Introduction
Stroke is a severe perioperative complication. Based
on the cause of injury, strokes can be classified into hemorrhagic
and ischemic, with ischemic stroke accounting for 87% of all
perioperative strokes (1). Brain
injury following cerebral ischemia includes the infarct core, which
is enveloped by the ischemic penumbra. In the ischemic penumbra,
mild inflammatory and excitotoxic mechanisms contribute to delayed
cell death, which displays the biochemical characteristics of
apoptosis. Moreover, brain cells stimulate innate protective
programs that activate signaling cascades. Signaling cascades
activated as part of innate protective programs not only determine
cell survival, but also influence the neurological deficit and the
mortality after stroke (2). It has
been reported that ischemic or hypoxic pretreatment can provide
significant protection against stroke-induced cerebral injury
(3). However, this treatment is not
suitable for elderly patients at high risk of stroke, as it is
difficult to predict the outcomes of patients receiving ischemic
pretreatment (4). Furthermore,
there are significant individual differences in the therapeutic
effects of ischemic pretreatment among patients (5). However, endogenous protection involves
distinct molecular targets that form the rational basis for the
development of neuroprotective drugs. Thus, current research aimed
at reducing perioperative stroke has focused on the neuroprotective
properties of anesthetics commonly used in the perioperative period
(6).
Morphine (Mor), a potent opioid analgesic, is widely
used for clinical anesthesia. Previous studies have reported that
pretreatment with opioid drugs may preserve cellular integrity
following acute cerebral hypoxia (7,8).
Moreover, Mor pretreatment (MP) can significantly improve the early
survival of animals after acute cerebral ischemia (9,10). It
has also been shown that endogenous opioid peptides may protect
against ischemic cerebral injury and have been considered as
potential targets for stroke therapy (11). However, the post-receptor signaling
mechanisms of MP-induced neuroprotection have yet to be fully
elucidated. Therefore, further investigation into the specific
signaling mechanisms of neuroprotection induced by MP is
necessary.
Protein kinase C (PKC) comprises a family of
phospholipid-dependent serine/threonine kinases that are involved
in a series of cellular functions, such as cell death and survival
mechanisms (12,13). Ischemic tolerance induced by
endogenous preconditioning agents is dependent on PKC activation,
suggesting that PKCs serve as key regulators of ischemic
preconditioning in the brain (14,15).
Based on the activation requirements, PKCs are divided into the
conventional (c)PKC (α, βI, βII and γ), novel (n)PKC (δ, ε, η and
θ) and atypical (a)PKC (ι, λ and ζ) isoforms. The activation of
cPKC requires Ca2+ and diacylglycerol, whereas the
activation of nPKC and aPKC only requires diacylglycerol and lipid
mediators, respectively. Accumulating evidence has indicated that
PKC participates in the initiation and the development of
ischemic/hypoxic preconditioning in the brain (16-19).
Our previous work revealed that activation of cPKCγ was involved in
the protective effect of hypoxic preconditioning against cerebral
ischemic injury (20). Mor
functions via three different types of opioid receptors, namely μ,
κ and δ receptors. As the main opioid receptor, the signaling
efficiency of μ receptors is tightly regulated and is ultimately
limited by the coordinated phosphorylation of intracellular serine
and threonine residues (21). The
phosphorylation of μ receptors occurs primarily at T370, and
phosphorylation of T370 stimulated by phorbol esters or
heterologous activation of Gq-coupled receptors is mediated by PKC
(22). Therefore, it was
hypothesized that cPKCγ may be involved in the protective effect of
MP against cerebral ischemic injury. Thus, a cPKCγ antagonist,
Go6983, was used to determine whether cPKCγ participated in the
protective effects of MP against cerebral ischemic injury in the
in vivo and in vitro experiments.
Materials and methods
In vivo experiments
Ethics. The experimental protocol was
designed in accordance with the Animal Protection Law of the
People's Republic of China and was approved by the Animal Ethics
Committee of Beijing Tongren Hospital Affiliated with Capital
Medical University (approval nos. AEEI-2014-114, AEEI-2018-141 and
AEEI-2019-115).
