further
evaluate the impact of STK10 on the apoptosis of PCa cells.
Compared with the control cells, the percentage of early apoptotic
cells (Annexin V+/PI-, 1.36±0.39 vs.
3.93±0.64%; P<0.01) and late apoptotic cells (Annexin
V+/PI+, 0.43±0.03 vs. 0.76±0.05%; P<0.001)
were decreased in STK10-KO cells (Fig.
2B and E). However, these low
percentages (<10%) of apoptotic rates are not physiologically
significant in vitro (21).
These results suggested that STK10 KO has no significant biological
effects on the apoptosis of PCa cells.
To explore the role of STK10 on the tumorigenesis
and metastasis of PCa, the migratory ability of DU145 PCa cells was
detected in vitro. The Transwell assays showed that the
number of cells that had migrated were decreased in the
STK10-KO group compared with that in the control group
(P<0.0001; Fig. 2F and G). These results demonstrated that
knockdown of STK10 could inhibit the migration of PCa cells, and
indicated that STK10 may serve an essential role in the metastasis
of PCa.
Target deletion of STK10 facilitates
the growth of tumor xenografts in vivo
After it was determined that STK10 deletion promoted
the proliferation and suppressed the apoptosis of PCa cells in
vitro, the effect of STK10 KO on xenograft tumors in
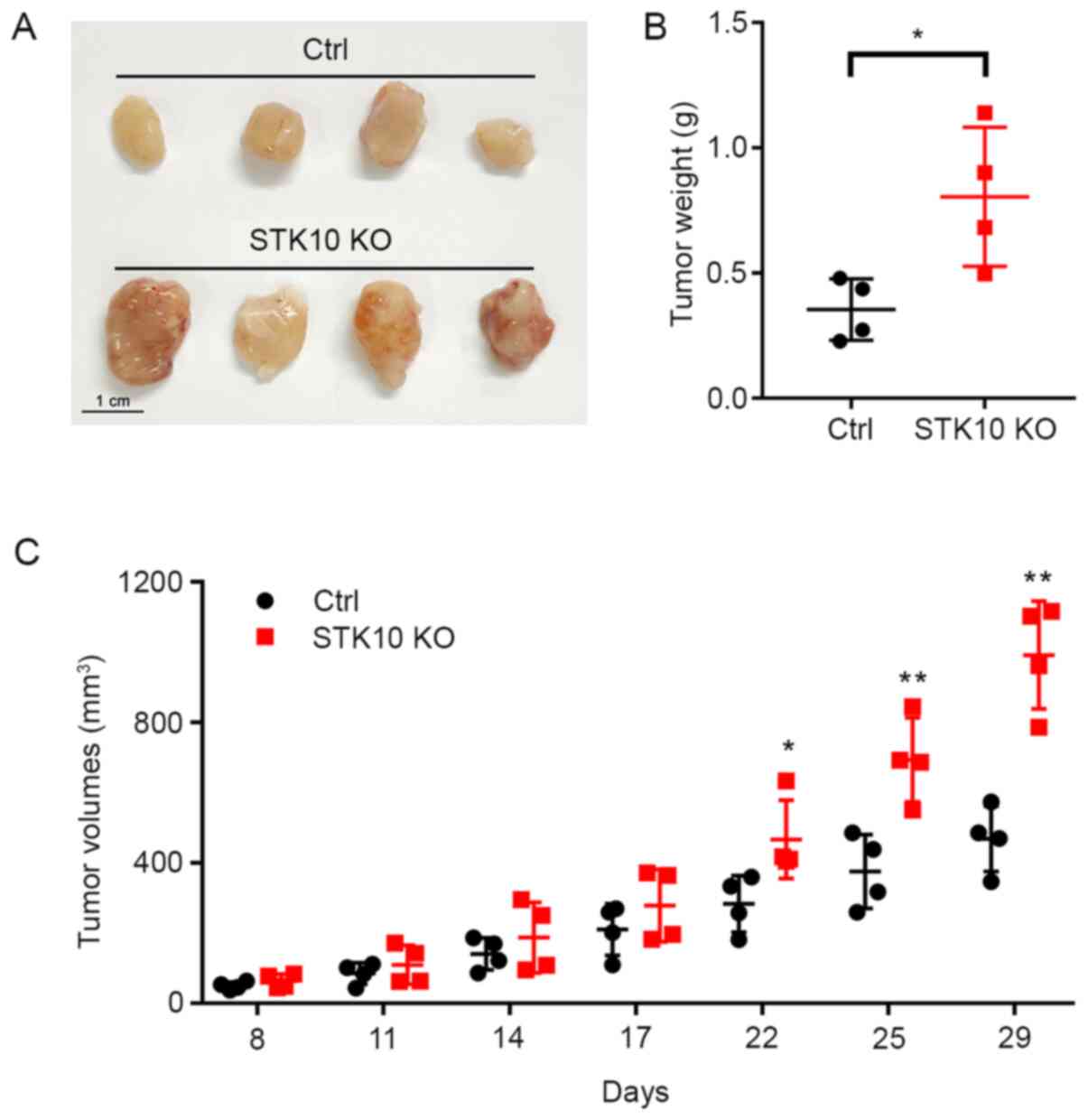
immunodeficient mice were investigated. As shown in Fig. 3A and B, the tumor weights were significantly
increased in mice bearing DU145 STK10-KO cells (0.805±0.278
vs. 0.354±0.122 g; P<0.05). Tumor progression was more rapid in
the DU145 STK10-KO group than those inoculated with DU145
control cells (Fig. 3C). The
difference in tumor volume was significant after 22 days of
subcutaneous injection, especially on days 25 and 29 (P<0.01).
These results demonstrated that STK10 deletion facilitated
tumorigenesis in a xenograft tumor model in vivo.
