Introduction
Neuroinflammation is a protective mechanism of the
central nervous system (CNS) following injury. However, if
neuroinflammation persists, it can lead to many neurodegenerative
diseases, such as Alzheimer's disease (AD) (1), Huntington's disease (HD) (2) and Parkinson's disease (PD) (3). Neuroinflammation is characterized by
abnormal activation of glial cells and the presence of inflammatory
mediators in the CNS microenvironment (4). Microglia are tissue macrophages in the
CNS, which have unique origins and functions (5). Under normal conditions, microglia
protect neurons by immune defense, phagocytic activity, antigen
presentation and secretion of immunomodulatory factors (6). However, in response to injury,
infection or inflammation, microglia are readily activated and
release various pro-inflammatory factors, including IL-6, TNF-α,
C-C motif chemokine ligand 2 and reactive oxygen species (ROS),
which increase the expression of inducible nitric oxide synthase
(iNOS) and cause accumulation of excessive nitric oxide (NO)
(7).
As a neurotransmitter and second messenger molecule,
NO mediates a variety of neuronal functions in the brain (8). NO is synthesized by iNOS, which is
detected at low levels in healthy brains and spinal cords (8). However, continuous activation of
microglia induces abnormal expression of iNOS proteins, leading to
a large accumulation of NO (9). The
latter can damage the cell membrane structure, affect DNA
transcription and protein synthesis, and directly damage neurons
(10); moreover, it can be further
oxidized by oxygen free radicals to form highly toxic
peroxynitrite, which can cause nitration of tyrosine residues in
cells and damage neurons (11). In
addition, NO causes neuronal degeneration by changing the
intracellular Fe2+ concentration (12). Knockdown of iNOS genes in primary
glial cells inhibits the production of NO, thus reducing the
degeneration of CNS (13). A
previous study suggested that iNOS- and NO-induced
neuroinflammatory responses in the CNS could play an important role
in the development of neurodegenerative lesions (13).
Hispidin is a polyphenolic substance that was first
isolated from the fruiting body of hispidus in 1889 by Zopf,
where its structure was later identified by Edwards et al
(14) as
6-(3,4-dihydroxyphenyl)-4-hydroxy-2-pyrone in 1961(15). Recent, hispidin were isolated from
ethanolic extracts and fermentation products of medicinal mushroom
Phellinus linteus (16). In
addition, a previous study demonstrated that hispidin has
anti-inflammatory, anti-bacterial, anti-oxidant, anti-cancer and
hypoglycemic regulatory functions (17). However, the inhibitory effect of
hisplidin on NO production by the lipopolysaccharide (LPS)-induced
microglia remains unclear. Therefore, the present study aimed to
investigate the effects of hispidin on NO production and iNOS
expression in BV-2 microglial cells, along with its underlying
mechanism.
Materials and methods
Chemicals and antibodies
Hispidin (cat. no. H5257),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and LPS (cat. no. L4391; from Escherichia coli serotype
0111:B4) were purchased from Sigma-Aldrich; Merck KGaA. Fetal
bovine serum (FBS, cat. no. SH30070.03) and DMEM (cat. no.
SH30003.01) were purchased from HyClone; Cytiva. Hoechst 33258
(cat. no. B8030), penicillin/streptomycin (P/S, cat. no. P1400),
N-acetylcysteine (NAC, cat. no. IA0050) were purchased from Beijing
Solarbio Science & Technology Co., Ltd. PD98059 (ERK signal
inhibitor; cat. no. S1805), SB203580 (P38 signal inhibitor; cat.
no. S1863), SP600125 (JNK signal inhibitor; cat. no. S1876),
CM-H2DCFDA (cat. no. S0033S) and Dihydroethidium (DHE;
cat. no. S0063) were obtained from Beyotime Institute of
Biotechnology. Rabbit monoclonal antibodies for phosphorylated
(p)-STAT3 (cat. no. bsm-52210R, dilution: 1:1,000), STAT3 (cat. no.
bsm-52235R, dilution: 1:500), p-JNK (cat. no. bsm-52462R, dilution:
1:1,000), IκB-α (cat. no. bsm-52169R, dilution: 1:500) and rabbit
polyclonal antibodies for p-JAK1 (cat. no. bs-3238R dilution:
1:1,000), JAK1 (cat. no. bs-1439R, dilution: 1:1,000), p-ERK (cat.
no. bs-3238R, dilution: 1:1,000), JNK (cat. no. bs-10562R,
dilution: 1:1,000), p-P38 (cat. no. bs-5477R, dilution: 1:1,000)
and P38 (cat. no. bs-0637R, dilution: 1:1,000) were purchased from
BIOSS. Mouse monoclonal antibodies for β-actin (cat. no. ab8226,
dilution: 1:1,000) were purchased from Abcam. Horseradish
peroxidase (HRP)-conjugated goat anti-rabbit IgG (cat. no. D111018,
dilution: 1:5,000) and anti-mouse IgG (cat. no D110097, dilution:
1:5,000) were purchased from Sangon Biotech Co., Ltd.. Cell culture
dishes were obtained from Wuxi NEST Biotechnology Co., Ltd.
TRIzol® reagent (cat. no. 10296028) was purchased from
Invitrogen; Thermo Fisher Scientific, Inc. PrimeScript™
reagent Kit with gDNA Eraser (cat. no. RR047A) was purchased from
Takara Bio, Inc. The primers for iNOS and GAPDH were synthesized by
Sangon Biotech Co., Ltd. ECL Western Blotting Substrate (cat. no.
PE0010) was obtained from Beijing Solarbio Science & Technology
Co., Ltd.
Cell culture
BV-2 microglial cells were obtained from the
American Type Culture Collection and cultivated in DMEM
supplemented with 10% FBS and 1% P/S (100 U/ml and 100 µg/ml,
respectively) at 37˚C and 5% CO2 under saturated
humidity. Cells were passaged when they were ~80% confluent.
According to different experimental arrangements, cells were
inoculated into the cell culture dish based on the required number
of cells, and a follow-up study was conducted after the cells
adhered to the wall.
Cell viability assay
BV-2 microglial cells were seeded in 96-well plates
at a concentration of 4x103 cells per well and cultured
in DMEM. The cells were treated with various concentrations of
hispidin (0-20 µg/ml) for 24 h. A solution of 5 mg/ml MTT was
subsequently added to each well and the cells were incubated for 4
h at 37˚C in an atmosphere containing 5% CO2.
Subsequently, the supernatant was removed and formazan was
dissolved in DMSO. Absorbance was measured at 490 nm using a UV Max
Kinetic Microplate Reader (Molecular Devices, LLC).
Detection of NO production by Griess
reagent
NO production was assessed based on the accumulation
of nitrite in the medium using a colorimetric reaction with Griess
reagent [0.1% N-(1-naphthyl) ethylenediamine dihydrochloride, 0.1%
sulfanilamide, and 2.5% H3PO4]. Culture
supernatants were collected and mixed with an equal volume of
Griess reagent. Absorbance was measured at 540 nm using the UV Max
Kinetic Microplate Reader.
