Introduction
Asthma is a chronic inflammatory disease of the
conducting airways and affects up to 334 million individuals
globally (1,2). Increasing experimental evidence has
suggested that the airway epithelium, the first line of defense
against the exposure of airway tissue to an inflammatory reaction,
plays a pivotal role in asthma pathogenesis (3). Airway epithelial cell (AEC)
inflammation is one of the characteristics of asthma (4,5). Given
that AEC injury is involved in asthma pathophysiology, maintaining
AEC survival and function may be an effective therapeutic strategy
for asthma (6). However, the
molecular mechanisms of AEC inflammation in asthma are still
unknown.
Micro(mi)RNAs are short endogenous RNAs that are
able to regulate gene expression by binding to their target mRNA to
alter mRNA degradation and translation (7-9).
The majority of asthma-related protein-coding genes have been found
to be regulated by miRNAs, suggesting that miRNAs could serve as
novel biomarkers of asthma (10-12).
Recently, a number of studies has revealed that miR-200b/c played a
role in asthma, and a decrease in miR-200b/c may be an underlying
cause of the asthmatic phenotype (13,14).
However, the regulatory mechanism of miR-200c-3p in asthma requires
further investigation.
A diverse microbial community exists in the
intestinal environment and mediates the composition of chemical
signals and metabolites within the gut (15). The human intestinal microflora,
where millions of genes are expressed, regulates numerous host
physiological processes, including energy consumption, nutritional
homeostasis and immunity (16,17).
Notably, a previous study supported the possibility of
microbiota-directed therapies for asthma (18). Metabolites of intestinal microflora
(MIM) have been reported to inhibit colorectal cancer cell
migration by increasing miR-200c expression levels (19). However, the association between MIM
and miR-200c-3p remains unclear in asthmatic AECs.
The present study investigated the regulation of
miR-200c-3p by MIM in asthmatic inflammation and provided novel
insights for the targeted therapy of asthma.
Materials and methods
Ethical statement
The present study was approved by the Ethics
Committee of The First Affiliated Hospital of Fujian Medical
University. All efforts were taken to minimize the suffering of the
animals.
Culture of intestinal microflora and
collection of MIM from mice
A total of three healthy male C57BL/c mice (8 weeks
old; 20-25 g) were purchased from Hunan SJA Laboratory Animal Co.,
Ltd. All the animals were housed in a specific pathogen-free
environment, with free access to food and water, and were provided
with 12-h shifts of light/dark cycles. The intestinal metabolites
were obtained from one of the three mice each time. An intestinal
microflora suspension was prepared by mixing 0.25 g fresh mouse
feces with 1 ml 0.9% NaCl in a sterile tube. Every 1 ml intestinal
microflora suspension was mixed with 9 ml anaerobic medium (Gifu
Anaerobic Broth medium; Nissui Pharmaceutical Co., Ltd.) and placed
in a culture bag. The culture bag was sealed after the addition of
an anaerobic bag and an oxygen indicator and was incubated at 36˚C
for 48 h, during which the culture bag was oscillated at 100 r/min
for the first 24 h and maintained at a still position for the
remaining 24 h. Thereafter, the MIM were collected and centrifuged
twice for 15 min at 10,000 x g, 4˚C. The supernatant was preserved
and filtered using a 0.22-um membrane under sterile conditions.
Epithelial cell culture and
transfection
The human bronchial epithelial cells (16HBE) were
purchased from Cell Resource Center, Peking Union Medical College
and cultured according to the manufacturer's instructions. The
cells in the logarithmic phase were treated with saline or 400 U/ml
HDMs (Sigma-Aldrich; Merck KGaA) for 24 h and correspondingly
divided into the Control or HDM groups. 16HBE cells, which were
pretreated with MIM before HDM induction were assigned to the MIM
group. Additional 16HBE cells, also in the logarithmic growth
phase, were transfected with miR-200c-3p mimics (sense,
5'-UAAUACUGCCGGGUAAUGAUGGA-3'; antisense,
5'-UCCAUCAUUACCCGGCAGUAUUA-3'), miR-200c-3p inhibitor
(5'-UCCAUCAUUACCCGGCAGUAUUA-3'), miR-200c-3p mimics negative
control (NC; sense, 5'-GUAGCUUAUCCGGAAUCUAAGUC-3'; antisense,
5'-GACUUAGAUUCCGGAUAAGCUAC-3') or miR-200c-3p inhibitor NC
(5'-GACUUAGAUUCCGGAUAAGCUAC-3') (Guangzhou RiboBio Co., Ltd.)
before HDM induction, and were divided into the following groups:
miR-200c-3p mimics, miR-200c-3p inhibitor, NC mimics and NC
inhibitor, respectively. A portion of the cells in the miR-200c-3p
inhibitor group or the NC inhibitor group were treated with MIM
before HDM induction and correspondingly termed the MIM +
miR-200c-3p inhibitor and MIM + NC inhibitor groups. The final
concentration of miR-200c-3p mimics, miR-200c-3p inhibitor or NCs
in the culture medium was 50 nM.
All the transfections were performed using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
transfection was sustained at 37˚C for 4 h. The following
experiments were performed 48 h after the transfection. The
transfected cells were suspended in DMEM (10% FBS) and seeded in
24-well plates at a density of 1x105 cells/well and
cultured at 37˚C in humidified incubator with 5% CO2.
Cells pretreated with 20 µM SP600125 (JNK inhibitor) for 1 h before
HDM (400 U/ml) stimulation for 24 h were termed as the SP600125
group, while the cells pretreated with 100 µM S3I-201 (STAT3
inhibitor) for 1 h before HDM stimulation were termed as the
S3I-201 group.
MTT assay
After the cells were incubated for 24, 48 and 72 h,
20 µl MTT solution (5 mg/ml; Merck KGaA) was added to each group
and the cells were cultured at 37˚C with 5% CO2 for 4 h.
Thereafter, the culture medium was discarded and replaced with 150
µl dimethyl sulfoxide. The plate was gently shaken for 10 min to
facilitate crystal dissolution. The optical density (OD) of each
well was measured at 570 nm using an enzyme-linked immunometric
meter. The MTT curve was created using OD as the ordinate variable
and time as the abscissa. The OD was obtained from three
independent experiments and each sample was calculated three times
and averaged to obtain the average value.