Disruption of STK10 downregulates the
phosphorylation of p38 MAPK and ERM, and influences the expression
levels of proteins related to cell migration and proliferation in
PCa cells
To investigate the underlying mechanism through
which STK10 regulates the biological characteristics of PCa cells,
the expression and phosphorylation levels of ERM proteins were
determined, as STK10 is one of the kinases responsible for the
phosphorylation of ERM proteins (18). The results of western blotting
showed that the phosphorylation levels of the ERM-family of
proteins were significantly decreased in STK10-KO cells
compared with that in the control cells (Fig. 4A). Numerous studies have
demonstrated that MAPKs are critical regulators of the survival and
migration of cancer cells. Next, the effects of STK10 deletion on
the modulation of ERK-MAPK, PI3K and p38 MAPK pathways were
assessed. Amongst all the MAPK signaling proteins that were tested
in this study, the levels of phosphorylated p38 MAPK was notably
decreased following targeted deletion of STK10, whereas the levels
of PI3K and phosphorylated ERK were mostly unaffected (Fig. 4A). Since STK10 KO promoted cell
proliferation, but inhibited cell migration, the expression levels
of proteins that are closely related to cell proliferation and
migration were then examined (Fig.
4A). The results showed that Cyclin D1 and Cyclin D3, which
promote cell cycle progression, were upregulated in STK10-KO
cells. However, the transcription factor ZEB1 was downregulated in
STK10-KO cells, which has been demonstrated to have a
significant effect on the expression levels of E-Cadherin proteins
in numerous studies (22). Thus,
compared with the control cells, STK10-KO cells exhibited
higher E-Cadherin protein expression levels, which may have effects
on the migratory ability of cells.