Detection of iNOS mRNA expression by
reverse transcription-semi quantitative PCR
Total cellular RNA was prepared using the
TRIzol® reagent, followed by cDNA synthesis using by the
reverse transcription kit in accordance with the manufacturer's
protocols. The reverse transcription reaction conditions were set
at 37˚C for 15 min, followed by 85˚C for 5 sec and finally reduced
to 4˚C. The cDNA was amplified (SuperScript™ IV One-Step
RT-PCR System with ezDNase™; cat. no. 12595025; Thermo
Fisher Scientific, Inc.) using the following PCR primers: iNOS
forward, 5'-CCCTTCCGAAGTTTCTGGCAGCAGC-3' and reverse,
5'-GGCTGTCAGAGCCTCGTGGCTTTGG-3', GAPDH forward,
5'-TGTGTCCGTCGTGGATCTGA-3' and reverse,
5'-CCTGCTTCACCACCTTCTTGA-3'. Thermocycling was performed using an
initial 94˚C hold step for 5 min. This hold step was followed by
25-30 cycles of 94˚C for 30 sec, 65˚C (iNOS) or 52˚C (GAPDH) for 30
sec and 72˚C for 30 sec and a final extension step for 5 min at
72˚C. The amplified samples were separated in 1% agarose gels with
ethidium bromide and images were taken with the Alpha Gel Imaging
System (Alpha Imager HP; Version 3.5.0; Protein Simple).
Western blotting
Following treatment with LPS or hispidin, cells were
washed twice with PBS, lysed with protein lysis buffer [20 mM
HEPES-OH (pH 7.0), 50 mM NaCl, 10% glycerol and 0.5% Triton X-100]
and incubated with 0.5 µg/ml leupeptin, 0.7 µg/ml pepstatin A, 0.1
mM 4 (2 aminoethyl) benzenesulfonyl fluoride and 2 µg/ml aprotinin
for 30 min at 4˚C. The concentration of the obtained proteins was
determined using Coomassie blue staining. The protein assay system
was prepared (800 µl DDW + 200 µl Coomassie Blue + 1 µl protein
sample). The OD value was detected at 595 nm using a UV
spectrophotometer and the obtained OD value was subsequently
substituted into a standard curve to obtain the protein
concentration. The proteins were then denatured for 5 min at 100˚C
and 20 µg protein was separated using 12% SDS-PAGE and transferred
onto nitrocellulose membranes (EMD Millipore). The membranes were
blocked with 5% skimmed milk for 30 min at room temperature. They
were then incubated with primary antibodies at 4˚C overnight.
Membranes were washed five times with Tris buffered saline (TBS)
containing Tween [10 mM Tris HCl (pH 7.5), 150 mM NaCl and 0.2%
Tween-20] and subsequently incubated with HRP-conjugated goat
anti-rabbit IgG or anti-mouse IgG (dilution, 1:5,000) for 1 h at
room temperature. After five washes with TBS to remove excess
antibody, an appropriate amount of ECL Plus (cat. no. PE0010; from
Beijing Solarbio Science & Technology Co., Ltd.) was added to
each membrane and specific binding was then detected using a
chemiluminescence detection system (Amersham Imager 600; Cytiva).
All quantification of bands was done using ImageJ 1.52a (National
Institutes of Health).
Measurement of ROS by flow cytometry
and fluorescence microscopy
To determine ROS levels, the BV-2 microglial cells
were incubated at 37˚C for 15 min with 10 mM CM-H2DCFDA.
The CM-H2DCFDA fluorescence intensity of 10,000 cells
was evaluated by flow cytometry (BDFACSCalibur™, BD
Biosciences). The results were analyzed using the WinMDI (Version
2.9, BD Biosciences) software.
Measurement of ROS by fluorescence
microscopy
Changes in cellular ROS levels were determined using
1 µM DHE and 2 µg/ml Hoechst 33258 (to observe the nucleus) at 37˚C
for 15 min and washed with PBS to detect changes in cellular ROS
levels. After washing with PBS, the magnification of the microscope
was adjusted to x200 and images were taken with a fluorescent core
cell culture microscope (EVOS® XL core cell culture
microscope; Thermo Fisher Scientific, Inc.) to qualitatively
observe the fluorescence intensity.
Statistical analysis
Data are presented as the means ± standard error of
the mean. Differences between experimental groups were analyzed by
one-way analysis of variance and a Tukey's test. GraphPad Prism
software version 4.0 (GraphPad Software, Inc.) was used to analyze
all results and P<0.05 was considered to indicate a
statistically significant difference.
Results
Hispidin inhibits the production of NO
in LPS-treated BV-2 microglial cells
To determine the cytotoxic effects of hispidin on
BV-2 microglial cells, the BV-2 microglial cells were treated with
various concentrations (0, 1, 5, 10 and 20 µg/ml) of hispidin for
24 h. As presented in Fig. 1A, 20
µg/ml hispidin had no effect on the viability of BV-2 microglial
cells, hence indicating that the inhibitory effect of hispidin on
LPS-induced inflammation was not due to cytotoxicity. Therefore, in
subsequent experiments, 20 µg/ml was used as the maximum treatment
concentration. To determine whether hispidin could exert any
anti-inflammatory effect, the levels of NO and ROS were measured in
BV-2 microglial cells after LPS treatment. Cells were pretreated
with hispidin (1, 5, 10 or 20 µg/ml) for 30 min and subsequently
treated with LPS for the indicated times (0, 1, 3, 6, 12 or 24 h).
The results demonstrated that hispidin decreased the production of
NO in LPS-treated BV-2 microglial cells in a dose- and
time-dependent manner (Fig. 1B and
C). Intracellular ROS levels were
detected by flow cytometry and fluorescence microscopy, and the
results demonstrated that LPS treatment increased the ROS level of
BV-2 microglial cells, while hispidin pretreatment significantly
reduced the intracellular ROS levels (Fig. 1D-F). Together, the results indicated
that hispidin regulates the activity of BV-2 microglial cells by
inhibiting the production of inflammatory mediators.
