Introduction
Primary liver cancer is the second leading cause of
cancer-associated mortality worldwide (1,2).
Primary liver cancer is one of the few tumors with a steady
increasing incidence and mortality. It is therefore a major public
health challenge (3). Liver cancer
normally comprises a heterogeneous group of malignant tumors,
including hepatocellular carcinoma (HCC), intrahepatic
cholangiocarcinoma (iCCA) and other rare tumors (4). The incidence of liver cancer is
strongly correlated with cirrhosis, which is caused by alcohol
abuse, hepatitis B and/or hepatitis C infection, aflatoxin
exposure, smoking or other chronic liver injuries, such as
metabolic liver diseases and autoimmune diseases (5,6).
Sorafenib is a multiple kinase inhibitor targeting cell surface and
intracellular kinases and that has been found to restrict tumor
growth via inducing apoptosis and inhibiting angiogenesis (7-9).
Furthermore, sorafenib activates liver cancer cell autophagy by
regulating PI3K/AKT/mammalian target of rapamycin (mTOR) (10-12),
Ras/RAF/MEK/ERK (13) and JAK-STAT
(14) signaling pathways and
induces the autophagic cell death (14). In clinical application, the efficacy
of sorafenib in advanced liver cancer has been confirmed by two
independent clinical trials (15,16)
and sorafenib has been approved as a systemic agent for the
treatment of the cancer. However, the underlying mechanisms of
sorafenib in liver cancer are still being investigated.
Hypoxia commonly occurs in liver cancer due to
intense oxygen metabolism (17).
Tumor hypoxia has been linked to poor patient outcomes (18,19).
Several techniques for assessing tumor hypoxia have allowed
clinicians to determine the extent of hypoxia in order to adopt
differential treatment strategies (20-22).
In addition, various methods have been established to simulate
hypoxic conditions in in vivo pre-clinical research, such as
cell treatment with cobalt chloride or cell culture in hypoxic
chamber (23,24). Hypoxia-inducible factors (HIFs) are
transcription factors responding to hypoxia. They are composed of
an α subunit and a β subunit and can be classified into the three
isoforms HIF 1α, HIF 2α and HIF 3α (25,26).
Degradation of HIFα by prolyl hydroxylase (PHD) can suppress the
activity of HIF under normoxia. However, the HIF commonly remains
active due to the deactivation of PHD under hypoxia (25). Upregulation of HIFs in liver cancer
cells contributes to cell adaption to the hypoxic environment via
regulation of proliferation, metabolism, inflammation and
angiogenesis (27,28). Activation of HIF-1α is a marker for
acute hypoxia since HIF-1α accumulation increases the expression of
PHD by positive feedback regulation, which leads to HIF-1α
degradation under chronic hypoxia (28). Multiple signaling pathways of
hypoxia-induced autophagy, including the HIF-1/mTOR signaling
pathway, have been reported (29).
However, the effects of hypoxia in liver cancer have not yet been
reported.
Autophagy is a highly conserved catabolic process
designed to deliver cellular materials to autophagosome-liposome
infused complex for the degradation of damaged organelles and
energy recycling (30,31). In mammalian cells, autophagy is
typically divided into the principal step-initiation, nucleation of
the autophagosome, elongation of the autophagosome membrane, fusion
with the lysosome and degradation (32). Multiple signaling pathways of
autophagy have been reported, including the P53(33), PI3K (34), Ras (35,36)
and JAK-STAT (32,37) signaling pathways. Chloroquine (CQ)
is frequently used as an inhibitor of autophagy in both cell
culture and in vivo (38).
The direct effects of CQ on autophagy are related to the expression
of HIF-1 and LC3II/I (39).
Therefore, CQ is usually employed to study the mechanism of
autophagy (40). It is hypothesized
that autophagy might serve a complicated role in tumorigenesis,
such as in liver tumorigenesis (41). However, whether sorafenib can
influence the malignant behavior of liver cancer by regulating
autophagy remains unknown.
In the present study, the effects of sorafenib and
hypoxia on the proliferation, autophagy and apoptosis of liver
cancer cells were investigated. Furthermore, the role of sorafenib
on the expression of hypoxia and autophagy-related proteins in
liver cancer cells was also evaluated.
Materials and methods
Materials
HepG2 cells and GFP-LC3 plasmid were kindly donated
by the Department of Biochemistry and Molecular Biology of the
Third Military Medical University. DMEM cell culture medium, FBS,
penicillin G, streptomycin and trypsin were purchased from Gibco;
Thermo Fisher Scientific, Inc. The transfection reagent
Lipofectamine™ 2000 was purchased from Omega Bio-Tek, Inc. Cell
Counting Kit-8 (CCK8-kit) was acquired from Abmole BioScience Inc.
Developer kit for western blotting was obtained from Pierce; Thermo
Fisher Scientific, Inc. The BCA protein concentration test kit
Pierce BCA Protein Assay kit was purchased from Pierce; Thermo
Fisher Scientific, Inc. Tris-base, glycine, SDS, acrylamide, DTT,
Tween-20 and BIS-Acrylamide reagents for western blotting were
obtained from Sangon Biotech Co., Ltd. PVDF membranes were
purchased from EMD Millipore. X-ray films were purchased from
Guangxi Yestar Medical System Co. Ltd. Methanol, glycerol, ethanol,
isopropanol and concentrated hydrochloric acid used for western
blotting were acquired from Guangzhou Chemical Reagent Factory. RNA
extraction kit was purchased from Ambion; Thermo Fisher Scientific,
Inc. CQ was provided by Sigma-Aldrich; Merck KGaA. Annexin
V-FITC/propidium iodide (PI) apoptosis kit was purchased from
eBioscience; Thermo Fisher Scientific, Inc. The following
antibodies were used in the present study: i) primary antibodies
against HIF-1 (cat. no. 3716S; 1:1,000; Cell Signaling Technology,
Inc.), LC3-I (cat. no. 12741S; 1:1000; Cell Signaling Technology,
Inc.), LC3-II (cat. no. 12741S; 1:1,000; Cell Signaling Technology,
Inc.), mTOR (cat. no. 2972S; 1:1,000; Cell Signaling Technology,
Inc.), p70 ribosomal S6 kinase (p70s6k; cat. no. 9202S; 1:1,000;
Cell Signaling Technology, Inc.), GAPDH (cat. no. 2118S; 1:1,000;
Cell Signaling Technology, Inc.); and ii) HRP-conjugated anti-mouse
(1:1,000; Santa Cruz Biotechnology, Inc.) or anti-rabbit secondary
antibody (1:1,000; Cell Signaling Technology, Inc.).