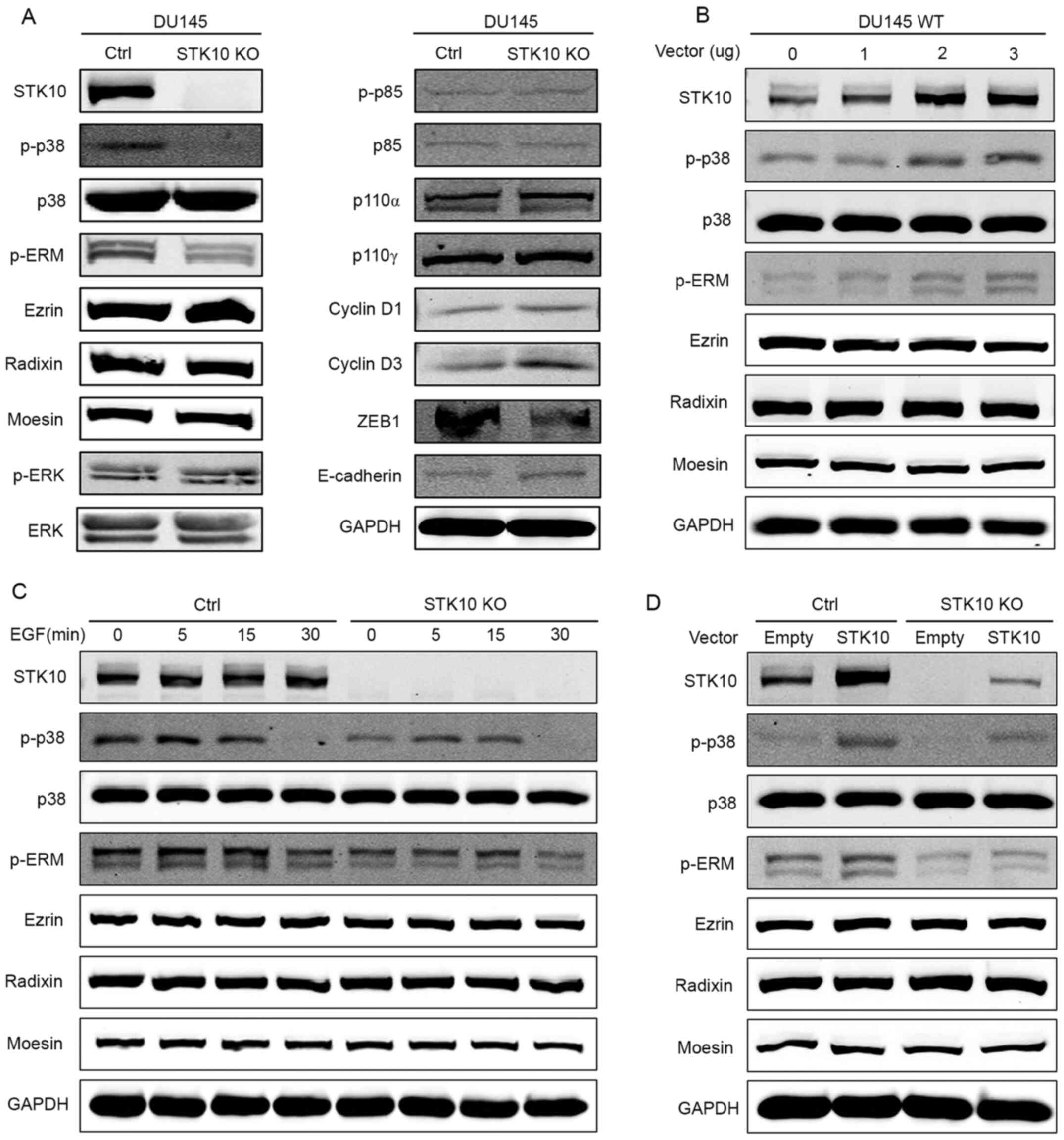 | Figure 4STK10 KO downregulates the
phosphorylation level of p38 MAPK and ERM. (A) Western blot
analysis showed decreased p38 MAPK and ERM phosphorylation, and
increased levels of ZEB1, E-Cadherin, Cyclin D1 and Cyclin D3, but
no change in activation of ERK or PI3K in the STK10-KO DU145
cells. (B) DU145 wild-type cells were transfected with the
indicated quantity of STK10 expression vectors (0, 1, 2 and
3 µg) or empty vectors (the total quantity of vectors was 3 µg).
The phosphorylation levels of p38 MAPK and ERM were assayed using
western blotting. (C) DU145 control and STK10-KO cells were
treated with EGF (50 ng/ml) for the indicated times (0, 5, 15 and
30 min). The phosphorylation levels of p38 MAPK and ERM were
assayed using western blotting. (D) DU145 control and
STK10-KO cells were transfected with 3 µg STK10 expression
vectors or empty vectors. The phosphorylation levels of p38 MAPK
and ERM were assayed using western blotting. STK10, serine
threonine kinase 10; KO, knockout; ERM, ezrin-radixin-moesin; ZEB1,
zinc finger E-box binding homeobox 1; EGF, epidermal growth factor;
p-, phospho-; ZEB, zinc finger E-box-binding homeobox 1; p110,
phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic
subunit. |
It is well known that EGFR and its downstream
pathways are associated with the pathological development and
progression of PCa. As shown in Fig.