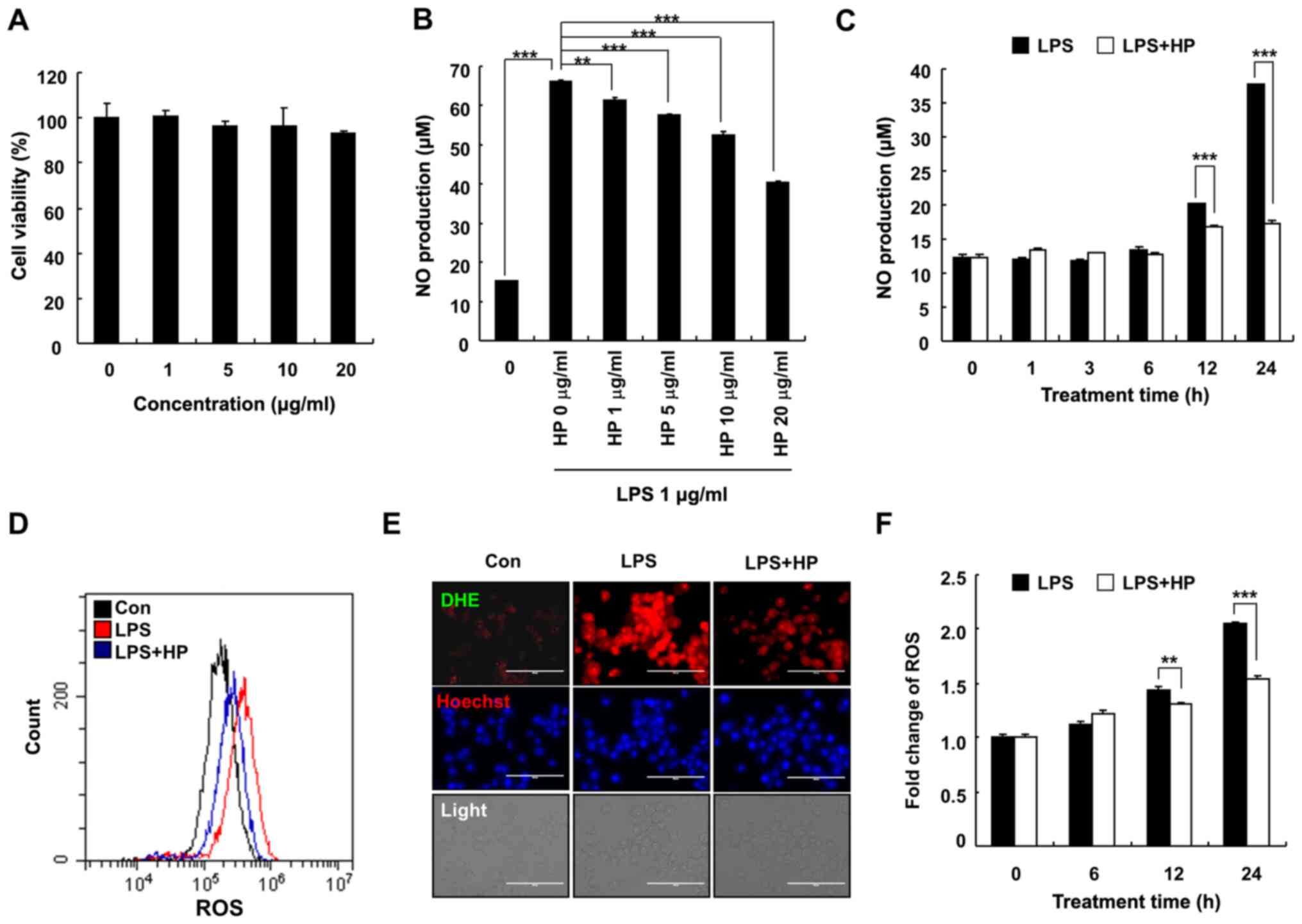 | Figure 1HP inhibits the production of NO in
LPS-treated BV-2 microglial cells. (A) Cells were treated with
various concentrations of HP and cultivated for 24 h. Cell
viability was evaluated by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
(B) The level of NO was analyzed in the culture medium using Griess
reagent. (C) Cells were pre-treated with designated concentrations
of HP for 30 min prior to LPS treatment (1 µg/ml) and were
incubated for 24 h. The cells were pretreated with 20 µg/ml HP for
30 min, followed by treatment with LPS (1 µg/ml) for the indicated
durations. Cells were pre-treated with 20 µg/ml HP for 30 min,
followed by treatment with LPS (1 µg/ml) for 24 h. The level of
cellular ROS was detected using (D) flow cytometry when stained
with CM-H2DCFDA and (E) fluorescence microscopy when
stained with Hoechst/DHE (blue/red). Scale bar, 200 µm. (F)
Following treatment with LPS (1 µg/ml) for different durations, the
level of cellular ROS was detected using flow cytometry after
staining with CM-H2DCFDA. Data are presented as the
means ± standard error of the mean for three different samples.
**P<0.01, ***P<0.001. HP, hispidin; NO,
nitric oxide; LPS, lipopolysaccharide; ROS, reactive oxygen
species; DHE, dihydroethidium; Con, control. |
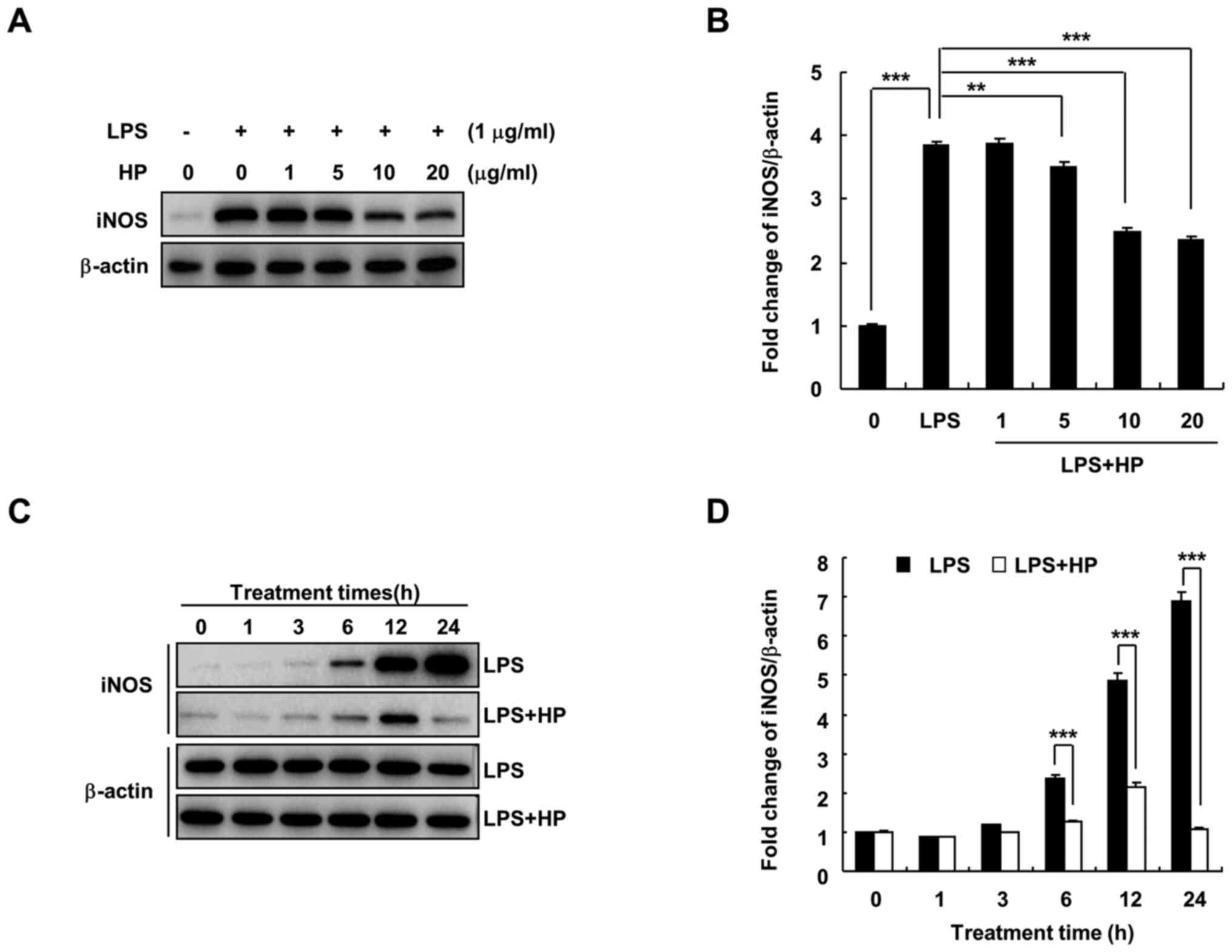
Hispidin inhibits the expression of
iNOS protein in LPS-treated BV-2 microglial cells
Under the pathological conditions of inflammation
and hypoxia and in the presence of tumors, iNOS expression is
increased and catalyzes the production of NO (7). When BV-2 microglial cells are
stimulated by substances, such as LPS, the expression of iNOS
increases, which in turn induces the production of large quantities
of NO and promotes the development of inflammation and other
diseases (18). As shown in
Fig. 2, hispidin treatment was
found to significantly decrease LPS-induced iNOS protein expression
in a dose- and time-dependent manner.