Cell culture and cell treatment
HepG2 cells were cultured in DMEM supplemented with
10% FBS, 100 U/ml penicillin G and 100 mg/ml streptomycin placed at
37˚C in a humidified incubator containing 5% CO2. The
cells (2.5x103) were transferred into a 6-well plate and
were exposed to a hypoxic (0.1% oxygen) or a normoxic (~20% oxygen)
environments for 24 h. The cells were subsequently treated with
sorafenib at the specified concentrations and for the indicated
durations. Cells in each group were then collected for further
experiments. For CQ treatment group, cells exposed to 50 µM CQ and
cultured at 37˚C with 5% CO2 for 24 h were subsequently
treated according indicated conditions.
Autophagosome evaluation
The HepG2 cells (2.5x103) were seeded
evenly in a 6-well plate and cultured at 37˚C with 5%
CO2 for two days. Then cells were transfected with
GFP-LC3 plasmid for 48 h at 37˚C (2 µg/well, GFP-LC3 was subcloned
from pEGFPC1-LC3 into the SnaB1 and Sal1 sites of pBABEpuro) using
Lipofectamine 2000 according to the manufacturers' instructions.
After treatment with various concentration of sorafenib (10, 20, 30
and 40 µM) at 37˚C for 12 h, cells were fixed in 4% formaldehyde at
20 min in room temperature. The autophagosomes were detected and
photographed under a fluorescence microscope (magnification, x100).
The green fluorescence of cells were quantified by ImageJ version
2.1.0 (National Institutes of Health) Cells with more than 5 puncta
were defined as autophagic cells (42).
CCK-8 assay
HepG2 cells (2x103 cells/100 µl) were transferred
into a 96-well plate and were exposed to hypoxic (0.1% oxygen) or
normoxic (~20% oxygen) environments for 24 h. Cells were then
treated with 30 µΜ sorafenib for 0, 24, 48 and 72 h, and cells were
incubated with 10 µl CCK-8 for 3h at 37℃. The absorbance at 450 nm
was read using a microplate reader.
Flow cytometer
HepG2 cells were treated as described above. Cells
were washed with PBS (Invitrogen; Thermo Fisher Scientific, Inc.)
and adjusted to a density of 1x106 cells/ml. The cells
were subsequently resuspended in 100 µl binding buffer, 5 µl
Annexin V-FITC and 5 µl PI and incubated in the dark at 4˚C for 15
min. Early + late apoptotic cells were washed with PBS and then
analyzed by BD LSR Fortessa flow cytometer (Becton, Dickinson and
Company) and FlowJo software version 7.6.1 (FlowJo LLC).
Western blotting
HepG2 cells were washed three times with PBS and
diluted in Tris-HCl buffer. RIPA buffer (Beyotime Institute of
Biotechnology) was added to HepG2 cells at 4˚C for 20 min. The
samples were centrifuged at 13,500 x g at 4˚C for 10 min and the
supernatants containing all proteins were collected and stored at
-80˚C. Protein concentration was determined using Pierce BCA
Protein assay kit. Proteins (20 µg) were separated by 10% SDS-PAGE
and were transferred onto PVDF membranes. Membranes were blocked
with 5% skimmed milk in TBS buffer overnight at 4˚C and washed
three times with TBS buffer. Membranes were subsequently incubated
in primary antibodies diluted into skimmed milk for 1 h at room
temperature. After washing three times with TBS buffer, membranes
were incubated with horseradish peroxidase-labeled secondary
antibodies for 45 min at room temperature. Membranes were imaged
using chemiluminescence (EMD Millipore). Relative expression levels
were normalized to endogenous control GAPDH using software
Image-Pro Plus 6.0 (National Institutes of Health).
Reverse transcription quantitative
(RT-q) PCR
RNA extraction kit was utilized to extract total RNA
from the treated HepG2 cells. RNA was reverse transcribed into cDNA
using the cDNA Synthesis according to the manufacturers'
instructions (PrimeScript RT reagent kit, Takara Bio, Inc.).
RT-qPCR reactions were conducted using SYBR Green qPCR SuperMix
(Invitrogen; Thermo Fisher Scientific, Inc.). The mRNA level of HIF
was evaluated using RT-qPCR, GAPDH was used as the reference gene.
The gene-specific primer sequences were: HIF-1-F:
TATGAGCCAGAAGAACTTTTAGGC; HIF-1-R: CACCTCTTTTGGCAAGCATCCTG;
GAPDH-F: CGGAGTCAACGGATTTGGTCGTAT; GAPDH-R:
AGCCTTCTCCATGGTGGTGAAGAC. The conditions of PCR were:
Pre-denaturation at 95˚C for 3 min, followed by 40 cycles of
denaturation for 10 sec at 95˚C, annealing at 58˚C for 30 sec and
then 95˚C for 10 sec. Melt Curve 65-95˚C with increments of 0.5˚C
for 5 sec. Following this, the expression levels of HIF-1 was
assessed. The relative quantification was performed using the
comparative 2-∆∆Cq method (43). ∆∆Ct=[Ct(target gene)-Ct(internal
gene)] experimental group-[Ct(target gene)-Ct(internal gene)]
control group.