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted from 16HBE cells using 1 ml
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. After quantification
by NanoDrop 2000 (Thermo Fisher Scientific, Inc.), the RNA was
reverse transcribed into cDNA using One Step PrimeScript RT-PCR Kit
(Takara Bio, Inc.). The RT reaction was maintained at 37˚C for 60
min and then at 85˚C for 5 sec. qPCR was conducted using a SYBR
Green fluorescent qPCR kit (Takara Biotechnology Co., Ltd.) and the
CFX96 real-time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The following thermocycling conditions were
used: Initial denaturation at 95˚C for 10 min, followed by 40
cycles of denaturation at 95˚C for 10 sec, annealing at 60˚C for 20
sec and extension at 72˚C for 34 sec. The threshold value was
selected manually at the lowest point of parallel logarithmic
amplification curves, and the Cq value (threshold cycle) of each
reaction was obtained. The data was analyzed using the
2-∆∆Cq method (20),
where 2-∆∆Cq represents the ratio of gene expression
between the experimental and control groups: ∆∆Cq=[Cq (target
gene)-Cq (reference gene)] experimental group-[Cq (target gene)-Cq
(reference gene)] control group. Cq refers to the cycle number when
the real-time fluorescence intensity of the reaction reaches the
threshold and the amplification shows a logarithmic manner. The PCR
experiments were repeated three times. The internal references for
miRNA and mRNA were U6 and GAPDH, respectively. The primer
sequences of each gene are listed in Table I.
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Name | Sequence (5'-3') |
|---|
| miR-200c F |
GGTAATACTGCCGGGTAAT |
| miR-200c R |
CAGTGCGTGTCGTGGAGT |
| U6 F |
CTCGCTTCGGCAGCACATATACT |
| U6 R |
ACGCTTCACGAATTTGCGTGTC |
| IL6ST F |
GTGTTTAGGATTCGCTGTATGA |
| IL6ST R |
CTGTAGCCTTGAGTATGGGATG |
| GAPDH F |
GTCAGTGGTGGACCTGACCT |
| GAPDH R |
TGCTGTAGCCAAATTCGTTG |
Western blot analysis
Cells were washed with PBS and lysed in RIPA buffer
containing phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology) on ice for 30 min. Afterwards, the cell lysates were
centrifuged at 12,000 x g for 10 min at 4˚C. The supernatant was
then transferred to 0.5-ml centrifuge tubes and stored at -20˚C or
quantified using a BCA kit. Next, 6X SDS loading buffer was added
to the tubes for protein denaturation at 100˚C for 5 min. The
proteins (40 µg per lane) were then separated using 12% SDS-PAGE
and transferred onto a PVDF membrane with pre-cooled transfer
buffer at 4˚C for 1.5 h. The membrane was blocked with 5% skimmed
milk in TBS-0.05% Tween-20 (TBST) buffer for 1 h. TBST-diluted
primary antibodies were then incubated with the membrane overnight
at 4˚C. The membrane was then washed with TBST 3 times each for 10
min. Thereafter, the secondary goat anti-rabbit IgG antibody
(1:5,000; cat. no. CW0103S; Beijing ComWin Biotech Co., Ltd.) was
incubated with the membrane for 2 h at room temperature. After the
incubation, the membrane was washed with TBST. The protein
expression levels of IL6ST, phosphorylated (p)-STAT3, STAT3, p-JNK
and JNK were detected by densitometry using an enhanced ECL
chemiluminescence substrate kit (cat. no. CW0049; Beijing ComWin
Biotech Co., Ltd.) and ImageJ software (version 1.46; National
Institutes of Health). The following primary antibodies were used:
Anti-IL6ST (1:1,000; cat. no. ab202850; Abcam), anti-STAT3
(1:1,000; cat. no. 12640S; Cell Signaling Technology, Inc.),
anti-p-STAT3 (1:1,000; cat. no. 9145T; Cell Signaling Technology,
Inc.), anti-JNK (1:1,000; cat. no. ab179461; Abcam), and anti-p-JNK
(1:1,000; cat. no. ab124956; Abcam). Anti-GAPDH antibody (1:500;
5174T; Cell Signaling Technology, Inc.) was used as the internal
reference.
ELISA
The supernatant of each group was collected after
the 16HBE cells were stimulated with HDM for 24 h. ELISA kits were
used to determine the IL-5 (Human IL-5 Quantikine ELISA Kit; cat.
no. D5000B; R&D Systems, Inc.) and IL-6 (Human IL-6 Quantikine
ELISA kit; cat. no. D6050; R&D Systems, Inc.) contents in the
supernatant. All the experiments were performed according to the
manufacturer's instructions.
Dual-luciferase reporter gene
assay
The target binding sites between IL6ST and
miR-200c-3p were predicted by the online tool StarBase v3.0
(http://starbase.sysu.edu.cn/). According
to the prediction, wild-type (WT) and mutant-type (MT) sequences of
the binding sites between IL6ST and miR-200c-3p were designed. MT
and WT fragments of IL6ST 3'-untranslated region (UTR) were cloned
and ligated into a luciferase vector (pGL3-Promoter; Promega
Corporation) and co-transfected with miR-200c-3p mimics,
miR-200c-3p inhibitors or their respective NCs (sequences as
aforementioned) into 16HBE cells, and the groups were termed as: MT
+ mimics group, MT + NC mimics group, MT + inhibitors group, MT +
NC inhibitors group, WT + mimics group, WT + NC mimics group, WT +
inhibitors group, or WT + NC inhibitors group. In addition, pRL-TK
vector (Renilla luciferase) was co-transfected into the
cells as internal reference. After transfection for 48 h, the
luciferase activity of each group was measured using a
dual-luciferase reporter assay kit (Generay Biotech Co., Ltd.). The
ratio of firefly luciferase activity to Renilla luciferase
activity was the relative luciferase activity. The relative
luciferase activity of the control groups was set to 1, and the
data of the other groups were represented as the ratio to the
control group.
Statistical analysis
Statistical analysis was performed using the SPSS
v17.0 (SPSS, Inc.) and GraphPad Prism v6.0 (GraphPad software,
Inc.). Data are presented as the mean ± standard deviation, and all
experiments were repeated three times. An unpaired Student's t-test
was used to compare two groups, while one-way analysis of variance
was used for comparisons among multiple groups, followed by a
Tukey's post hoc test. P<0.05 indicates a statistically
significant difference.