Hispidin inhibits NO production and
iNOS expression by inhibiting MAPK signaling pathway in LPS-treated
BV-2 microglial cells
To investigate the mechanism of inhibition of
LPS-induced NO production and iNOS expression by hispidin in BV-2
microglial cells, phosphorylation of MAPK signaling pathway-related
proteins (JNK, P38 and ERK) by hispidin was investigated. BV-2
microglial cells were pretreated with 20 µg/ml hispidin for 30 min
and then treated with LPS (1 µg/ml) for the indicated times. As
expected, LPS treatment increased the phosphorylation levels of
JNK, P38 and ERK. However, after pretreatment with hispidin, the
phosphorylation levels were downregulated and the P38 signaling
pathway was inhibited to the greatest extent (Fig. 3A-D). Next, to clarify whether
hispidin inhibits LPS-induced NO production in BV-2 microglial
cells by inhibiting the MAPK signaling pathway, the inhibitory
effects of MAPK inhibitors (P38, SB203580; ERK, PD98059; and JNK,
SP600125), ROS scavenger (NAC) and selective inhibitor of iNOS
(S-methylisothiourea sulfate; SMT) on LPS-induced NO production and
iNOS protein expression were compared. The results indicated that
all reagents, except NAC, reduced NO production and iNOS protein
expression in LPS-treated BV-2 microglial cells (Fig. 3E-G).
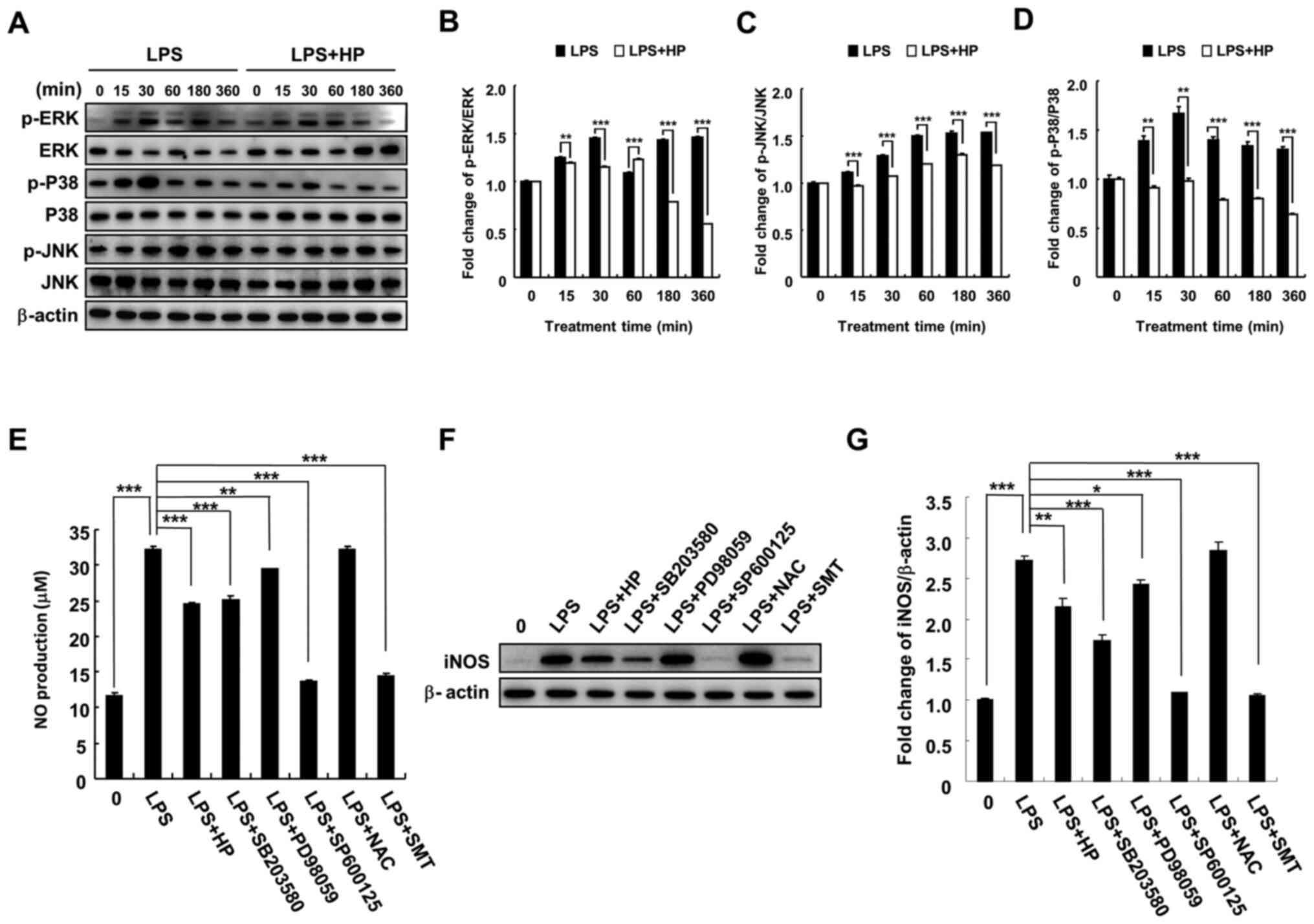 | Figure 3HP inhibits NO production and iNOS
expression via inhibition of the MAPK signaling pathway in
LPS-treated BV-2 microglial cells. Cell were pre-treated with 20
µg/ml HP for 30 min, followed by treatment with LPS (1 µg/ml) as
well as inhibitors (P38, SB203580; ERK, PD98059; and JNK, SP600125)
for the indicated durations. (A) Protein expression levels of
p-ERK, ERK, p-P38, P38, p-JNK and JNK were detected by western
blotting. (B-D) The associated protein expression levels are
represented as the means ± SD. (E) Level of NO was evaluated in the
culture medium using Griess reagent. (F) Protein expression of iNOS
was detected by western blotting. (G) Associated protein expression
levels are presented as means ± SD. *P<0.05,
**P<0.01 and ***P<0.001. HP, hispidin;
NO, nitric oxide; iNOS, inducible NO synthase; LPS,
lipopolysaccharide; p, phosphorylated; NAC, N-acetylcysteine; SMT,