Statistical analysis
Each experiment was performed at least three times
independently and data were presented as the means ± standard
deviation. One-way ANOVA followed by Tukey's post-hoc test was used
for multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
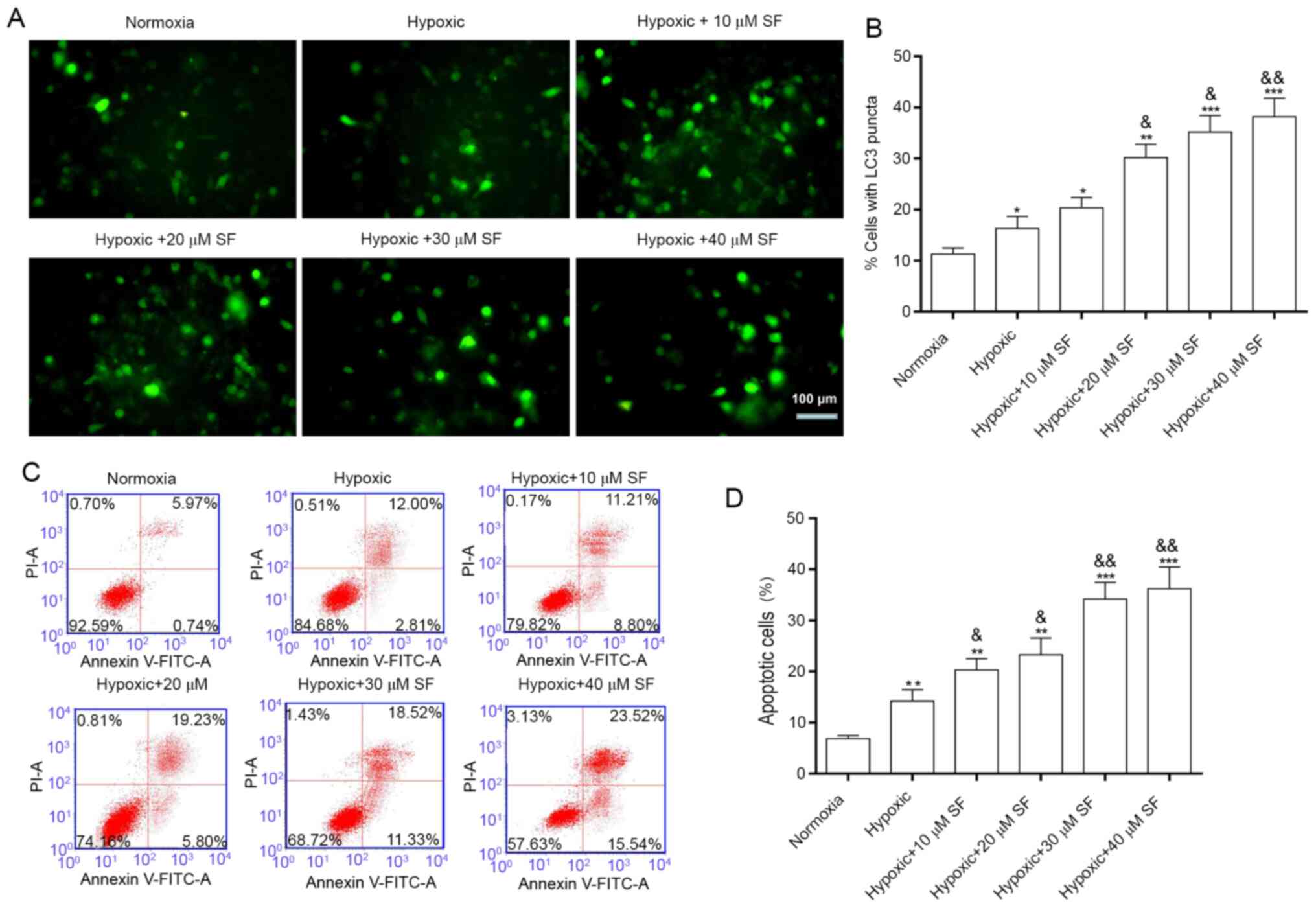
Sorafenib induces the autophagy and
apoptosis of HepG2 cells under hypoxia
LC3 is a transmembrane protein that is abundant in
the cytoplasm and located on the membrane of autophagosomes during
autophagosome formation (44,45).
The present study demonstrated that LC3 fluorescence intensity was
significantly enhanced in HepG2 cells under hypoxia compared with
cells under normoxia. Furthermore, treatment with sorafenib
significantly enhanced LC3 fluorescence intensity in a
concentration-dependent manner (P<0.05, P<0.01 and
P<0.001; Fig. 1A and B). In addition, sorafenib and
hypoxia-induced apoptosis was examined by flow cytometry. The
results demonstrated that the percentage of apoptotic cells
increased significantly in HepG2 cells under hypoxia compared with
cells in the normoxia group (14.81 vs 6.71%). Furthermore, the
percentage of apoptotic cells was significantly increased following
treatment of HepG2 cell under hypoxia with sorafenib in a
concentration-dependent manner (P<0.05, P<0.01 and
P<0.001; Fig. 1C and D), up to 39.06%, for 40 µM sorafenib.
Taken together, these findings demonstrated that sorafenib may
enhance hypoxia-induced autophagy and apoptosis in HepG2 cells.
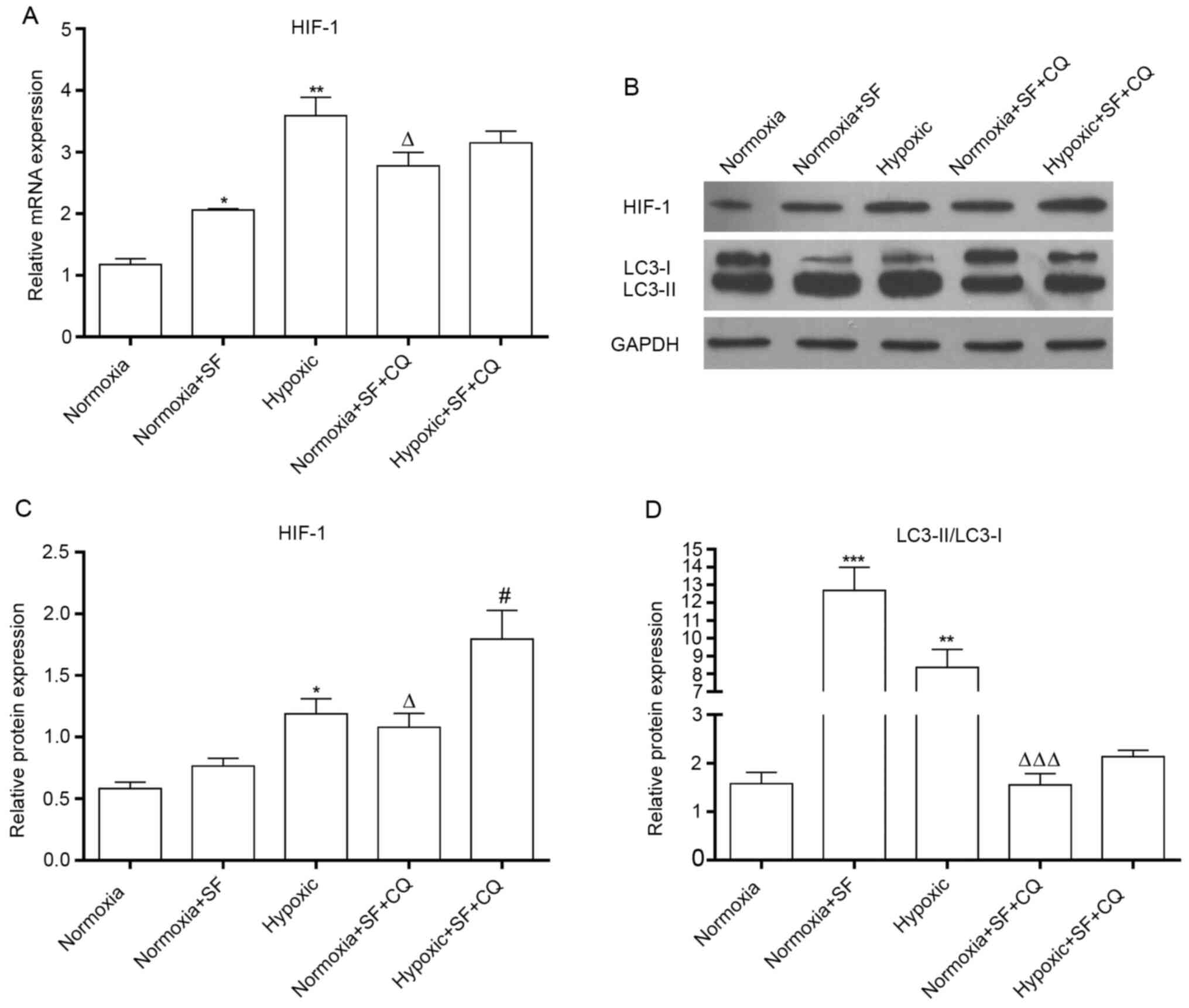
Sorafenib significantly upregulates
HIF-1 and LC3II/I and downregulated mTOR and p70s6K in HepG2 cells
under hypoxia
Due to the promotion of autophagy by Sorafenib, the
signaling pathways that might be related to autophagy in hypoxic
HepG2 cells were further investigated. The results demonstrated
that hypoxia could upregulate HIF-1 at the mRNA and protein levels
in HepG2 cells, and that hypoxia-induced HIF-1 overexpression was
further enhanced by sorafenib (P<0.05; Fig. 2A and B). Subsequently, further investigation
demonstrated that mTOR and p70s6K expression was significantly
downregulated and that LC3II/LC3I ratio was significantly
upregulated in HepG2 cells treated with SF under hypoxia.