Results
Low miR-200c-3p expression and high
IL6ST expression in HDM-induced cells
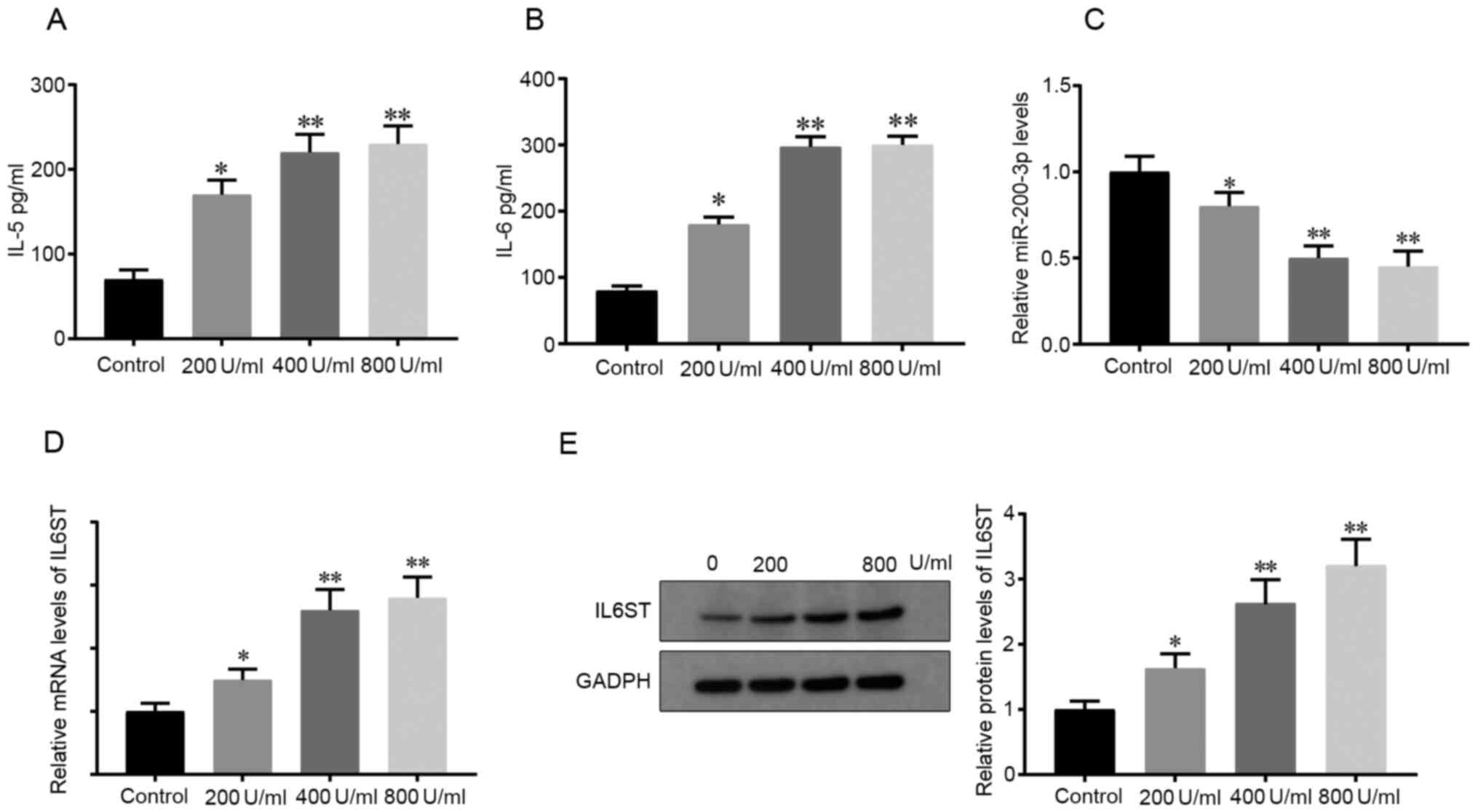
First, 200, 400 and 800 U/ml HDM was used to treat
the 16HBE cells in vitro for 24 h and the results from ELISA
showed that the concentrations of the inflammatory mediators, IL-5
and IL-6, were elevated in contrast to those in the Control group,
indicating that HDM successfully induced an inflammatory response
in the 16HBE cells (P<0.05; Fig.
1A and B). In addition, the
secretion of IL-5 and IL-6 levelled off at 800 U/ml HDM (Fig. 1A and B). Thus, 400 U/ml HDM was used for the
following experiments.
Next, the mRNA expression levels of miR-200c-3p and
IL6ST in the HDM-induced cells was determined. RT-qPCR showed that
compared with that in the control cells, the mRNA expression level
of miR-200c-3p was decreased in the HDM-induced cells (P<0.05;
Fig. 1C), indicating that HDM
suppressed miR-200c-3p mRNA expression level. In addition, the mRNA
and protein expression level of IL6ST in the HDM-induced cells was
found to be higher compared with that in the Control group
(P<0.05; Fig. 1D and E, respectively). The aforementioned
findings indicated that miR-200c-3p and IL6ST could be involved in