S-methylisothiourea sulfate. |
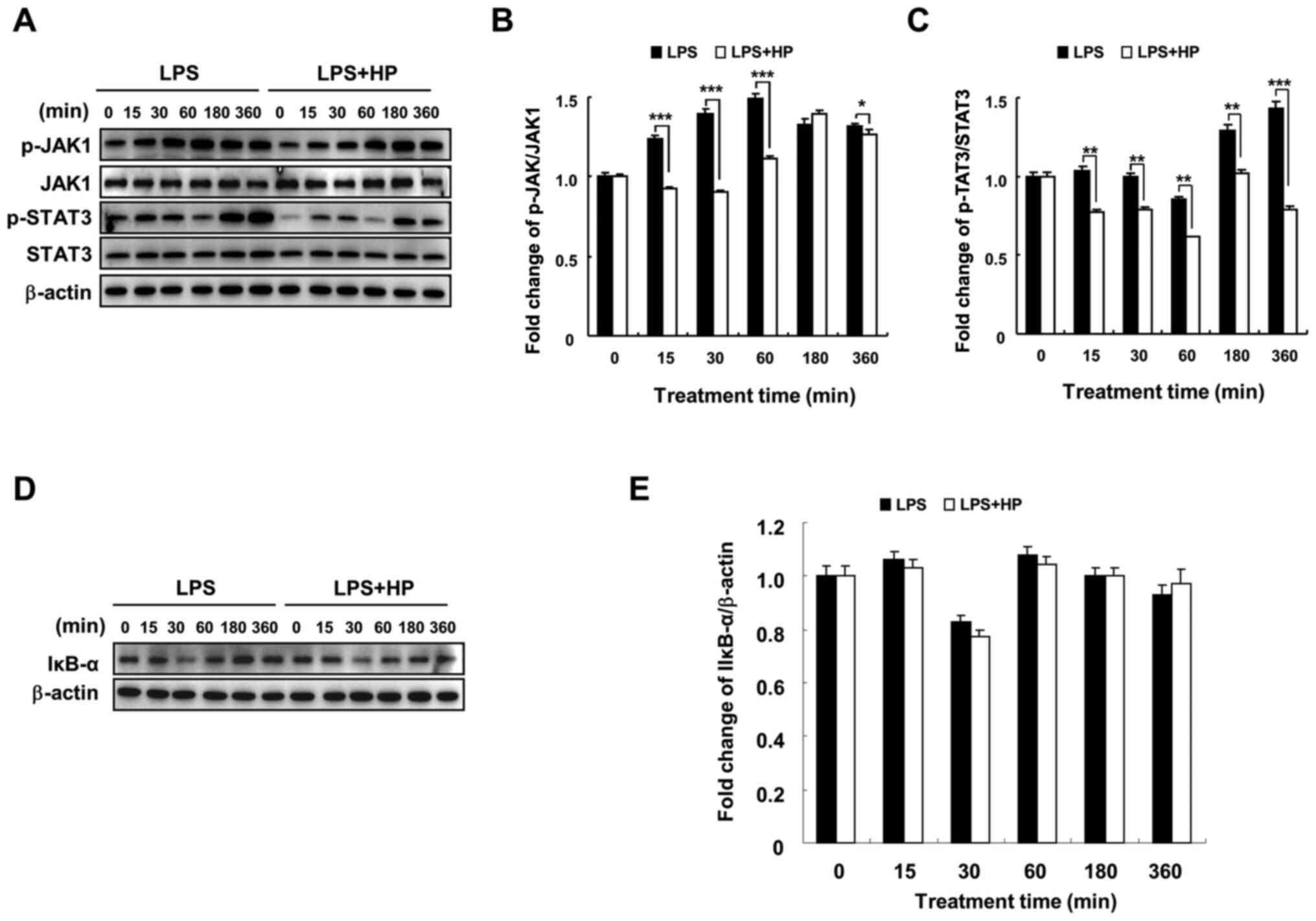
Hispidin inhibits the JAK1/STAT3
signaling pathway, but does not affect the NF-κB signaling pathway
in LPS-treated BV-2 microglial cells
In a previous study, hispidin was found to inhibit
NF-κB, MAPK and JAK1/STAT3 signaling pathways in RAW264.7
macrophages and played an anti-inflammatory role (17). The current results indicated that
hispidin could inhibit the MAPK signaling pathway in LPS-treated
BV-2 microglial cells (Fig. 3) and
changes were detected in two other signaling pathways by western
blotting. Hispidin treatment was observed to downregulate the
expression of p-JAK1 and p-STAT3 compared to that in the
LPS-treated group; however, it had no significant effect on the
protein expression of IκB-α (Fig.
4). These results indicate that although hispidin inhibited the
LPS-induced JAK1/STAT3 signaling pathway in BV-2 microglial cells,
it had no effect on the NF-κB signaling pathway.
Discussion
Microglia are resident immune cells in the CNS and
the literature has confirmed that they have a macrophage-like
phenotype, with regard to morphological and quantitative changes
during microglial cell activation (5). Microglia form one of the main factors
in the occurrence and development of neuroinflammation (19). Undifferentiated, resting microglia
are responsible for monitoring and patrolling the brain
environment, as well as activating a response to injury and
infection (20). Activated
microglia trigger the inflammatory immune response in the nervous
system by releasing cytokines, including IL-1, IL-6 and TNF-α,
which clears damaged and necrotic cells or neurons from the lesion
(21). However, when microglial
cells remain activated, they secrete a large number of neurotoxic
mediators, including inflammatory cytokines, inflammatory
chemokines, ROS, NO and the excitotoxic amino acid glutamate, which
disrupt synapses and neural connections in the brain, triggering a
series of neuroinflammatory responses that result in
neurodegenerative diseases, such as AD (22). These cytotoxic mediators interact
with each other to amplify their own toxicity, induce neuronal
death, damage the nervous system and increase the risk of central
neurodegenerative diseases (23).