4C, DU145 cells were treated with EGF (50 ng/ml) for 0, 5, 15
and 30 min. The phosphorylation levels of p38 MAPK and ERM
increased and decreased as the time changed. However,
phosphorylation levels of p38 MAPK and ERM in STK10-KO cells
were consistently lower than that in the control cells, and the
time required to reach peak ERM phosphorylation was delayed. To
further explore the relationship between STK10 and p38,
overexpression experiments were conducted. Firstly, 0, 1, 2 or 3 µg
of the STK10 expression vectors were transfected into DU145
wild-type cells, and the phosphorylation levels of p38 MAPK and ERM
were determined using western blotting. The results showed that the
phosphorylation levels of p38 MAPK and ERM increased with the
increase in STK10 protein expression levels (Fig. 4B). Next, an STK10 expression vector
(3 µg) or an empty vector were transfected into DU145 control cells
and STK10-KO cells. The results showed that when STK10
protein was expressed in STK10-KO cells, phosphorylation
levels of p38 MAPK and ERM increased (Fig. 4D). These data indicated that the
changes seen in STK10-KO DU145 PCa cells may be due to
dysregulated activation of the p38 MAPK and ERM proteins signaling
pathways.
Discussion
Being an extremely heterogeneous malignant disease,
PCa is characterized by abnormal proliferation of cells in the
prostate gland and metastasis of tumor cells to other tissues. It
has been demonstrated that age, dietary habits, family history,
race and some genetic factors are associated with the etiology of
PCa (23). However, the exact
pathogenesis of this disease is still not fully understood.
Therefore, it is necessary to identify novel risk factors and
clarify the underlying molecular mechanisms involved in the
development and progression of PCa.
In the present study, the role of STK10 in the
regulation of the biological characteristics of PCa were assessed
using a CRISPR-Cas9-mediated STK10-KO DU145 cell line. The
proliferation, cell cycle progression, apoptosis, migration and
tumorigenic activity in these cells were detected using CCK-8 and
BrdU incorporation assays, Annexin V/PI staining, a Transwell assay
and a xenograft tumor model in vivo. The results showed that
STK10 depletion promoted proliferation and cell cycle progression,
and facilitated the growth of tumor xenografts in vivo;
however, it inhibited the apoptosis and migration of PCa cells.
Next, the phosphorylation and expression levels of ERM proteins,
which are substrates of STK10, were determined. As
cytoskeleton-plasma membrane linker proteins, ERM proteins are
essential in the metastasis of various types of cancer via
regulation of cell morphogenesis, cell survival, cell motility and
signaling transduction (11,15,24,25).
The present data showed that STK10 KO resulted in decreased
ERM phosphorylation, which indicated that STK10 is involved in the
migration of PCa cells partially through regulating the activity of
ERM proteins.
Given the widespread role of MAPK signaling pathways
in the cellular functions of cancer cells, the expression and
phosphorylation levels of ERK-MAPK, PI3K and p38 MAPK were
detected. Western blotting revealed that STK10 knockdown did not
affect the expression and activation of ERK or PI3K, but
significantly suppressed the activation of p38 MAPK. A wide range
of studies have demonstrated that p38 MAPK signaling inhibits cell
proliferation and promotes cell migration in various malignant
types of cancer (26-28).
ZEB1 is an epithelial-mesenchymal-transition
(EMT)-related transcription factor that is located downstream of
the MAPK pathway; it can increase EMT progression in PCa (29). ZEB1 can downregulate E-Cadherin
levels and promote cancer invasion (22). Cyclin D1 and Cyclin D3 encode the
regulatory subunit of a holoenzyme and promote progression through
the G1-S phase of the cell cycle, which serve pivotal roles in the
development of a subset of cancers, including PCa, and are
regulated by p38 and other kinases (30-32).
The results of the present study showed that the protein expression
levels of E-Cadherin, Cyclin D1 and Cyclin D3 were upregulated in
STK10-KO cells, whereas the transcription factor ZEB1 was
downregulated in STK10-KO cells. Thus, the changes in cell
proliferation in vitro, tumorigenesis in vivo and
migration of STK10-KO DU145 PCa cells, could be attributed
to the low activity of p38 MAPK. Although several reports have
revealed that p38 MAPK is indirectly implicated in the
phosphorylation of ERM proteins (33,34),
the relationship between p38 MAPK and ERM kinase STK10 remains to
be further elucidated. Nonetheless, the present study demonstrated
a novel role of STK10 in the regulation of the p38 MAPK signaling
pathway.