Animal housing and grouping. A total of 100
adult male C57BL/6 mice (age, 12-14 weeks; weight, 18-22 g) were
purchased from Beijing Vital River Laboratory Animal Technology
Co., Ltd. The mice were housed under standard conditions
(temperature, 21±1˚C; relative humidity, 60±10%), with a 12-h
light/dark cycle and free access to food and water. Using a random
number table, the mice were divided into four groups in order to
receive different treatments (n=25 per group) as follows: i) Sham;
ii) ischemia; iii) Mor + ischemia (Mor + I); and iv) Mor + Go6983 +
ischemia (Mor + Go6983 + I) groups. All the animals were included
in the data analysis and assigned to different tests. Not all the
animals in each group were included in each test. The specific
animal numbers are indicated in the figure legends.
Animal model. The middle cerebral artery
occlusion (MCAO)-induced permanent focal cerebral ischemia mouse
model was established as previously described (20). For the MCAO surgery, mice were
anesthetized with an intraperitoneal (IP) injection of 2% sodium
pentobarbital (60 mg/kg). The left common artery and ipsilateral
external carotid artery were exposed and ligated through a ventral
midline neck incision. Following common carotid arteriotomy, a 4-0
surgical monofilament with a blunt tip (0.23 mm diameter; Guangzhou
Jialing Biotechnology Co., Ltd.) was inserted into the common
carotid artery until a mild resistance was felt, which was ~12 mm
distal to the carotid bifurcation, thereby occluding the origin of
the MCA. In the sham group, the common carotid artery was exposed,
but not occluded. The operative time was 10-15 min. If more time
was required, an additional 2% sodium pentobarbital dose (20-30% of
the initial dose) was administered to the mice, if needed, to
maintain the surgical plane of anesthesia. Throughout the
procedure, the body temperature was maintained at 37˚C using a
heating lamp and thermal blanket. After the surgery, the mice were
kept warm in an undisturbed environment for a minimum of 2 h for
observation, and cerebral blood flow was monitored using the laser
Doppler Flowmetry (Perimed PeriFlux system 5000; Perimed AB).
Drugs. Mor (Sigma-Aldrich; Merck KGaA) was
dissolved in normal saline (0.9% NaCl) and injected IP at
concentration of 10 mg/1 ml to achieve doses of 10 mg/kg. The Mor
dose was selected on the basis of previous research showing
dose-related effects on changes in the behavior and central nervous
system of mice (23-25).
The drug dose used in mice was 10-20 fold that used in humans
(26). Go6983 (Sigma-Aldrich; Merck
KGaA) was dissolved in DMSO and mixed with normal saline (0.9%
NaCl). The final concentration of DMSO was <0.1%. The total
volume of Go6983 was 5 µl (6 nM), and the drug was introduced in
advance into the polyethylene tube connecting the microinjection
cannula and the microsyringe. The microinjection cannula was kept
in the guide cannula for 5 min after drug administration to avoid a
backflow of drug solution. The dose of the inhibitor used in the
present study was appropriate for the microinjection method
(27).
Drug administration. Mice in the Mor + I
group were treated with 10 mg/kg Mor via intraperitoneal injection
at 24 h prior to cerebral ischemia induction. Under anesthesia with
sodium pentobarbital, mice in the Mor + Go6983 + I group were
injected with 5 µl Go6983 (6 nM) or DMSO (Go6983 can only be
dissolved in DMSO and it was necessary to exclude the extra effect
of DMSO on the results) into the intracerebral ventricle. Go6983
treatment was performed in a blinded manner, in which statisticians
and examiners were blinded to animal grouping and drug treatments.
Go6983 is a general cPKC inhibitor at low concentrations, but more
specifically inhibits cPKCγ activation at a concentration of 6 nM
(28). Drug administration into the
intracerebral ventricle was performed as previously described by
Muñoz et al (29). Animals
were positioned in a stereotaxic frame, and a cannula (28-gauge;
stainless steel; inner diameter, 0.18 mm; outer diameter, 0.36 mm)
was lowered stereotaxically into the left cerebral ventricle to a
position defined by the following coordinates: 0.5 mm posterior and
1.0 mm lateral to the bregma, and 3.5 mm below the skull surface.