Furthermore, expression of mTOR and p70s6K was decreased in
sorafenib-treated hypoxic HepG2 cells. These results showed that
the combination of sorafenib and hypoxia resulted in a
significantly higher HIF-1 expression and LC3II/LCI ratio and a
significantly stronger downregulation of mTOR and p70s6K
(P<0.05, P<0.01 and P<0.001; Fig. 2C and D). Taken together, these findings
indicated that sorafenib could strengthen the autophagy of normal
and hypoxic HepG2 cells.
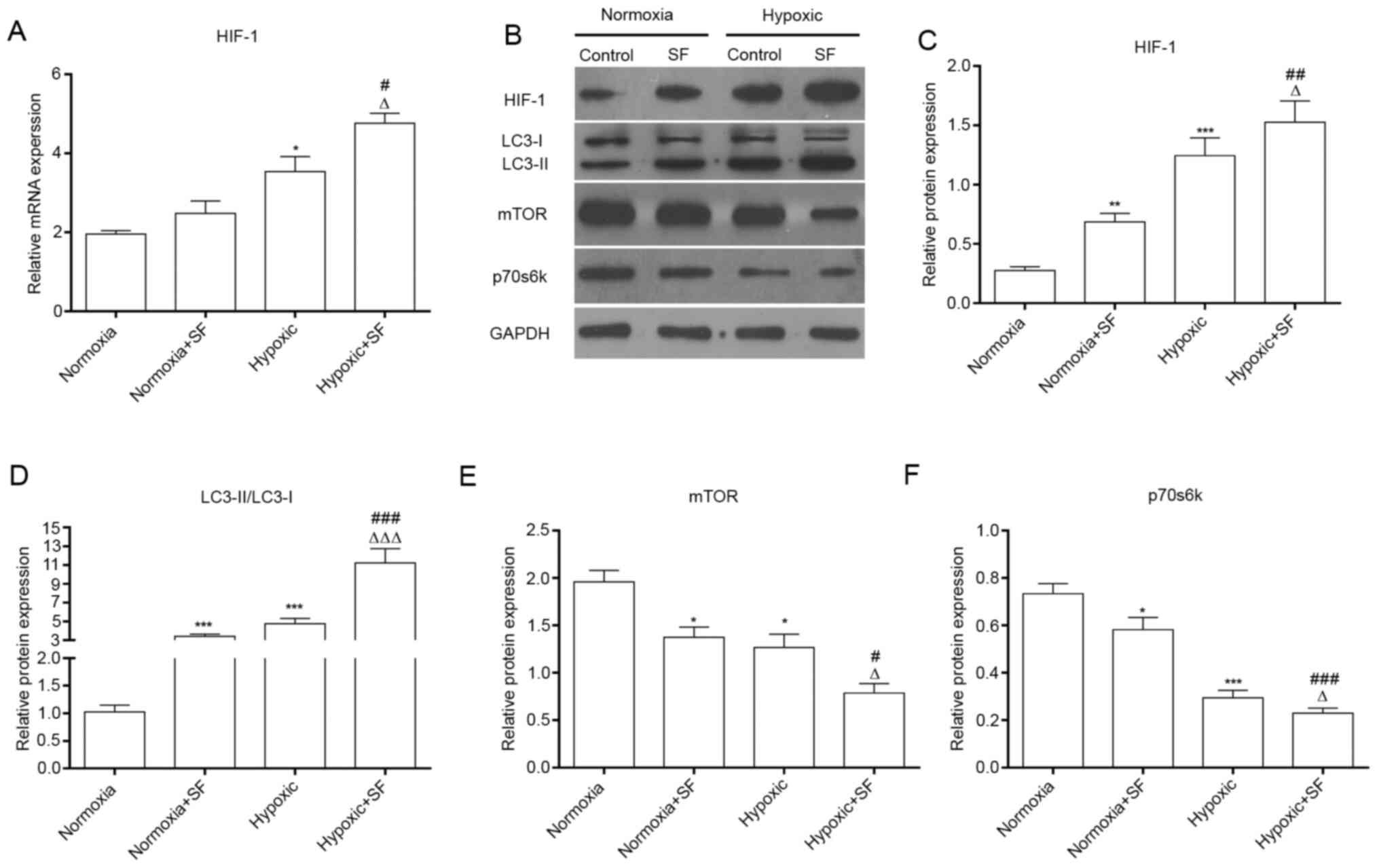 | Figure 2SF upregulates HIF-1, LC3II/I and
downregulates mTOR and p70s6K in HepG2 cells under hypoxia. (A)
After treatment with SF, total mRNA was collected and expression
level of HIF-1 was measured by reverse transcription quantitative
PCR in HepG2 cells under normoxia and hypoxia. (B) Proteins from
treated HepG2 cells were collected and the expression of HIF-1,
LC3, mTOR and p70s6K was determined using western blotting. (C-F)
Relative protein expression of HIF-1, LC3II/I, mTOR and p70s6K was
quantified. *P<0.05, **P<0.01 and
***P<0.001 vs. normoxia group. #P<0.05,
##P<0.01 and ###P<0.001 vs. normoxia +
SF group. ∆P<0.05, ∆∆∆P<0.001 vs.