Taken together, these data suggest that STK10 KO
promotes PCa cell proliferation by inhibiting p38 MAPK activation,
and suppresses PCa cell migration mainly via inhibiting P38 MAPK
signaling and ERM protein activation. A limitation of this study is
the use of only one cell line. The conclusions drawn would be more
convincing if similar results were observed in different PCa cell
lines. Although more precise studies are required to clarify the
functional role of STK10 in the pathogenesis of PCa, to the best of
our knowledge, the present study is the first to demonstrate a
possible role of STK10 in the proliferation and metastasis of PCa,
and provides novel information that may be useful for the
prevention, diagnosis and treatment of this disease. In addition,
this information may prove to be important in elucidating the
biology underlying PCa, which will be of great benefit to patients
in the future.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by grants from the National
Natural Science Foundation of China (grant nos. 81671538 and
81971462), the Shanghai Municipal Bureau of Health for researchers
(grant nos. 201740191 and 20174Y0120) and the grant from Science
and Technology Commission of Shanghai Municipality (grant no.
18ZR1423500).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ and HZ conceived and designed the study. LZ, RG,
JM, JW and CS performed the experiments. SL and LT generated cell
lines. LL, ZW, JL and HZ analyzed data. JL and HZ wrote the paper.
ZW and HZ were responsible for research supervision, coordination
and strategy. LZ and HZ confirm the authenticity of all the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental manipulations were approved by the
Animal Ethics Committee of Ruijin Hospital Affiliated to Shanghai
Jiao Tong University School of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Schatten H: Brief overview of prostate
cancer statistics, grading, diagnosis and treatment strategies. Adv
Exp Med Biol. 1095:1–14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Shah ET, Upadhyaya A, Philp LK, Tang T,
Skalamera D, Gunter J, Nelson CC, Williams ED and Hollier BG:
Repositioning ‘old’ drugs for new causes: Identifying new
inhibitors of prostate cancer cell migration and invasion. Clin Exp
Metastasis. 33:385–399. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Liu G, Ren F and Song Y: Upregulation of
SPOCK2 inhibits the invasion and migration of prostate cancer cells
by regulating the MT1-MMP/MMP2 pathway. PeerJ.
7(e7163)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tang C, Liu T, Wang K, Wang X, Xu S, He D
and Zeng J: Transcriptional regulation of FoxM1 by HIF-1α mediates
hypoxia induced EMT in prostate cancer. Oncol Rep. 42:1307–1318.
2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wang G, Zhao D, Spring DJ and DePinho RA:
Genetics and biology of prostate cancer. Genes Dev. 32:1105–1140.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sartor O and de Bono JS: Metastatic
prostate cancer. N Engl J Med. 378:645–657. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nguyen DX, Bos PD and Massagué J:
Metastasis: From dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Massagué J and Obenauf AC: Metastatic
colonization by circulating tumour cells. Nature. 529:298–306.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Arpin M, Chirivino D, Naba A and
Zwaenepoel I: Emerging role for ERM proteins in cell adhesion and
migration. Cell Adhes Migr. 5:199–206. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
McClatchey AI: ERM proteins. Curr Biol.
22:R784–R785. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ponuwei GA: A glimpse of the ERM proteins.
J Biomed Sci. 23(35)2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Louvet-Vallée S: ERM proteins: From
cellular architecture to cell signaling. Biol Cell. 92:305–316.
2000.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Clucas J and Valderrama F: ERM proteins in
cancer progression. J Cell Sci. 128(1253)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
McClatchey AI: ERM proteins at a glance. J
Cell Sci. 127:3199–3204. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kuramochi S, Moriguchi T, Kuida K, Endo J,
Semba K, Nishida E and Karasuyama H: LOK is a novel mouse
STE20-like protein kinase that is expressed predominantly in
lymphocytes. J Biol Chem. 272:22679–22684. 1997.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Belkina NV, Liu Y, Hao JJ, Karasuyama H
and Shaw S: LOK is a major ERM kinase in resting lymphocytes and
regulates cytoskeletal rearrangement through ERM phosphorylation.