To confirm that the solutions were administered exactly into the
cerebral ventricle, some mice were injected with 5 ml of diluted
1:10 India ink to examine their brains macroscopically after
sectioning.
Neurobehavioral tests. To evaluate the effect
of MP on neurological recovery following MCAO-induced ischemia, a
number of neurobehavioral tests were performed 6 h after MCAO
surgery. At this time, the ischemic penumbra or peri-infarct region
is more apparent, although the neurological deficits and infarct
volume continue to progress (30).
Using the neurological disability status scale (NDSS) reported by
Rodriguez et al (31), the
neurological deficits of mice were scored as follows: 0, no
neurological dysfunction; 2, slight decrease in mobility and
presence of passivity; 4, moderate neurological dysfunction and
additional alterations, including moderate hypomobility, flattened
posture, lateralized posture, hunched back, ataxic gait, decreased
body tone and muscular strength, as well as slight motor
incoordination; 6, disabled but able to walk, with more marked
hypomobility, circling, tremor, jerks and/or convulsions, forelimb
flection, and moderate motor incoordination; 8, respiration
distress and total incapacity to move/coordinate; and 10, death. If
the criteria for a precise grade given in the scoring list were not
met, the nearest appropriate number was utilized: 1, 3, 5, 7 or 9.
Before the experiment, the examiners were trained in NDSS
evaluation and were proficient in the NDSS criteria.
Evaluation of ischemic infarct and edema.
Immediately after the evaluation of the neurological deficits, a
double-blind measurement of the infarct volume was conducted. Mice
were anesthetized at 6 h via an IP injection of 2% sodium
pentobarbital (60 mg/kg) after MCAO surgery, and the brain was
quickly removed and cut into 1.5-mm coronal sections. The brain
sections were incubated for 20 min in a solution of 0.5%
2,3,5-triphenyltetrazolium chloride (TTC) in 10 mM PBS at 37˚C, and
then scanned into a computer. The infarct size was analyzed
according to an evaluation procedure reported by Wexler et
al (32). The edema ratio (E)
was calculated using the following equation: E =
(∑VL-∑VR)/(∑VL+∑VR) x100%, where ∑VL and ∑VR are the volumes of the
left (ischemic) and right (non-ischemic) hemispheres, respectively.
The background (B) was calculated using the following equation:
B=∑VS/∑VT x100%, where ∑VS is the volume of the unstained white
matter in the sham group and ∑VT is the total brain volume. To
account for the effect of edema and background, the infarct size
(I) was indirectly estimated and expressed as a percentage of the
total brain using the following equation: I = [∑VI x (1-E)/∑VT x
(1-B)] x100%, where VI is the volume of the tissue of MCAO model
mice not stained with TTC.
Nissl staining. Nissl staining was used to
assess neuronal cell damage in brain sections. Mice were deeply
anesthetized with an IP injection of 2% sodium pentobarbital (60
mg/kg) and were transcardially perfused with 100 mM PBS containing
4% paraformaldehyde. Brains were removed and postfixed in 4%
paraformaldehyde at 4˚C for 24 h, followed by incubation in graded
sucrose solutions (20% for 24 h and 30% for another 24 h) for
dehydration at 4˚C. The brains were cut into 20-µm sections, washed
in fresh PBS, stained with 0.04% cresyl violet (Sigma-Aldrich;
Merck KGaA) and dissolved in acetate buffer for 1 h at room
temperature. Images were captured under a Nikon 50i light
microscope with a x40 objective (Nikon Corporation). Nissl-stained
sections in the ipsilateral side to the MCAO were used for
counting, and cell numbers in five random high-power fields were
averaged in this area for each section. The images were analyzed
using ImageJ software version 1.8.0 (National Institutes of
Health).