hypoxic group. Data are presented as the means ± standard deviation
(n=3). SF, sorafenib; mTOR, mammalian target of rapamycin; HIF-1,
hypoxia-inducible factor-1; p70s6k, p70 ribosomal S6 kinase. |
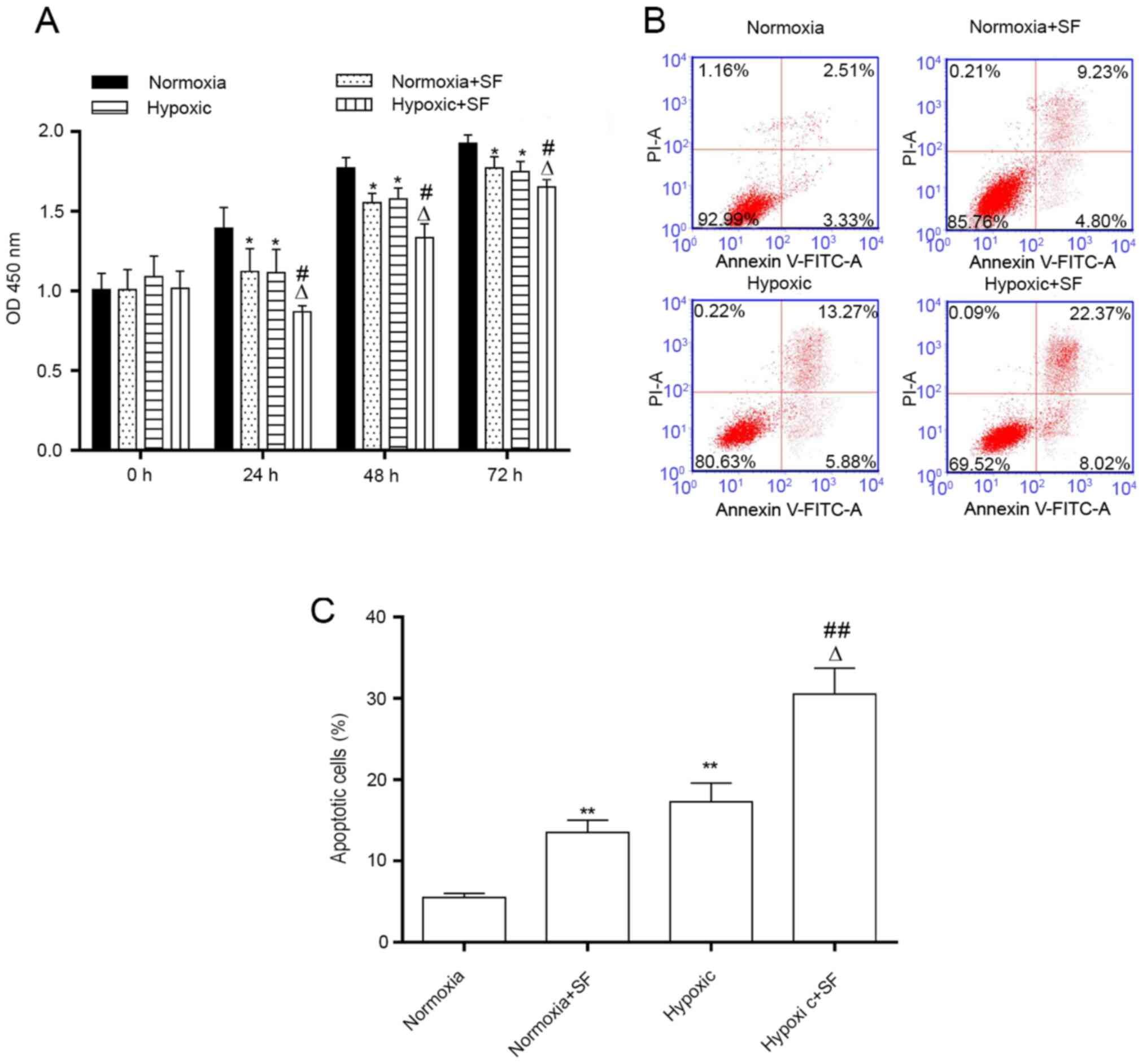
Sorafenib inhibits the proliferation
and stimulates the apoptosis of HepG2 cells under normoxia and
hypoxia
Consistently with the aforementioned findings, the
results demonstrated that cell proliferation was significantly
decreased in hypoxic HepG2 cells compared with cells in the
normoxia group, and that sorafenib further inhibited the
proliferation of HepG2 cells under hypoxia after 24, 48 and 72 h
(P<0.05; Fig. 3A). Furthermore,
the number of apoptotic cells was increased significantly in the
hypoxic HepG2 cells compared with those in the normoxia group
(19.15 vs 5.84%). This phenomenon was further enhanced following
HepG2 cell treatment with 30 µM sorafenib for 24 h (30.39 vs.
19.12%; P<0.05 and P<0.01; Fig.
3B and C). The results
demonstrated that sorafenib could inhibit the proliferation and
promote the apoptosis of HepG2 cells under normoxia and
hypoxia.
CQ reverses the sorafenib-induced
increase in HIF-1 and LC3II/I expression in HepG2 cells under
normoxia and hypoxia
To further determine whether autophagy pathway might
contribute to the apoptosis of liver cancer induced by sorafenib
and hypoxia, the autophagy inhibitor CQ was used to evaluate the
autophagy with or without sorafenib in HepG2 cells under normoxia
and hypoxia. The results demonstrated that CQ could partially
reverse the upregulation of HIF-1 induced by sorafenib and/or
hypoxia in HepG2 cells (P<0.05 and P<0.01; Fig. 4A and B). In addition, CQ significantly inhibited
autophagy by decreasing LC3II/LC3I ratio in HepG2 cells treated
with sorafenib and/or hypoxia (P<0.05 and P<0.001; Fig. 4B-D). Taken together, these results
suggested that hypoxia and sorafenib could upregulate HIF-1
expression via regulating autophagy.