HDM-induced inflammation of the 16HBE cells.
MIM increases miR-200c-3p mRNA
expression and decreases IL6ST mRNA and protein expression levels
to inhibit airway inflammation in the epithelial cells
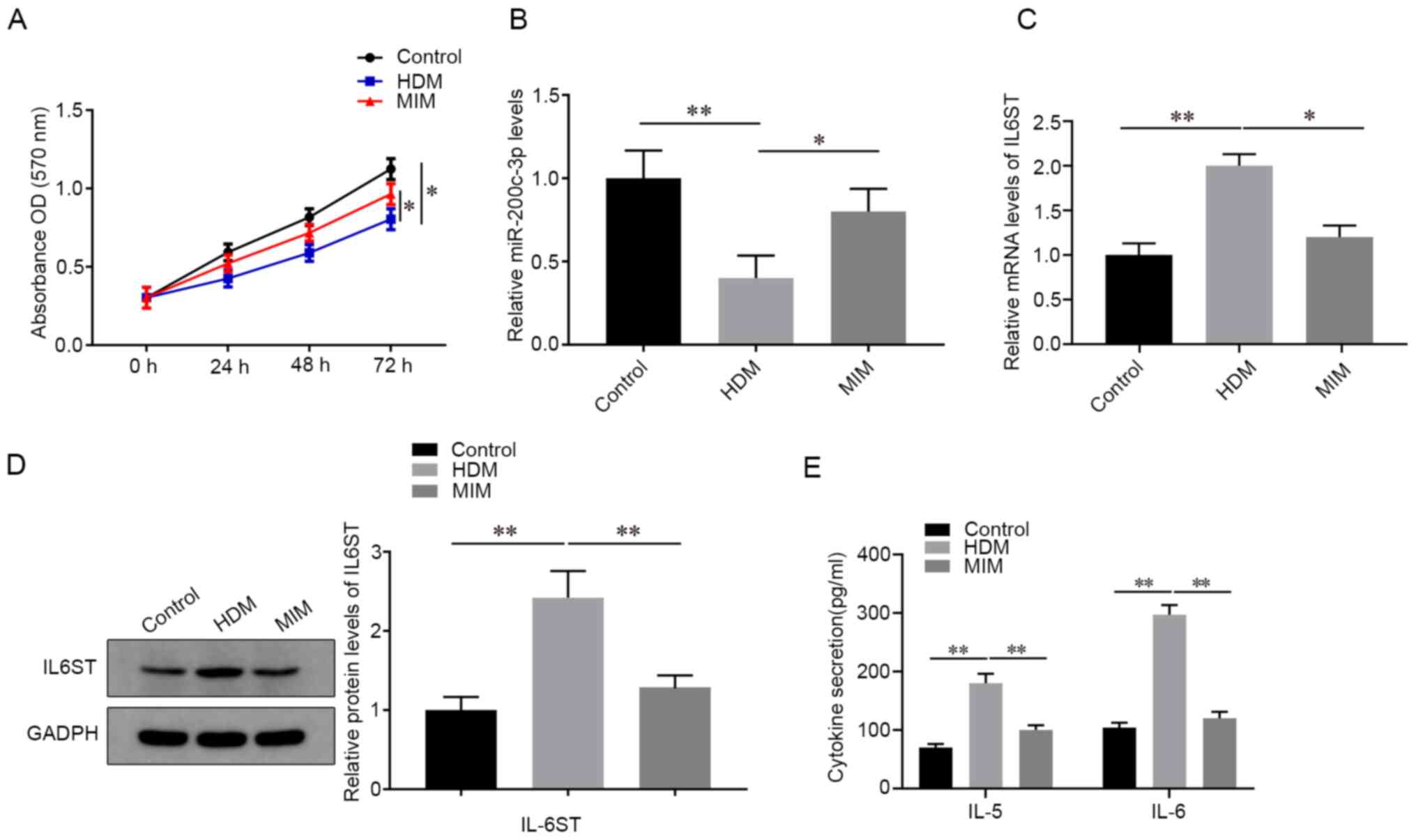
Next, the 16HBE cells were pretreated with MIM and
induced with HDM for 24 h to determine the effect of MIM on the
mRNA expression level of miR-200c-3p and IL6ST. As shown in
Fig. 2A, the MIM group showed
enhanced cell viability compared with that in the HDM group
(P<0.05). In addition, compared with that in the HDM group, the
MIM group showed increased miR-200c-3p mRNA expression levels
(P<0.05; Fig. 2B) and decreased
IL6ST mRNA and protein expression levels (P<0.05; Fig. 2C and D, respectively), and decreased
concentrations of IL-5 and IL-6 (P<0.05; Fig. 2E).
miR-200c-3p blocks HDM-induced
elevation of IL-5 and IL-6 levels by regulating IL6ST
expression
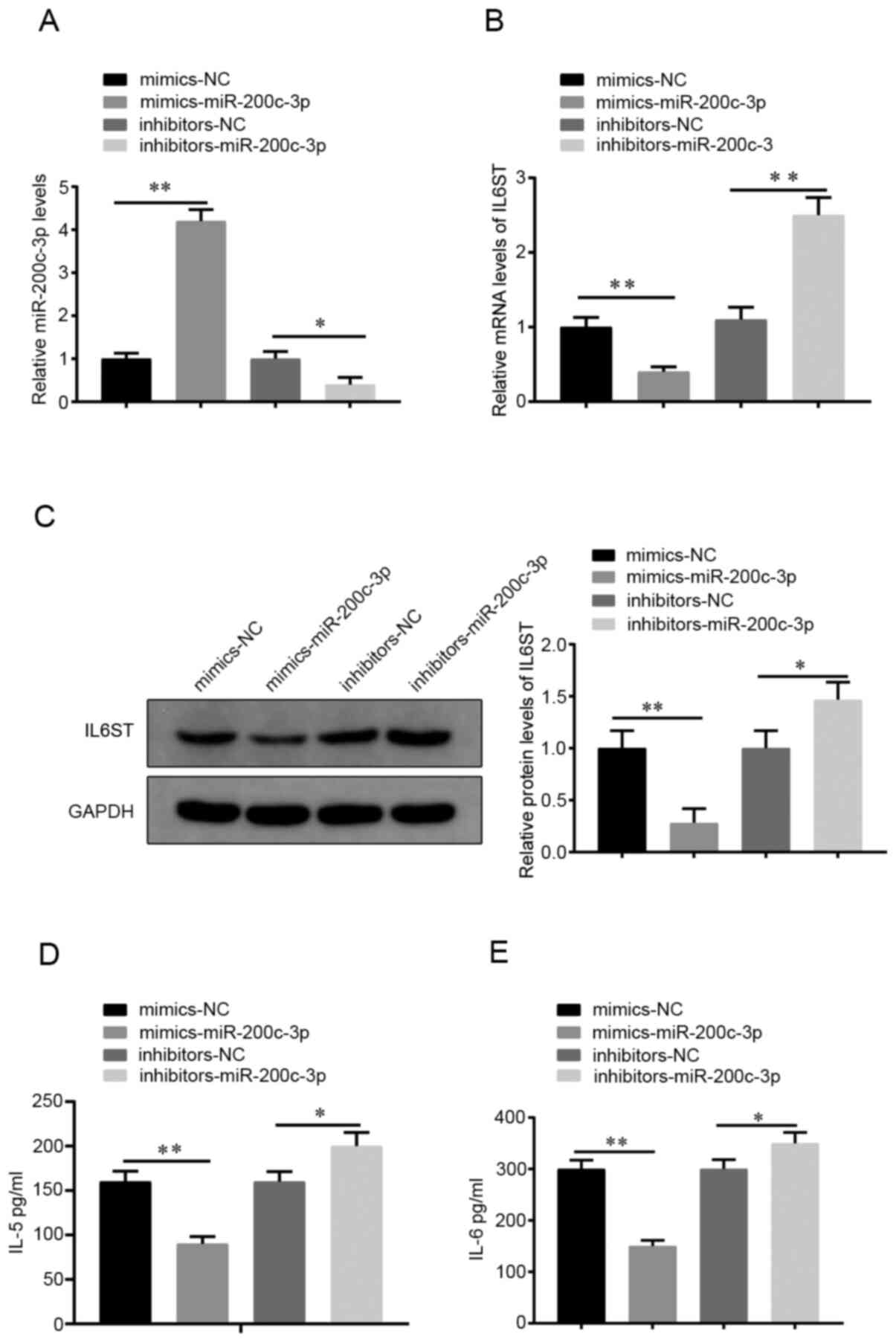
To investigate the effect of miR-200c-3p on the
biological activities of the HDM-induced cells, the miR-200c-3p
mimics or -miR-200c-3p inhibitor were transfected into the 16HBE
cells, after which the cells were stimulated by HDM for 24 h.