This pathological process further activates the surrounding
microglia, stimulates the excessive release of inflammatory
mediators, induces central neuroinflammatory responses and forms a
vicious cycle of central inflammatory cascade responses (24). Therefore, the aforementioned studies
have collectively indicated that inflammation of brain tissue due
to continuous activation of microglia is the key to the induction
of neurodegenerative diseases.
NO is a highly unstable biological free radical,
which is widely distributed in various tissues of mammals,
particularly the nervous tissue (25). It is critical in various important
physiological functions and is one of the main neurotransmitters in
memory formation (26). NO is
catalyzed by NOS to generate L-arginine; there are three main types
of NOS in the body, namely, eNOS, present in endothelial cells,
nNOS, present in neuronal cells, and iNOS, present in macrophages,
hepatocytes and glial cells (27).
NOS is present in almost all tissues, and multiple subtypes coexist
in the same tissue, for example, all three subtypes are present in
the nervous system (28). As an
intercellular second messenger, NO plays an important role in
cardiovascular, neurological, immunomodulatory, anti-inflammatory
and antitumor processes (29). In
the CNS, NO plays a dual role as an intercellular messenger for
neuroprotection and neurotoxicity (11). NO produced under normal
physiological conditions is beneficial to the functioning of the
nervous system; studies have shown that low concentrations of NO
plays an important role in brain neuromodulation, neurotransmission
and synaptic plasticity (8,30). When endogenous or exogenous NO is
overproduced and released, it becomes a strong neurotoxic substance
that can be involved in a variety of neurodegenerative and
inflammatory pathological processes, leading to neuronal apoptosis
and disease induction (8). A large
volume of evidence has indicated excessive NO and its mediated
apoptosis in organs and tissues to be associated with neurological
diseases, such as epilepsy, AD, PD, amyotrophic lateral sclerosis
and HD, as well as cerebral ischemia/reperfusion injury and Down's
syndrome (18,31-34).
This is of particular interest to neurodegenerative conditions,
such as AD and PD, which increasingly are the focus of research on
the pathogenesis and treatment of such diseases (3,35).
Therefore, effective inhibition of NO production by activated
microglia may be an important strategy to control
neuroinflammation. In the present study, hispidin treatment was
found to significantly decrease NO production and iNOS protein
expression levels in LPS-treated BV-2 microglial cells. The effect
of hispidin on iNOS mRNA was also investigated and hispidin
treatment did not affect the mRNA content of iNOS (Fig. S1); hence, it was speculated that
hispidin inhibits LPS-induced NO production in BV-2 cells by
affecting iNOS protein synthesis, although the probable underlying
mechanism requires further investigation. The current results
indicate that hispidin may play an important role in the
suppression of neuroinflammation.
In addition, the present results demonstrated the
inhibitory effect of hispidin on ROS levels in LPS-treated
microglia. ROS includes oxygen-containing radicals or oxygen atoms
involved in oxidation reactions in the body, as well as peroxides
and superoxides that produce oxygen radical electrons (36,37).
ROS plays a vital role in regulating the process of
neurodegenerative diseases (38,39).
As an upstream regulator of inflammation, ROS increase the
expression of pro-inflammatory genes and induce neurotoxicity
through lipid peroxidation (38,40).
Moreover, oxygen free radicals catalyze the formation of neurotoxic
substances with NO, such as peroxynitrite (36). A pharmacological studies have shown
that oxidative stress damage is closely associated with
neuroinflammatory response, and jointly promotes the occurrence and
development of CNS-degenerative diseases (41,42).
The present findings on the inhibitory effect of hispidin on ROS
production suggested that hispidin could be a potential
anti-inflammatory drug candidate for the treatment of LPS-induced
neuroinflammation.
In our previous study, it was reported that hispidin
exerts anti-inflammatory effects by extensively inhibiting the
activation of three signaling pathways in LPS-induced RAW264.7
macrophages, including NF-κB, MAPK and JAK1/STAT3(17). However, whether hispidin plays a
role against neuroinflammation by inhibiting the signaling pathways
in microglia remains unknown. The NF-κB signaling pathway is
heavily involved in the inflammatory response and consists of
p65/p50 and IκB. Of these, IκB inhibits NF-κB, and the
transcription of hundreds of genes, such as inflammatory factors,
chemical chemokines, enzymes and adhesion molecules can be
regulated only when this inhibition is removed (41). Typically, NF-κB, consisting of
p65/p50 and IκB, is present in the cytoplasm in an inactive state
(43). When a pathogen infects the
cell, it acts as a pattern of pathogen-associated molecules that is
recognized by a pattern recognition receptor on the surface of the
cell membrane and binds to it (44). This receptor-ligand binding
transmits an extracellular infection signal into the cell, which in
turn recruits signal adapter proteins in the cytoplasm to
phosphorylate, ubiquitinate and degrade IκB through activation of
the IκB kinase complex (45). It
can enter the cell nucleus, bind to DNA, and regulate the
transcription and expression of inflammatory cytokines (45). The cytokines produced bind to
receptors on the cell membrane, phosphorylate JAK, which in turn
phosphorylates downstream molecules of STAT, ultimately inducing a
transcriptional response and increasing gene expression of relevant
inflammatory mediators (46).
Notably, the present results demonstrated that LPS decreased the
expression of IκB-α while hispidin had no effect on IκB-α. This
result was inconsistent with the previous finding in RAW264.7, in
which hispidin increased the expression of IκB-α and inhibited
activation of the NF-κB signaling pathway. The appearance of this
discrepancy was of interest and subsequently it was noted that
hispidin had previously been reported as an inhibitor of cell
permeability against protein kinase C (PKC) (47). During macrophage activation, PKC
acts as an intermediate mediator of MAPK signaling pathway
activation (48); while it has been
shown that when PKC binds to a foreign stimulus, such as LPS, it
leads to phosphorylation of IκB-α (49). Therefore, the present study
speculated that the reason why hispidin has different regulatory
effects on the activated signaling pathway in the BV-2 microglial
cells and RAW264.7 is most likely due to the difference in the
origin of the two cells, which leads to differences in PKC protein
expression on the membranes of these two cells. However, this
hypothesis requires further investigation. Meanwhile, the present
study identified that hispidin significantly downregulated the
phosphorylation of JAK1 and STAT3 and inhibited activation of the
JAK1/STAT3 signaling pathway. Although the effect of hispidin on
cytokine production by LPS-activated BV-2 microglial cells was not
examined in the present study, inhibition of the JAK1/STAT3
signaling pathway implied that hispidin influences cytokine
production.