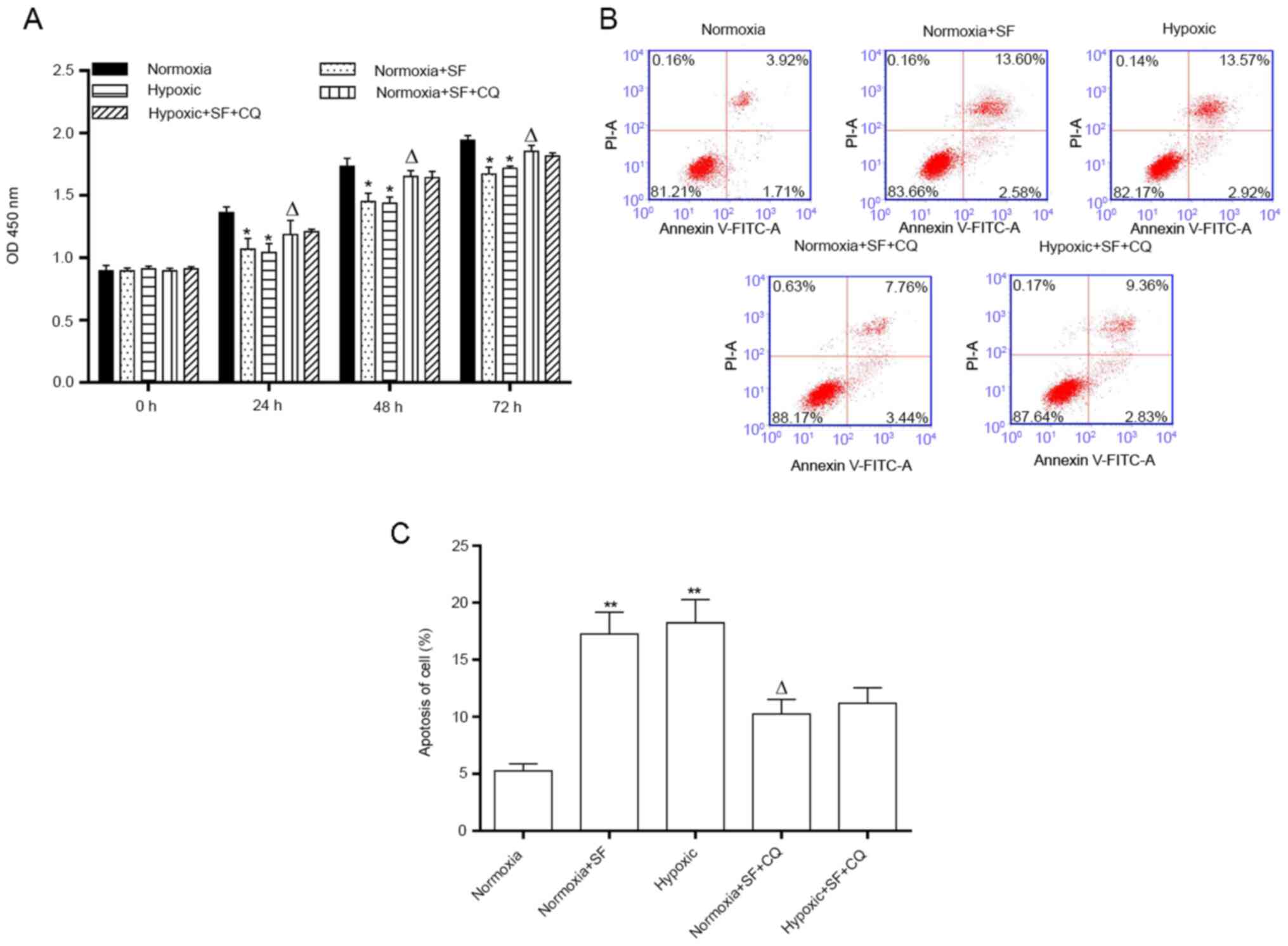
CQ attenuates the inhibition of
proliferation and induction of apoptosis mediated by sorafenib in
HepG2 cells under normoxia and hypoxia
The present study demonstrated that the inhibition
of HepG2 cell proliferation, which was mediated by sorafenib and/or
hypoxia, could also be weakened by CQ (P<0.05; Fig. 5A). Furthermore, this study reported
that the promoting effect of sorafenib and/or hypoxia on apoptosis
could also be partially reversed by CQ in HepG2 cells (11.2 vs.
16.18% and 12.19 vs. 16.49%; P<0.05 and P<0.01; Fig. 5B). However, the percentage of
necrotic cells was not significantly changed. These findings
suggested that sorafenib and hypoxia may prevent the proliferation
and accelerate the apoptosis of HepG2 cells by regulating the
autophagy signaling pathway.
Discussion
The present study reported the underlying mechanism
of sorafenib-induced liver cancer cell autophagy and apoptosis. The
results demonstrated that sorafenib could promote cell autophagy
and apoptosis under hypoxia. Furthermore, according to the effect
of sorafenib on autophagy under hypoxia, we hypothesized that
sorafenib may share a similar autophagic mechanism with hypoxia.
The results demonstrated that both sorafenib and hypoxia induced
cell autophagy via the HIF/mTOR-related signaling pathway. In
addition, to address the crucial role of sorafenib-mediated
autophagy in liver cancer cell, autophagy was inhibited using CQ.
The results showed that CQ inhibited the apoptosis and promoted the
proliferation of liver cancer cells, which were mediated by
sorafenib or hypoxia. These findings strongly suggested that
autophagy may contribute to the inhibition of proliferation in
sorafenib-treated cells via the HIF/mTOR signaling pathway.
Although regorafenib and lenvatinib have been
approved as second-line treatments to improve the clinical outcomes
of patients with advanced liver cancer, these treatments only
extend the median overall survival by ~3 months compared with
patients treated with sorafenib (46). It is therefore crucial to determine
the underlying mechanism of sorafenib in order to extend the
overall survival of patients with advanced-stage liver cancer.
Previous studies have reported that sorafenib combined with various
autophagy-inducing drugs, such as metformin (13), capsaicin (47) and pemetrexed (48), can suppress the proliferation of
liver cancer cells in vitro. Conversely to studies on
autophagy-promoting cell death, sorafenib-induced autophagy can
also be considered as a survival mechanism for liver cancer cells.
It has been reported that liver cancer cells treated with sorafenib
can be killed by inhibiting autophagy when combined with certain
inhibitors, such as siRNA specific for ATG7(11) and CQ (49,50),
compared with cells not treated with these inhibitors. The results
from the present study demonstrated that autophagy induction via
the HIF/mTOR signaling pathway may contribute to the suppression of
the proliferation of cancer cells. Liver cancer cells are often in
a state of moderate hypoxia (0.1% oxygen) due to impaired hepatic
blood oxygen exchange and metabolic pressure. The present study
also confirmed that chronic and moderate autophagy had a protective
effect on cancer cells; however, severe hypoxia (<0.1% oxygen)
could activate the HIF-1/mTOR signaling pathway and induce cell
death (29). We performed the
autophagy experiment in a normal state of hypoxia (0.1%
oxygen).