TUNEL staining. Mice were anesthetized with
an i.p. injection of 2% sodium pentobarbital (60 mg/kg) and were
transcardially perfused with 100 mM PBS containing 4%
paraformaldehyde. Brains were removed and postfixed in 4%
paraformaldehyde at 4˚C for 24 h, followed by incubation in graded
sucrose solutions (20% for 24 h and 30% for another 24 h) for
dehydration at 4˚C. The brains (n=6) were cut into 10-µm sections
for TUNEL staining. The TUNEL staining kit (DeadEnd Fluorometric
TUNEL system; Promega Corporation) was used to assess neuronal
apoptosis. According to the manufacturer's instructions, the brain
sections were placed in equilibration buffer and incubated with
nucleotide mix and rTdT enzyme at 37˚C for 1 h. The reaction was
stopped using a termination buffer (300 mM NaCl; 30 mM sodium
citrate) for 15 min at room temperature. Then, neuronal nuclei were
stained with Hoechst 33258 and the slices were sealed by mounting
medium (cat. no. S2100, Beijing Solarbio Science & Technology
Co., Ltd.). The images were visualized by fluorescence microscopy
with a x40 objective. The cell numbers in five random high-power
fields were averaged in this area for each section (Leica DM4000B;
Leica Microsystems GmbH).
Western blot analysis. Mice were deeply
anesthetized by intraperitoneal injection of 2% sodium
pentobarbital (60 mg/kg) and decapitated. The mouse brains were
removed 6 h after MCAO surgery and immediately placed into ice-cold
artificial cerebral spinal fluid (125.0 mM NaCl; 2.5 mM KCl; 2.0 mM
CaCl2; 26.0 mM NaHCO3; 1.25 mM
NaH2PO4; 1.0 mM MgCl2; 5.0 mM
glucose; pH 7.4) bubbled with 95% O2/5% CO2
and were then dissected, as described in a previous report
(33). In brief, tissues 2 mm from
the anterior tip of the frontal lobe were removed, and the
remaining brain tissues were cut into four 2-mm sections. Each
hemisphere was longitudinally cut 1 mm from the midline to remove
the tissue supplied by the anterior cerebral artery. A 45˚
transverse diagonal cut was then made to separate the ischemic core
and peri-infarct region. The corresponding regions from the
non-ischemic hemisphere were dissected as the contralateral
controls. All tissues were frozen in liquid nitrogen and kept at
-70˚C for subsequent analysis.
To obtain whole cell lysates, the tissue samples
were rapidly thawed, homogenized at 25˚C in buffer C (5 mM Tris-Cl;
pH 7.5; containing 2 mM dithiothreitol, 2 mM EDTA, 1 mM EGTA, 5
µg/ml each of leupeptin, aprotinin, pepstatin A and chymostatin, 50
mM potassium fluoride, 50 µM okadaic acid, 5 mM sodium
pyrophosphate and 2% sodium dodecyl sulfate); the homogenates were
centrifuged at 30,000 x g for 30 min at 4˚C and the supernatants
were collected . The protein concentration was determined using a
BCA kit (Pierce; Thermo Fisher Scientific, Inc.) with albumin
diluted in buffer C as the standard.
Samples loaded with equal amounts of protein (50 µg)
were electrophoresed on 10% SDS-polyacrylamide gels and then
transferred onto PVDF membranes (Cytiva) at 4˚C. After several
rinses with TBS/Tween-20 (TBST; 20 mM Tris-Cl; pH 7.5; 0.15 M NaCl;
0.05% Tween-20), the PVDF membrane was blocked with 10% non-fat
milk in TBST for 1 h at room temperature. The blocked PVDF membrane
was then incubated with rabbit anti-caspase-3 (1:1,000; cat. no.
9662; Cell Signaling Technology, Inc.) for 3 h at 25˚C. To verify
equal loading of protein, the blots were reprobed with a primary
monoclonal antibody targeting rabbit anti-β-actin (1:1,000; cat.
no. 4970; Cell Signaling Technology, Inc.) for 3 h at room
temperature, and with the corresponding secondary antibody
AffiniPure mouse anti-rabbit IgG (H+L; 1:5,000, cat. no.