MAPKs play an important regulatory role in the
expression and secretion of a variety of inflammatory mediators
(50,51). In our previous study, it was shown
that inhibition of phosphorylation of JNK inhibits the production
of NO and expression of iNOS protein induced by LPS (17). In the present study, hispidin was
found to inhibit the phosphorylation of p38, ERK and JNK, with p38
demonstrating the most marked change. Furthermore, the inhibitory
effects of MAPK inhibitors, the ROS scavenger (NAC) and the
selective inhibitor of iNOS (SMT) on LPS-induced NO production and
iNOS protein expression were compared. The results demonstrated
that the addition of MAPK signaling pathway inhibitors decreased
LPS-induced NO production and western blot results showed the
reduction of iNOS protein expression. These results collectively
suggested that activation of the MAPK signaling pathway is
associated with the activation of BV-2 microglial cells by LPS.
Conversely, the inhibition of NO by the addition of other
inhibitors may suggest that hispidin could be a potential inhibitor
candidate of iNOS; however, the mechanism of this inhibition
requires further investigation. The scavenging of ROS by NAC did
not inhibit NO production and iNOS protein expression, indicating
that ROS produced by LPS-induced BV-2 microglial cells does not
promote the production of NO and that they may be relatively
independent in the development of neuroinflammation.
Thus, the present study indicated that hispidin
significantly inhibits NO production and iNOS expression by
suppressing the MAPK signaling pathway, though not the NF-κB
signaling pathway, in LPS-activated microglial cells. In addition,
hispidin was shown to inhibit activation of the JAK1/STAT3
signaling pathway, which may have an effect on cytokine release.
These findings may provide a novel insight into the possibility of
hispidin serving as a therapeutic target for clinical treatment of
neuroinflammation.
Supplementary Material
Effect of HP on iNOS mRNA content in
LPS-treated BV-2 microglial cells. Cells were pre-treated with
different concentrations of HP for 30 min prior to LPS treatment (1
μg/ml) and were incubated for 24 h. (A) The content of iNOS
mRNA was evaluated by quantitative PCR. (B) The associated mRNA
levels are presented as means ± SD. HP, hispidin; NO, nitric oxide;
iNOS, inducible NO synthase; LPS, lipopolysaccharide.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no.
2020R1I1A2052417), the Korean Research Institute of Bioscience and
Biotechnology Research Information System (grant no. RBM0112112)
and also supported by the project Sanzong of Heilongjiang Bayi
Agricultural University (grant no. ZRCPY202029).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MHJ, DQC, HNS and TK performed the experiments and
analyzed the data. YHJ prepared screen natural product, analyzed
the data and took part in the study design. YHH collected, analyzed
and interpreted the experimental data. MHJ, DQC and HNS confirm the
authenticity of all the raw data. HS and TK conceived and designed
the project. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
DiSabato DJ, Quan N and Godbout JP:
Neuroinflammation: The devil is in the details. J Neurochem. 139
(Suppl 2):S136–S153. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Walker FO: Huntington's disease. Lancet.
369:218–228. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang J, He C, Wu WY, Chen F, Wu YY, Li WZ,
Chen HQ and Yin YY: Biochanin A protects dopaminergic neurons
against lipopolysaccharide-induced damage and oxidative stress in a
rat model of Parkinson's disease. Pharmacol Biochem Behav.
138:96–103. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Estes ML and McAllister AK: Maternal
immune activation: Implications for neuropsychiatric disorders.
Science. 353:772–777. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Greenhalgh AD, David S and Bennett FC:
Immune cell regulation of glia during CNS injury and disease. Nat
Rev Neurosci. 21:139–152. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Streit WJ: Microglia as neuroprotective,
immunocompetent cells of the CNS. Glia. 40:133–139. 2002.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sun HN, Jin MH, Han B, Feng L, Han YH,
Shen GN, Yu YZ, Jin CH, Lian ZX, Lee DS, et al:
16α,17α-epoxypregnenolone-20-oxime prevent LPS-induced NO
production and iNOS expression in BV-2 microglial cells by
inhibiting JNK phosphorylation. Biol Pharm Bull. 37:1096–1102.
2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Calabrese V, Boyd-Kimball D, Scapagnini G
and Butterfield DA: Nitric oxide and cellular stress response in
brain aging and neurodegenerative disorders: The role of vitagenes.
In Vivo. 18:245–267. 2004.PubMed/NCBI
|
|
9
|
Hannibal L: Nitric oxide homeostasis in
neurodegenerative diseases. Curr Alzheimer Res. 13:135–149.
2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Meini A, Sticozzi C, Massai L and Palmi M:
A nitric oxide/Ca(2+)/calmodulin/ERK1/2 mitogen-activated protein
kinase pathway is involved in the mitogenic effect of IL-1beta in
human astrocytoma cells. Br J Pharmacol. 153:1706–1717.
2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ijomone OM, Aluko OM, Okoh COA and
Ebokaiwe AP: Nω-nitro-L-arginine, a nitric oxide synthase
inhibitor, attenuates nickel-induced neurotoxicity. Drug Chem
Toxicol. 1–10. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Majewski M, Kozlowska A, Thoene M,
Lepiarczyk E and Grzegorzewski WJ: Overview of the role of vitamins
and minerals on the kynurenine pathway in health and disease. J
Physiol Pharmacol. 67:3–19. 2016.PubMed/NCBI
|
|
13
|
Broom L, Marinova-Mutafchieva L, Sadeghian
M, Davis JB, Medhurst AD and Dexter DT: Neuroprotection by the
selective iNOS inhibitor GW274150 in a model of Parkinson disease.
Free Radic Biol Med. 50:633–640. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Edwards RL, Lewis DG and Wilson DV: 983.
Constituents of the higher fungi. Part I. Hispidin, a new
4-hydroxy-6-styryl-2-pyrone from polyporus hispidus (Bull.) Fr. J
Chem Soc: 4995-5002, 1961.
|
|
15
|
Edwards RL and Wilson DV: 984.
Constituents of the higher fungi. Part II. The synthesis of
hispidin. J Chem Soc (Resumed). 5003–5004. 1961.
|
|
16
|
Chandimali N, Huynh DL, Jin WY and Kwon T:
Combination effects of hispidin and gemcitabine via inhibition of
stemness in pancreatic cancer stem cells. Anticancer Res.
38:3967–3975. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Han YH, Chen DQ, Jin MH, Jin YH, Li J,
Shen GN, Li WL, Gong YX, Mao YY, Xie DP, et al: Anti-inflammatory
effect of hispidin on LPS induced macrophage inflammation through
MAPK and JAK1/STAT3 signaling pathways. Appl Biol Chem.
63(21)2020.
|
|
18
|
Oh YC, Li W and Choi JG: Saussureae radix
attenuates neuroinflammation in LPS-stimulated mouse BV2 microglia
via HO-1/Nrf-2 induction and inflammatory pathway inhibition.
Mediators Inflamm. 2021(6687089)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Takata K, Kitamura Y, Saeki M, Terada M,
Kagitani S, Kitamura R, Fujikawa Y, Maelicke A, Tomimoto H,
Taniguchi T and Shimohama S: Galantamine-induced amyloid-{beta}
clearance mediated via stimulation of microglial nicotinic
acetylcholine receptors. J Biol Chem. 285:40180–40191.