Autophagy can be activated by mTOR dependent
pathways and non-motor dependent pathways (51,52).
Among the various autophagy pathways, the mTOR-related autophagy
pathway is the most common one investigated (30). It has been demonstrated that LC3I
can be converted into LC3II and transferred to the autophagosome
membrane in the cytoplasm, which could also be induced by mTOR
downregulation or dephosphorylation (53). p70 ribosomal S6 kinase (p70S6K) is a
ribosomal protein downstream of mTOR that can regulate autophagy
via AMPK activation mediated by the transforming growth
factor-β-activated kinase 1(54).
Furthermore, it has been reported that CQ can promote the apoptosis
of liver cancer cells treated with sorafenib (49,50);
however, in these studies, sorafenib at lower concentration (5-10
µΜ) might not activate the HIF/mTOR autophagy pathway and reversely
leads to protective autophagy, which is inhibited by CQ. Of note,
sorafenib has been previously confirmed to significantly
downregulate mTOR expression in liver cancer, which is consistent
with the findings from the present study (47). It has also been shown that mTOR
phosphorylation could be markedly increased with sorafenib
treatment (55). Summarized, the
results from the present study might be explained by the fact that
various signaling pathways of autophagy can be activated by
different experimental conditions. The present study demonstrated
that sorafenib may prevent the proliferation and induce the
autophagy and apoptosis of liver cancer cells via the
HIF-1/mTOR-related signaling pathway.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Youth Development
Foundation of The First Affiliated Hospital of Hainan Medical
University (grant no. HYFYPY201704).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QZY and LHG designed all the experiments. LHG, XLH,
JW and YKC performed the experiments. QZY and SBL analyzed the
data. QSY collated the data, wrote and edited the manuscript. All
authors confirmed the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cong WM, Bu H, Chen J, Dong H, Zhu YY,
Feng LH, Chen J and Guideline Committee: Practice guidelines for
the pathological diagnosis of primary liver cancer: 2015 update.
World J Gastroenterol. 22:9279–9287. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yamashita T and Kaneko S: Liver cancer.
Rinsho Byori. 64:787–796. 2016.PubMed/NCBI(In Japanese).
|
|
3
|
Murray CJL, Vos T, Lozano R, Naghavi M,
Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S,
et al: Disability-adjusted life years (DALYs) for 291 diseases and
injuries in 21 regions, 1990-2010: A systematic analysis for the
Global Burden of Disease Study 2010. Lancet. 380:2197–2223.
2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhu XD and Sun HC: Emerging agents and
regimens for hepatocellular carcinoma. J Hematol Oncol.
12(110)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fattovich G, Stroffolini T, Zagni I and
Donato F: Hepatocellular carcinoma in cirrhosis: Incidence and risk
factors. Gastroenterology. 127 (Suppl 1):S35–S50. 2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Grandhi MS, Kim AK, Ronnekleiv-Kelly SM,
Kamel IR, Ghasebeh MA and Pawlik TM: Hepatocellular carcinoma: From
diagnosis to treatment. Surg Oncol. 25:74–85. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Keating GM: Sorafenib: A review in
hepatocellular carcinoma. Target Oncol. 12:243–253. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Méndez-Blanco C, Fondevila F,
García-Palomo A, González-Gallego J and Mauriz JL: Sorafenib
resistance in hepatocarcinoma: Role of hypoxia-inducible factors.
Exp Mol Med. 50:1–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Haga Y, Kanda T, Nakamura M, Nakamoto S,
Sasaki R, Takahashi K, Wu S and Yokosuka O: Overexpression of c-Jun
contributes to sorafenib resistance in human hepatoma cell lines.
PLoS One. 12(e0174153)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gedaly R, Angulo P, Hundley J, Daily MF,
Chen C and Evers BM: PKI-587 and sorafenib targeting PI3K/AKT/mTOR
and Ras/Raf/MAPK pathways synergistically inhibit HCC cell
proliferation. J Surg Res. 176:542–548. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Shimizu S, Takehara T, Hikita H, Kodama T,
Tsunematsu H, Miyagi T, Hosui A, Ishida H, Tatsumi T, Kanto T, et
al: Inhibition of autophagy potentiates the antitumor effect of the
multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J
Cancer. 131:548–557. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang CZ, Wang XD, Wang HW, Cai Y and Chao
LQ: Sorafenib inhibits liver cancer growth by decreasing mTOR, AKT,
and PI3K expression. J BUON. 20:218–222. 2015.PubMed/NCBI
|
|
13
|
Ling S, Song L, Fan N, Feng T, Liu L, Yang
X, Wang M, Li Y, Tian Y, Zhao F, et al: Combination of metformin
and sorafenib suppresses proliferation and induces autophagy of
hepatocellular carcinoma via targeting the mTOR pathway. Int J
Oncol. 50:297–309. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS,
Cheng AL, Chen PJ and Chen KF: Mcl-1-dependent activation of Beclin
1 mediates autophagic cell death induced by sorafenib and SC-59 in
hepatocellular carcinoma cells. Cell Death Dis.
4(e485)2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: SHARP Investigators Study Group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390.
2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and safety of
sorafenib in patients in the Asia-Pacific region with advanced
hepatocellular carcinoma: A phase III randomised, double-blind,
placebo-controlled trial. Lancet Oncol. 10:25–34. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xiong XX, Qiu XY, Hu DX and Chen XQ:
Advances in hypoxia-mediated mechanisms in hepatocellular
carcinoma. Mol Pharmacol. 92:246–255. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo Y, Xiao Z, Yang L, Gao Y, Zhu Q, Hu L,
Huang D and Xu Q: Hypoxia-inducible factors in hepatocellular
carcinoma (Review). Oncol Rep. 43:3–15. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vaupel P and Mayer A: Hypoxia in cancer:
Significance and impact on clinical outcome. Cancer Metastasis Rev.
26:225–239. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Walsh JC, Lebedev A, Aten E, Madsen K,
Marciano L and Kolb HC: The clinical importance of assessing tumor
hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic
opportunities. Antioxid Redox Signal. 21:1516–1554. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Koch CJ and Evans SM: Optimizing hypoxia
detection and treatment strategies. Semin Nucl Med. 45:163–176.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Höckel M and Vaupel P: Tumor hypoxia:
Definitions and current clinical, biologic, and molecular aspects.