RT-qPCR results showed that the miR-200c-3p mimics group had an
increased miR-200c-3p mRNA expression level (vs. NC mimics) and the
miR-200c-3p inhibitor group had a decreased miR-200c-3p mRNA
expression level (vs. NC inhibitor group) (P<0.05; Fig. 3A), indicating effective miR-200c-3p
overexpression or silencing in the HDM-induced cells.
Compared with that in the NC mimics group, the
miR-200c-3p mimics group had lower mRNA and protein IL6ST
expression levels (P<0.01; Fig.
3B and C) and suppressed the
secretion of IL-5 and IL-6 (P<0.01; Fig. 3D and E, respectively). The opposite effect was
found for the mRNA and protein expression levels of IL6ST
(P<0.05; Fig. 3B and C) and the concentration of IL-5 and IL-6
in the miR-200c-3p inhibitor group compared with that in the NC
inhibitor group (P<0.05; Fig. 3D
and E). The aforementioned results
indicated that miR-200c-3p could inhibit HDM-induced inflammation
in 16HBE cells.
miR-200c-3p inhibitor reverses the
suppression of HDM-induced inflammation by MIM
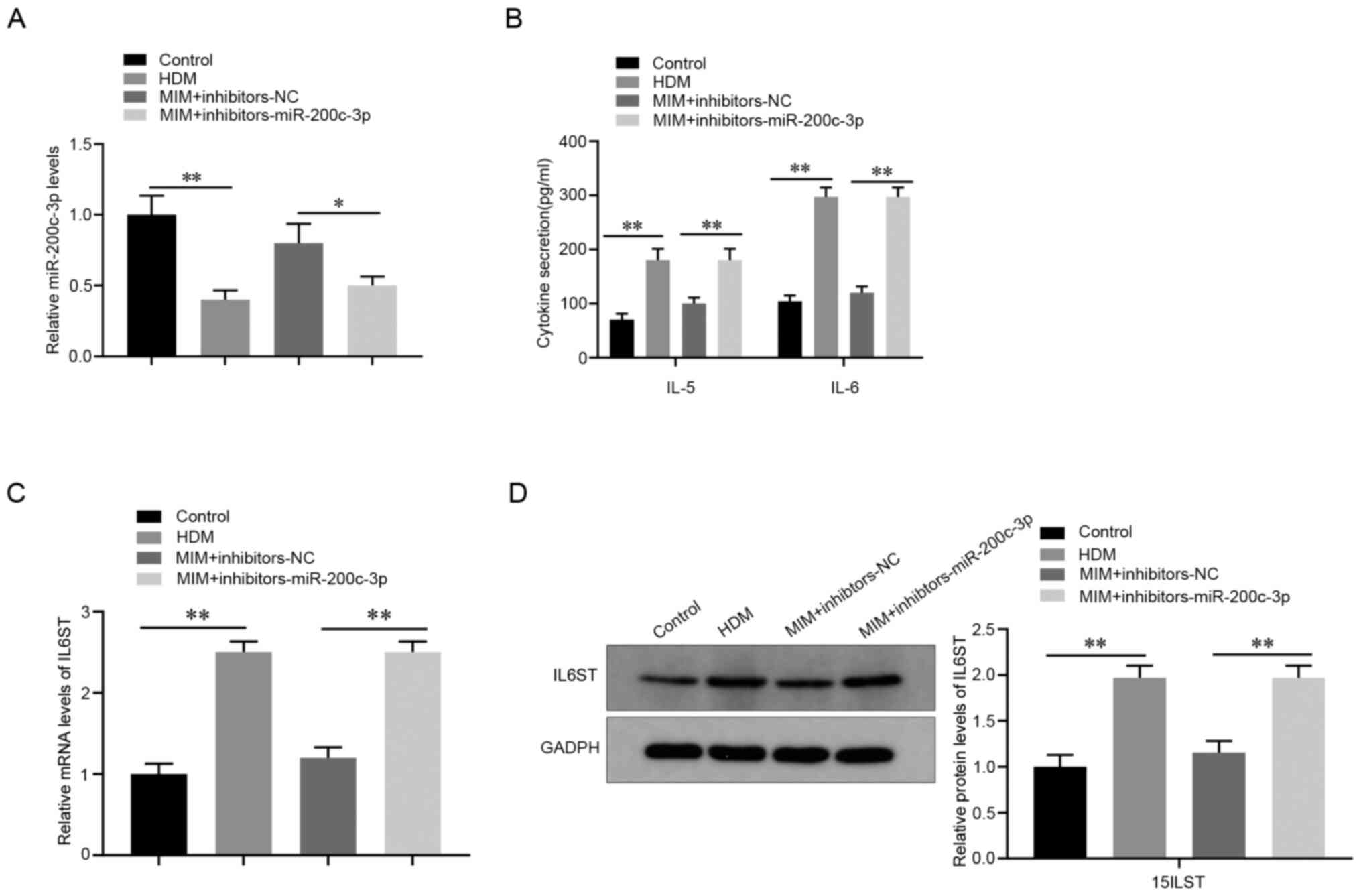
A rescue experiment was designed to further validate
the effect of miR-200c-3p on HDM-induced inflammation. Compared
with that in the MIM + NC inhibitor group, the miR-200c-3p
inhibitor reduced the mRNA expression level of miR-200c-3p in
HDM-induced 16HBE cells treated with MIM (P<0.05; Fig. 4A) and increased the secretion of
IL-5 and IL-6 (P<0.01; Fig. 4B).
Furthermore, the mRNA and protein expression levels of IL6ST were
also increased in the MIM + miR-200c-3p inhibitor group compared
with that in the MIM + NC inhibitor group (P<0.01; Fig. 4C and D). Collectively, these data showed that
the miR-200c-3p inhibitor reversed the suppression of HDM-induced
inflammation by MIM in AECs.