2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lawson LJ, Perry VH, Dri P and Gordon S:
Heterogeneity in the distribution and morphology of microglia in
the normal adult mouse brain. Neuroscience. 39:151–170.
1990.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yang I, Han SJ, Kaur G, Crane C and Parsa
AT: The role of microglia in central nervous system immunity and
glioma immunology. J Clin Neurosci. 17:6–10. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kaur D, Sharma V and Deshmukh R:
Activation of microglia and astrocytes: A roadway to
neuroinflammation and Alzheimer's disease. Inflammopharmacology.
27:663–677. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
González-Scarano F and Baltuch G:
Microglia as mediators of inflammatory and degenerative diseases.
Annu Rev Neurosci. 22:219–240. 1999.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Voet S, Srinivasan S, Lamkanfi M and van
Loo G: Inflammasomes in neuroinflammatory and neurodegenerative
diseases. EMBO Mol Med. 11(e10248)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen K, Northington FJ and Martin LJ:
Inducible nitric oxide synthase is present in motor neuron
mitochondria and Schwann cells and contributes to disease
mechanisms in ALS mice. Brain Struct Funct. 214:219–234.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kelly MEM and Barnes S: Physiology and
pathophysiology of nitric oxide in the retina. Neuroscientist.
3:357–360. 1997.
|
|
27
|
Marks JD and Schreiber MD: Inhaled nitric
oxide and neuroprotection in preterm infants. Clin Perinatol.
35:793–807. 2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ignarro LJ, Cirino G, Casini A and Napoli
C: Nitric oxide as a signaling molecule in the vascular system: An
overview. J Cardiovasc Pharmacol. 34:879–886. 1999.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Jaffrey SR and Snyder SH: Nitric oxide: A
neural messenger. Annu Rev Cell Dev Biol. 11:417–440.
1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tramutola A, Lanzillotta C, Perluigi M and
Butterfield DA: Oxidative stress, protein modification and
Alzheimer disease. Brain Res Bull. 133:88–96. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gjoneska E, Pfenning AR, Mathys H, Quon G,
Kundaje A, Tsai LH and Kellis M: Conserved epigenomic signals in
mice and humans reveal immune basis of Alzheimer's disease. Nature.
518:365–369. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Henkel JS, Beers DR, Zhao W and Appel SH:
Microglia in ALS: The good, the bad, and the resting. J Neuroimmune
Pharmacol. 4:389–398. 2009.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shaw PR and Haydar TF: Mitigating
cognitive deficits in down syndrome by managing microglia
activation. Neuron. 108:799–800. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ren Y, Jiang J, Jiang W, Zhou X, Lu W,
Wang J and Luo Y: Spata2 knockdown exacerbates brain inflammation
via NF-κB/P38MAPK signaling and NLRP3 inflammasome activation in
cerebral ischemia/reperfusion rats. Neurochem Res: Jun1, 2021 (Epub
ahead of print). doi: 10.1007/s11064-021-03360-8.
|
|
35
|
Law A, Gauthier S and Quirion R: Say NO to
Alzheimer's disease: The putative links between nitric oxide and
dementia of the Alzheimer's type. Brain Res Brain Res Rev.
35:73–96. 2001.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Estévez AG and Jordán J: Nitric oxide and
superoxide, a deadly cocktail. Ann N Y Acad Sci. 962:207–211.
2002.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Halliwell B and Gutteridge JM: Oxygen
toxicity, oxygen radicals, transition metals and disease. Biochem
J. 219:1–14. 1984.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liu B and Hong JS: Role of microglia in
inflammation-mediated neurodegenerative diseases: Mechanisms and
strategies for therapeutic intervention. J Pharmacol Exp Ther.
304:1–7. 2003.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wang T, Qin L, Liu B, Liu Y, Wilson B,
Eling TE, Langenbach R, Taniura S and Hong JS: Role of reactive
oxygen species in LPS-induced production of prostaglandin E2 in
microglia. J Neurochem. 88:939–947. 2004.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Farber JL: Mechanisms of cell injury by
activated oxygen species. Environ Health Perspect. 102 (Suppl
10):S17–S24. 1994.PubMed/NCBI View Article : Google Scholar
|
|
41
|
You MM, Chen YF, Pan YM, Liu YC, Tu J,
Wang K and Hu FL: Royal jelly attenuates LPS-induced inflammation
in BV-2 microglial cells through modulating NF-κB and p38/JNK
signaling pathways. Mediators Inflamm. 2018(7834381)2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Stephenson J, Nutma E, van der Valk P and
Amor S: Inflammation in CNS neurodegenerative diseases. Immunology.
154:204–219. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Solt LA and May MJ: The IkappaB kinase
complex: Master regulator of NF-kappaB signaling. Immunol Res.
42:3–18. 2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Chen J, Yin W, Tu Y, Wang S, Yang X, Chen
Q, Zhang X, Han Y and Pi R: L-F001, a novel multifunctional ROCK
inhibitor, suppresses neuroinflammation in vitro and in vivo:
Involvement of NF-κB inhibition and Nrf2 pathway activation. Eur J
Pharmacol. 806:1–9. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Karin M and Delhase M: The I kappa B
kinase (IKK) and NF-kappa B: Key elements of proinflammatory
signalling. Semin Immunol. 12:85–98. 2000.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Heinrich PC, Behrmann I, MüLler-Newen G,
Schaper F and Graeve L: Interleukin-6-type cytokine signalling
through the gp130/Jak/STAT pathway. Biochem J. 334:297–314.
1998.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Gonindard C, Bergonzi C, Denier C,
Sergheraert C, Klaebe A, Chavant L and Hollande E: Synthetic
hispidin, a PKC inhibitor, is more cytotoxic toward cancer cells
than normal cells in vitro. Cell Biol Toxicol. 13:141–153.
1997.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Dann SG, Golas J, Miranda M, Shi C, Wu J,
Jin G, Rosfjord E, Upeslacis E and Klippel A: p120 catenin is a key
effector of a Ras-PKCε oncogenic signaling axis. Oncogene.
33:1385–1394. 2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
van der Vorst EPC, Theodorou K, Wu Y,
Hoeksema MA, Goossens P, Bursill CA, Aliyev T, Huitema LFA, Tas SW,
Wolfs IMJ, et al: High-density lipoproteins exert pro-inflammatory
effects on macrophages via passive cholesterol depletion and
PKC-NF-κB/STAT1-IRF1 signaling. Cell Metab. 25:197–207.
2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Wu WY, Wu YY, Huang H, He C, Li WZ, Wang
HL, Chen HQ and Yin YY: Biochanin A attenuates LPS-induced
pro-inflammatory responses and inhibits the activation of the MAPK
pathway in BV2 microglial cells. Int J Mol Med. 35:391–398.
2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Cano E and Mahadevan LC: Parallel signal
processing among mammalian MAPKs. Trends Biochem Sci. 20:117–122.
1995.PubMed/NCBI View Article : Google Scholar
|