J Natl Cancer Inst. 93:266–276. 2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Rana NK, Singh P and Koch B: CoCl2
simulated hypoxia induce cell proliferation and alter the
expression pattern of hypoxia associated genes involved in
angiogenesis and apoptosis. Biol Res. 52(12)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Allen CB, Schneider BK and White CW:
Limitations to oxygen diffusion and equilibration in in vitro cell
exposure systems in hyperoxia and hypoxia. Am J Physiol Lung Cell
Mol Physiol. 281:L1021–L1027. 2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK,
Li LL, Koh HY, Tsang FH, Wei LL, Wong CM, et al: Hypoxia inducible
factor HIF-1 promotes myeloid-derived suppressor cells accumulation
through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun.
8(517)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kahraman DS, Diniz G, Sayhan S, Ayaz D,
Yemen M, Karadeniz T and Sanci M: The clinicopathologic
significance of hypoxia inducible factor 1 alpha expression in
ovarian serous tumors. Eur J Gynaecol Oncol. 40:242–245. 2019.
|
|
27
|
Lin C-A, Chang L-L, Zhu H, He Q-J and Yang
B: Hypoxic microenvironment and hepatocellular carcinoma treatment.
Hepatoma Res. 4(26)2018.
|
|
28
|
Wilson GK, Tennant DA and McKeating JA:
Hypoxia inducible factors in liver disease and hepatocellular
carcinoma: Current understanding and future directions. J Hepatol.
61:1397–1406. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Fang Y, Tan J and Zhang Q: Signaling
pathways and mechanisms of hypoxia-induced autophagy in the animal
cells. Cell Biol Int. 39:891–898. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36(52)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Onorati AV, Dyczynski M, Ojha R and
Amaravadi RK: Targeting autophagy in cancer. Cancer. 124:3307–3318.
2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Levy JMM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542.
2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
White E: Autophagy and p53. Cold Spring
Harb Perspect Med. 6(a026120)2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang J, Zhang JX and Zhang QL:
PI3K/AKT/mTOR-mediated autophagy in the development of autism
spectrum disorder. Brain Res Bull. 125:152–158. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kinsey CG, Camolotto SA, Boespflug AM,
Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap
JT, et al: Protective autophagy elicited by RAF→MEK→ERK inhibition
suggests a treatment strategy for RAS-driven cancers. Nat Med.
25:620–627. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhang X, Cheng Q, Yin H and Yang G:
Regulation of autophagy and EMT by the interplay between p53 and
RAS during cancer progression (Review). Int J Oncol. 51:18–24.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wu Q, Wu W, Fu B, Shi L, Wang X and Kuca
K: JNK signaling in cancer cell survival. Med Res Rev.
39:2082–2104. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455.
2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yang S, Qiang L, Sample A, Shah P and He
Y-Y: NF-κB signaling activation induced by chloroquine requires
autophagosome, p62 protein, and c-Jun N-terminal kinase (JNK)
signaling and promotes tumor cell resistance. J Biol Chem.
292:3379–3388. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Edelstein CL, Venkatachalam MA and Dong Z:
Autophagy inhibition by chloroquine and hydroxychloroquine could
adversely affect acute kidney injury and other organ injury in
critically ill patients with COVID-19. Kidney Int. 98:234–235.
2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Weiskirchen R and Tacke F: Relevance of
autophagy in parenchymal and non-parenchymal liver cells for health
and disease. Cells. 8(8)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bravo-San Pedro JM, Pietrocola F, Sica V,
Izzo V, Sauvat A, Kepp O, Maiuri MC, Kroemer G and Galluzzi L:
High-throughput quantification of GFP-LC3+ dots by
automated fluorescence microscopy. Methods Enzymol. 587:71–86.
2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Runwal G, Stamatakou E, Siddiqi FH, Puri
C, Zhu Y and Rubinsztein DC: LC3-positive structures are prominent
in autophagy-deficient cells. Sci Rep. 9(10147)2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lee YK and Lee JA: Role of the mammalian
ATG8/LC3 family in autophagy: Differential and compensatory roles
in the spatiotemporal regulation of autophagy. BMB Rep. 49:424–430.
2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Sasaki R, Kanda T, Fujisawa M, Matsumoto
N, Masuzaki R, Ogawa M, Matsuoka S, Kuroda K and Moriyama M:
Different mechanisms of action of regorafenib and lenvatinib on
Toll-like receptor-signaling pathways in human hepatoma cell lines.
Int J Mol Sci. 21(3349)2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Dai N, Ye R, He Q, Guo P, Chen H and Zhang
Q: Capsaicin and sorafenib combination treatment exerts synergistic
anti- hepatocellular carcinoma activity by suppressing EGFR and
PI3K/Akt/mTOR signaling. Oncol Rep. 40:3235–3248. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Bareford MD, Hamed HA, Tang Y,
Cruickshanks N, Burow ME, Fisher PB, Moran RG, Nephew KP, Grant S
and Dent P: Sorafenib enhances pemetrexed cytotoxicity through an
autophagy-dependent mechanism in cancer cells. Autophagy.
7:1261–1262. 2011.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Rong LW, Wang RX, Zheng XL, Feng XQ, Zhang
L, Zhang L, Lin Y, Li ZP and Wang X: Combination of wogonin and
sorafenib effectively kills human hepatocellular carcinoma cells
through apoptosis potentiation and autophagy inhibition. Oncol
Lett. 13:5028–5034. 2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke
AW, Wang XY, Dai Z, Peng YF, Gu CY, et al: Targeting autophagy
enhances sorafenib lethality for hepatocellular carcinoma via ER
stress-related apoptosis. Autophagy. 7:1159–1172. 2011.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32.
2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Fritzen AM, Frøsig C, Jeppesen J, Jensen
TE, Lundsgaard AM, Serup AK, Schjerling P, Proud CG, Richter EA and
Kiens B: Role of AMPK in regulation of LC3 lipidation as a marker
of autophagy in skeletal muscle. Cell Signal. 28:663–674.
2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Sun J, Mu Y, Jiang Y, Song R, Yi J, Zhou
J, Sun J, Jiao X, Prinz RA, Li Y, et al: Inhibition of p70 S6
kinase activity by A77 1726 induces autophagy and enhances the
degradation of superoxide dismutase 1 (SOD1) protein aggregates.
Cell Death Dis. 9(407)2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Gedaly R, Angulo P, Hundley J, Daily MF,
Chen C, Koch A and Evers BM: PI-103 and sorafenib inhibit
hepatocellular carcinoma cell proliferation by blocking
Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer Res.
30:4951–4958. 2010.PubMed/NCBI
|