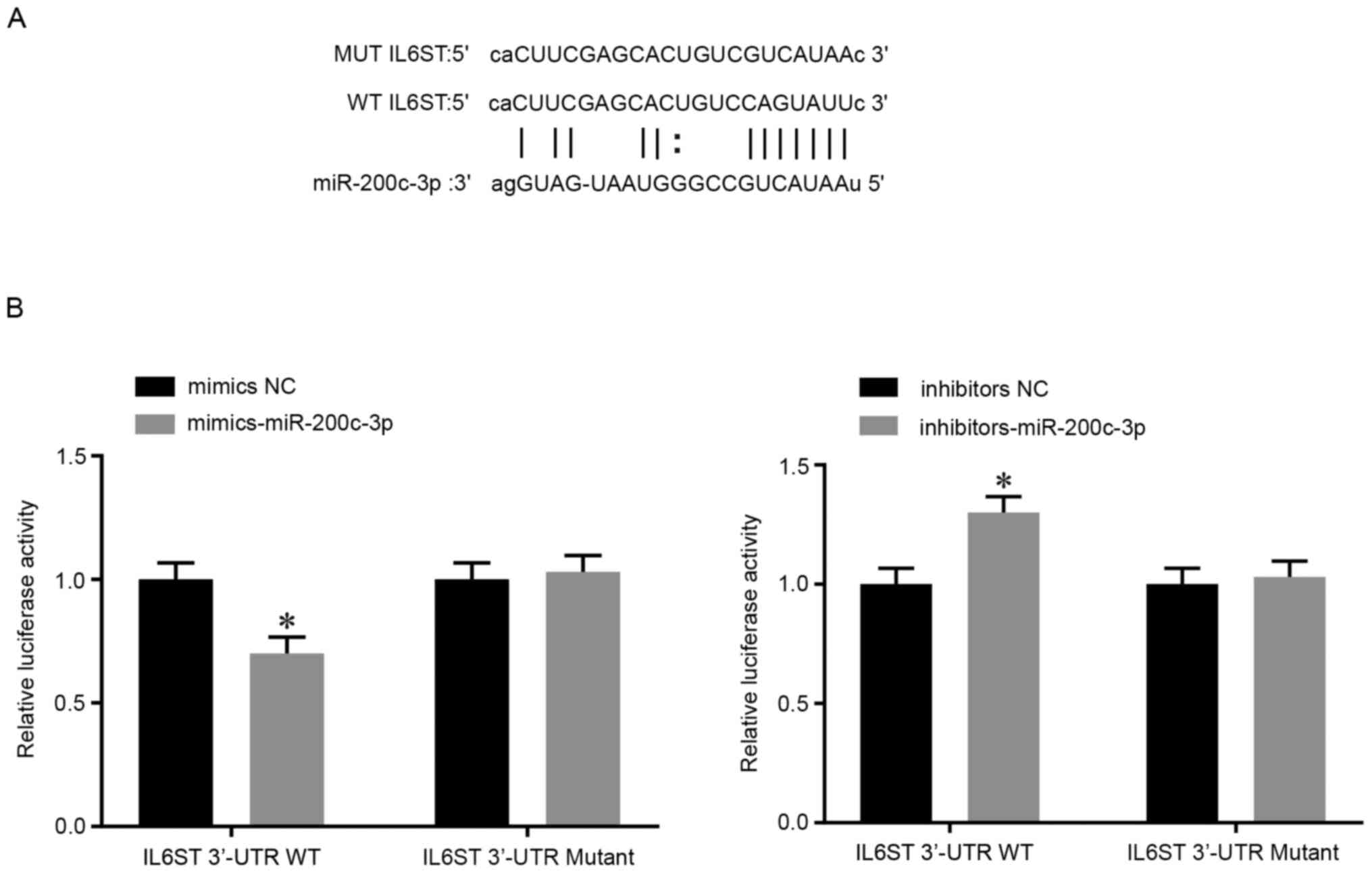
IL6ST is a functional target of
miR-200c-3p
Based on the prediction results from the StarBase
v3.0 database, the putative binding site of miR-200c-3p was located
on the 3'-UTR of IL6ST (Fig. 5A).
The results of the luciferase reporter gene assay showed that
compared with that in the WT + NC group, the luciferase activity in
the WT + mimics group was markedly inhibited, whereas the WT +
inhibitor group showed increased luciferase activity (P<0.05;
Fig. 5B). In addition, no visible
difference in luciferase activity was observed between the MT +
mimics group and MT + NC group or between the MT + inhibitors group
and MT + NC group. These results suggested that miR-200c-3p
targeted the 3'-UTR of IL6ST to inhibit the expression level of
IL6ST in asthmatic AECs.
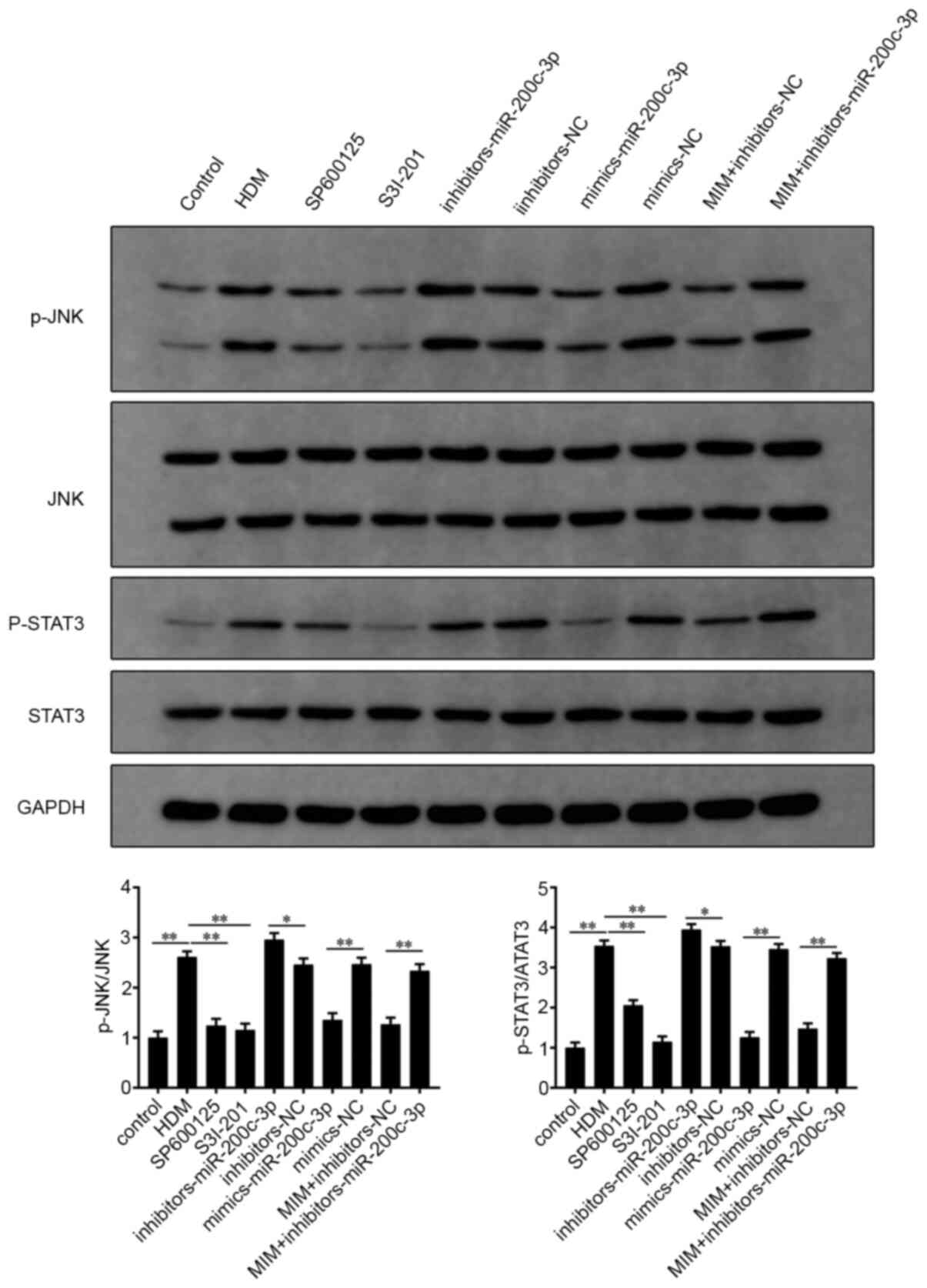
MIM suppress inflammation in asthmatic
AECs by regulating the JNK/STAT3 signaling pathway
As shown in Fig. 6,
p-JNK and p-STAT3 expression levels were significantly higher in
the HDM-induced 16HBE cells (P<0.01; HDM group vs. control
group). The addition of the JNK inhibitor, SP600125 and the STAT3
inhibitor, S3I-201, alone, suppressed HDM-induced JNK and STAT3
phosphorylation (P<0.01; SP600125 or S3I-201 group vs. HDM
group). Furthermore, the miR-200c-3p mimics group had decreased
p-JNK and p-STAT3 protein expression levels, whereas the
miR-200c-3p inhibitor group had elevated p-JNK and p-STAT3 protein
expression levels compared with that in their respective NC groups
(P<0.05). In addition, the expression levels of p-JNK and
p-STAT3 in the MIM + miR-200c-3p inhibitor group were significantly
higher compared with that in the MIM + NC inhibitor group
(P<0.01). The aforementioned results indicated the involvement
of the JNK/STAT3 signaling pathway in MIM-mediated inflammation in
asthmatic AECs.
Discussion
The prevalence of asthma has been increasing over
the last few decades and this has been accompanied by a substantial
economic burden (21). Therefore,
there is an urgent requirement to improve the precise
pathophysiology of asthma and achieve an optimal therapeutic effect
on this disease. In the present study, significantly lower
miR-200c-3p mRNA and higher IL6ST mRNA and protein expression
levels were detected in HDM-induced AECs. Further analyses showed
that MIM could upregulate miR-200c-3p mRNA expression levels to
inhibit the IL6ST/JNK/STAT3 signaling pathway; thus, decreasing
inflammation in HDM-induced AECs.
The important contribution of the intestinal
microflora to human health has been described in previous
literature. For example, the intestinal microflora plays an
irreplaceable role in promoting or preventing atherosclerotic
cardiovascular disease (22). The
intestinal microflora has also been associated in the development
of autism and mood disorders (23).
Recently, the significance of miRNAs in intestinal
microflora-mediated host physiology has been elucidated (24-27).
The findings of the present study revealed that MIM treatment
enhanced the viability and decreased the inflammatory responses of
asthmatic AECs. Notably, MIM exerted this protection by
upregulating the mRNA expression level of miR-200c-3p.
Several studies have reported that miRNAs modulate
inflammatory responses and airway remodeling in asthma (28,29).