Proc Natl Acad Sci USA. 106:4707–4712. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Endo J, Toyama-Sorimachi N, Taya C,
Kuramochi-Miyagawa S, Nagata K, Kuida K, Takashi T, Yonekawa H,
Yoshizawa Y, Miyasaka N, et al: Deficiency of a STE20/PAK family
kinase LOK leads to the acceleration of LFA-1 clustering and cell
adhesion of activated lymphocytes. FEBS Lett. 468:234–238.
2000.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang L, Lu SY, Guo R, Ma JX, Tang LY,
Shen Y, Shen CL, Lu LM, Wang ZG, Liu J, et al: Knockout of STK10
promotes the migration and invasion of cervical cancer cells.
Transl Cancer Res. 9:7079–7090. 2020.
|
|
21
|
Kupcho K, Shultz J, Hurst R, Hartnett J,
Zhou W, Machleidt T, Grailer J, Worzella T, Riss T, Lazar D, et al:
A real-time, bioluminescent annexin V assay for the assessment of
apoptosis. Apoptosis. 24:184–197. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Odero-Marah V, Hawsawi O, Henderson V and
Sweeney J: Epithelial-Mesenchymal Transition (EMT) and Prostate
Cancer. Adv Exp Med Biol. 1095:101–110. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Adjakly M, Ngollo M, Dagdemir A, Judes G,
Pajon A, Karsli-Ceppioglu S, Penault-Llorca F, Boiteux JP, Bignon
YJ, Guy L, et al: Prostate cancer: The main risk and protective
factors-Epigenetic modifications. Ann Endocrinol (Paris). 76:25–41.
2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gloerich M, Ponsioen B, Vliem MJ, Zhang Z,
Zhao J, Kooistra MR, Price LS, Ritsma L, Zwartkruis FJ, Rehmann H,
et al: Spatial regulation of cyclic AMP-Epac1 signaling in cell
adhesion by ERM proteins. Mol Cell Biol. 30:5421–5431.
2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lallemand D and Arpin M: Moesin/ezrin: A
specific role in cell metastasis? Pigment Cell Melanoma Res.
23:6–7. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Koul HK, Pal M and Koul S: Role of p38 MAP
kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bradham C and McClay DR: p38 MAPK in
development and cancer. Cell Cycle. 5:824–828. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Moriwaki K and Asahi M: Augmented TME
O-GlcNAcylation promotes tumor proliferation through the inhibition
of p38 MAPK. Mol Cancer Res. 15:1287–1298. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Krebs AM, Mitschke J, Lasierra Losada M,
Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D,
Reichardt W, Bronsert P, et al: The EMT-activator Zeb1 is a key
factor for cell plasticity and promotes metastasis in pancreatic
cancer. Nat Cell Biol. 19:518–529. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Fu M, Wang C, Li Z, Sakamaki T and Pestell
RG: Minireview: Cyclin D1: normal and abnormal functions.
Endocrinology. 145:5439–5447. 2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lee RJ, Albanese C, Stenger RJ, Watanabe
G, Inghirami G, Haines GK III, Webster M, Muller WJ, Brugge JS,
Davis RJ, et al: pp60(v-src) induction of cyclin D1 requires
collaborative interactions between the extracellular
signal-regulated kinase, p38, and Jun kinase pathways. A role for
cAMP response element-binding protein and activating transcription
factor-2 in pp60(v-src) signaling in breast cancer cells. J Biol
Chem. 274:7341–7350. 1999.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang B, Wang Z, Han L, Gong S, Wang Y, He
Z, Feng Y and Yang Z: Prognostic significance of cyclin D3
expression in malignancy patients: A meta-analysis. Cancer Cell
Int. 19(158)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang C, Wu Y, Xuan Z and Zhang S, Wang X,
Hao Y, Wu J and Zhang S: p38MAPK, Rho/ROCK and PKC pathways are
involved in influenza-induced cytoskeletal rearrangement and
hyperpermeability in PMVEC via phosphorylating ERM. Virus Res.
192:6–15. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Koss M, Pfeiffer GR II, Wang Y, Thomas ST,
Yerukhimovich M, Gaarde WA, Doerschuk CM and Wang Q:
Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha and
modulate permeability increases in human pulmonary microvascular
endothelial cells. J Immunol. 176:1218–1227. 2006.PubMed/NCBI View Article : Google Scholar
|