211-005-109; Jackson ImmunoResearch Laboratories, Inc.) for 1 h at
room temperature. After secondary antibody incubation, an ECL kit
(Cytiva) was used to detect the signals. Images were digitized and
analyzed by using ImageMaster 2D Platinum Software version 5.0
(Cytiva).
In vitro experiments
To further investigate the mechanisms underlying the
involvement of cPKCγ in the MP-induced neuroprotection, mouse N2a
neuroblastoma cells were exposed to conditions mimicking the in
vivo ischemic-like state, and caspase-3-positive cells were
analyzed via flow cytometry.
Cell treatment. Mouse N2a neuroblastoma cells
(kindly gifted by Dr Yun Wang, Peking University), were grown to
50% confluence in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% FBS in a 37˚C chamber (Thermo Electron LED GmbH;
Thermo Fisher Scientific, Inc.) under normoxic conditions (5%
CO2, 95% O2). To mimic the in vivo
ischemic-like conditions, N2a cells were exposed to oxygen-glucose
deprivation (OGD) treatment, in which the culture medium was
replaced by glucose-free DMEM and cells were maintained in a
hypoxic chamber (5% CO2, 1% O2, 94%
N2) for 3 h at 37˚C . After OGD exposure, cells were
returned to glucose-containing DMEM under normoxic conditions for
24 h of reoxygenation at 37˚C. MP was performed by incubating cells
with 3 µM Mor for 30 min followed by 30 min of washing before OGD.
Furthermore, 6 nM Go6983 (a cPKCγ antagonist) were added 30 min
before MP.
Flow cytometry. Activation of caspase-3 in
N2a cells by OGD was analyzed using flow cytometry with FITC active
caspase-3 Apoptosis kit (cat. no 550480; BD Pharmingen) according
to the manufacturer's instructions. After 24 h of reoxygenation,
cells were counted and the amount of antibody needed were
calculated. According to the kit instructions, these reagents had
been pre-diluted for use at the recommended volume per test and
1x106 cells in a 100 µl experimental sample was
recommended and with an antibody volume of 20 µl. The cells were
resuspended in BD Cytofix/Cytoperm solution and incubated for 20
min on ice. The cells were washed with BD Perm/Wash buffer and
incubated with anti-cleaved caspase-3 antibodies for 30 min at room
temperature. The cells were then washed and resuspended in BD
Perm/Wash buffer and analyzed via flow cytometry using a
FACSCalibur flow cytometer (BD Biosciences). A negative control was
performed with no anti-cleaved caspase-3 antibody. The software
used for analysis was BD CellQuest Pro version 6.0
(Becton-Dickinson and Company).
Statistical analysis
Statistical analysis was conducted using SPSS
software (version 25; IBM Corp.). One-way ANOVA followed by
pairwise multiple comparison procedures using the Bonferroni test
were adopted. The number of experiments repeats was two. All data
are presented as the mean ± SEM. P<0.05 was considered to
indicate statistically significant differences.