miR-200c-3p has been identified as a tumor inhibitor in numerous
types of human cancer, including breast cancer, prostate cancer and
clear cell renal cell carcinoma (30,31).
However, the potential role of miR-200c-3p has not been
investigated in asthma. In the present study, decreased mRNA
expression of miR-200c-3p and increased mRNA and protein expression
level of IL6ST was found in the HDM-induced AECs. The transfection
of miR-200c-3p mimics decreased the mRNA and protein expression
level of IL6ST, as well as decreasing the secretion of 2
inflammatory factors. Based on online software prediction, it was
found that miR-200c-3p could target IL6ST; thus, inhibiting the
transcription and translation of IL6ST. A rescue experiment was
designed to understand the involvement of miR-200c-3p in the
MIM-mediated suppression of the asthmatic inflammation of AECs. The
results showed that the miR-200c-3p inhibitor reversed the
suppression of HDM-induced AEC inflammation by MIM. Therefore, MIM
could upregulate miR-200c-3p to reduce IL6ST-dependent inflammation
in AECs.
However, the accurate mechanism by which miR-200c-3p
mediated asthmatic inflammation was unclear; thus, the functional
mechanism of miR-200c-3p was further investigated. A previous study
has shown that activated signaling pathways, including the
JNK/STAT3 signaling pathway, were associated with lung cancer cell
survival, migration, inflammation and proliferation (32). The current study revealed that the
protein expression levels of p-JNK and p-STAT3 were significantly
increased in the HDM-induced AECs. Notably, miR-200c-3p could
downregulate the phosphorylation levels of JNK and STAT3 and this
effect was enhanced when cells were treated with MIM. Previous
research suggested that the JNK/STAT3 signaling pathway was
associated with apoptosis and participated in the inflammation of
myocardial injury (33). Overall,
MIM could decrease the phosphorylation of JNK and STAT by promoting
miR-200c-3p, thereby enhancing the proliferation of asthmatic
AECs.
In summary, the present study uncovered the effect
of MIM in asthma and the underlying molecular mechanism and the
findings suggested that MIM suppressed AEC inflammation, which can
occur in asthma, indicating the possibility of MIM treatment as a
novel therapy for asthma. MIM exerted therapeutic effects by
regulating the miR-200c-3p/IL6ST axis and the JNK/STAT3 signaling
pathway. Nevertheless, there are limitations to the HDM-induced
asthma cell models, and there is a lack of animal and clinical
experiments to support the results of the cell experiments. In
addition, which component plays a role in intestinal metabolites
also requires further investigated.
Acknowledgements
Not applicable.
Funding
Funding: This research was funded by a grant from Startup Fund
for Scientific Research of Fujian Medical University (grant no.
2017XQ1079).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH conceived the ideas, designed the experiments and
supervised the study. HH and BW performed the experiments. LH, HH
and BW analyzed the data. LH and BW provided critical materials. HH
and BW wrote the manuscript. LH, HH and BW confirm the authenticity
of all the raw data. All authors have read and approved the final
version of the manuscript.
Ethics approval and consent to
participant
The present study was approved by the ethical
committee of First Affiliated Hospital of Fujian Medical
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lambrecht BN and Hammad H: The immunology
of asthma. Nat Immunol. 16:45–56. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Papi A, Brightling C, Pedersen SE and
Reddel HK: Asthma. Lancet. 391:783–800. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Gon Y and Hashimoto S: Role of airway
epithelial barrier dysfunction in pathogenesis of asthma. Allergol
Int. 67:12–17. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mertens TCJ, Karmouty-Quintana H, Taube C
and Hiemstra PS: Use of airway epithelial cell culture to unravel
the pathogenesis and study treatment in obstructive airway
diseases. Pulm Pharmacol Ther. 45:101–113. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mitchell PD and O'Byrne PM:
Epithelial-derived cytokines in asthma. Chest. 151:1338–1344.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhang H, Sun Y, Rong W, Fan L, Cai Y, Qu
Q, Gao Y and Zhao H: miR-221 participates in the airway epithelial
cells injury in asthma via targeting SIRT1. Exp Lung Res.
44:272–279. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kho AT, McGeachie MJ, Moore KG, Sylvia JM,
Weiss ST and Tantisira KG: Circulating microRNAs and prediction of
asthma exacerbation in childhood asthma. Respir Res.
19(128)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lam JK, Chow MY, Zhang Y and Leung SW:
siRNA versus miRNA as therapeutics for gene silencing. Mol Ther
Nucleic Acids. 4(e252)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ong J, Timens W, Rajendran V, Algra A,
Spira A, Lenburg ME, Campbell JD, van den Berge M, Postma DS, van
den Berg A, et al: Identification of transforming growth
factor-beta-regulated microRNAs and the microRNA-targetomes in
primary lung fibroblasts. PLoS One. 12(e0183815)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kivihall A, Aab A, Soja J, Sładek K, Sanak
M, Altraja A, Jakiela B, Bochenek G and Rebane A: Reduced
expression of miR-146a in human bronchial epithelial cells alters
neutrophil migration. Clin Transl Allergy. 9(62)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Liu Z, Zhang XH, Callejas-Diaz B and
Mullol J: MicroRNA in United airway diseases. Int J Mol Sci.
17(716)2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Svitich OA, Sobolev VV, Gankovskaya LV,
Zhigalkina PV and Zverev VV: The role of regulatory RNAs (miRNAs)
in asthma. Allergol Immunopathol (Madr). 46:201–205.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Taka S, Tzani-Tzanopoulou P, Wanstall H
and Papadopoulos NG: MicroRNAs in asthma and respiratory
infections: Identifying common pathways. Allergy Asthma Immunol
Res. 12:4–23. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tang X, Wu F, Fan J, Jin Y, Wang J and
Yang G: Posttranscriptional regulation of interleukin-33 expression
by MicroRNA-200 in bronchial asthma. Mol Ther. 26:1808–1817.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lustri BC, Sperandio V and Moreira CG:
Bacterial chat: Intestinal metabolites and signals in
host-microbiota-pathogen interactions. Infect Immun. 85:e00476–17.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tuddenham S and Sears CL: The intestinal
microbiome and health. Curr Opin Infect Dis. 28:464–470.
2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang G, Huang S, Wang Y, Cai S, Yu H, Liu
H, Zeng X, Zhang G and Qiao S: Bridging intestinal immunity and gut
microbiota by metabolites. Cell Mol Life Sci. 76:3917–3937.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ver Heul A, Planer J and Kau AL: The human
microbiota and asthma. Clin Rev Allergy Immunol. 57:350–363.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xu Z, Tao J, Chen P, Chen L, Sharma S,
Wang G and Dong Q: Sodium butyrate inhibits colorectal cancer cell
migration by downregulating Bmi-1 through enhanced miR-200c
expression. Mol Nutr Food Res. 62(e1700844)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Choi JY, Lee HY, Hur J, Kim KH, Kang JY,
Rhee CK and Lee SY: TRPV1 blocking alleviates airway inflammation
and remodeling in a chronic asthma murine model. Allergy Asthma
Immunol Res. 10:216–224. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jie Z, Xia H, Zhong SL, Feng Q, Li S,
Liang S, Zhong H, Liu Z, Gao Y, Zhao H, et al: The gut microbiome
in atherosclerotic cardiovascular disease. Nat Commun.
8(845)2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Mangiola F, Ianiro G, Franceschi F,
Fagiuoli S, Gasbarrini G and Gasbarrini A: Gut microbiota in autism
and mood disorders. World J Gastroenterol. 22:361–368.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dong J, Tai JW and Lu LF: miRNA-microbiota
interaction in gut homeostasis and colorectal cancer. Trends
Cancer. 5:666–669. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu S, da Cunha AP, Rezende RM, Cialic R,
Wei Z, Bry L, Comstock LE, Gandhi R and Weiner HL: The host shapes
the gut microbiota via fecal MicroRNA. Cell Host Microbe. 19:32–43.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Teng Y, Ren Y, Sayed M, Hu X, Lei C, Kumar
A, Hutchins E, Mu J, Deng Z, Luo C, et al: Plant-derived exosomal
MicroRNAs shape the gut microbiota. Cell Host Microbe.
24:637–652.e8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chang CS and Kao CY: Current understanding
of the gut microbiota shaping mechanisms. J Biomed Sci.
26(59)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bartel S, Schulz N, Alessandrini F,
Schamberger AC, Pagel P, Theis FJ, Milger K, Noessner E, Stick SM,
Kicic A, et al: Pulmonary microRNA profiles identify involvement of
Creb1 and Sec14l3 in bronchial epithelial changes in allergic
asthma. Sci Rep. 7(46026)2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Martinez-Nunez RT, Rupani H, Platé M,
Niranjan M, Chambers RC, Howarth PH and Sanchez-Elsner T:
Genome-wide posttranscriptional dysregulation by MicroRNAs in human
asthma as revealed by frac-seq. J Immunol. 201:251–263.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Danarto R, Astuti I, Umbas R and Haryana
SM: Urine miR-21-5p and miR-200c-3p as potential non-invasive
biomarkers in patients with prostate cancer. Turk J Urol. 46:26–30.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Maolakuerban N, Azhati B, Tusong H, Abula
A, Yasheng A and Xireyazidan A: MiR-200c-3p inhibits cell migration
and invasion of clear cell renal cell carcinoma via regulating
SLC6A1. Cancer Biol Ther. 19:282–291. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bowman B: FADD and its phosphorylation
mediate mitogenic signaling in mutant Kras tumors, 2015.
|
|
33
|
Fan S, Sun JB, Li R, Song X and Li J:
Lycopene protects myocardial ischemia injury through anti-apoptosis
and anti-oxidative stress. Eur Rev Med Pharmacol Sci. 23:3096–3104.
2019.PubMed/NCBI View Article : Google Scholar
|