Results
In vivo experiments
MP attenuates MCAO-induced neurological
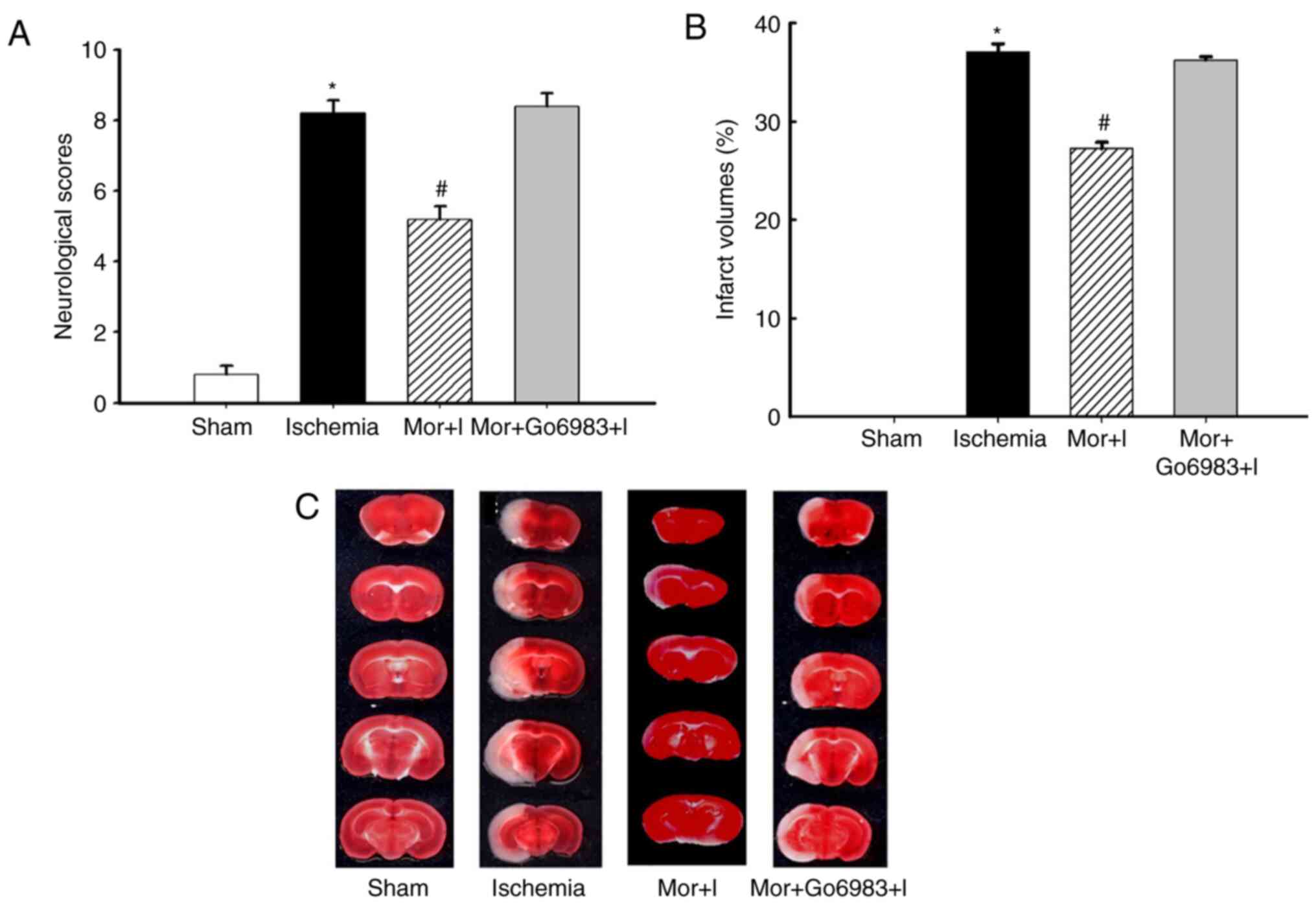
disability and cerebral ischemic injury. The neurological score
was 8.1±0.5 and 5.0±0.4 in the ischemia and MP groups,
respectively, with a statistically significant difference
(P<0.05, n=10 per group; Fig.
1A). Furthermore, the cerebral infarct volume was significantly
decreased in the MP group compared with that in the ischemia group
(ischemia 38.3±0.2% vs. MP 28.1±0.5%, P<0.05; Fig. 1B). An illustrated sample of cerebral
infarction is shown in Fig. 1C.
Inhibition of cPKCγ activation abolishes
MP-induced neuroprotection. The neurological score (8.0±0.4)
and cerebral infarct volume (37.1±0.3%) in the Mor + I + Go6983
group were not significantly different from those in the ischemia
group (8.1±0.5% and 38.3±0.2%, respectively) but were significantly
increased compared with those in the MP group (5.0±0.4% and
28.1±0.5%, respectively; Fig.
1A-C), indicating that cerebral ventricular injection of the
cPKCγ inhibitor Go6983 attenuated the MP-induced
neuroprotection.
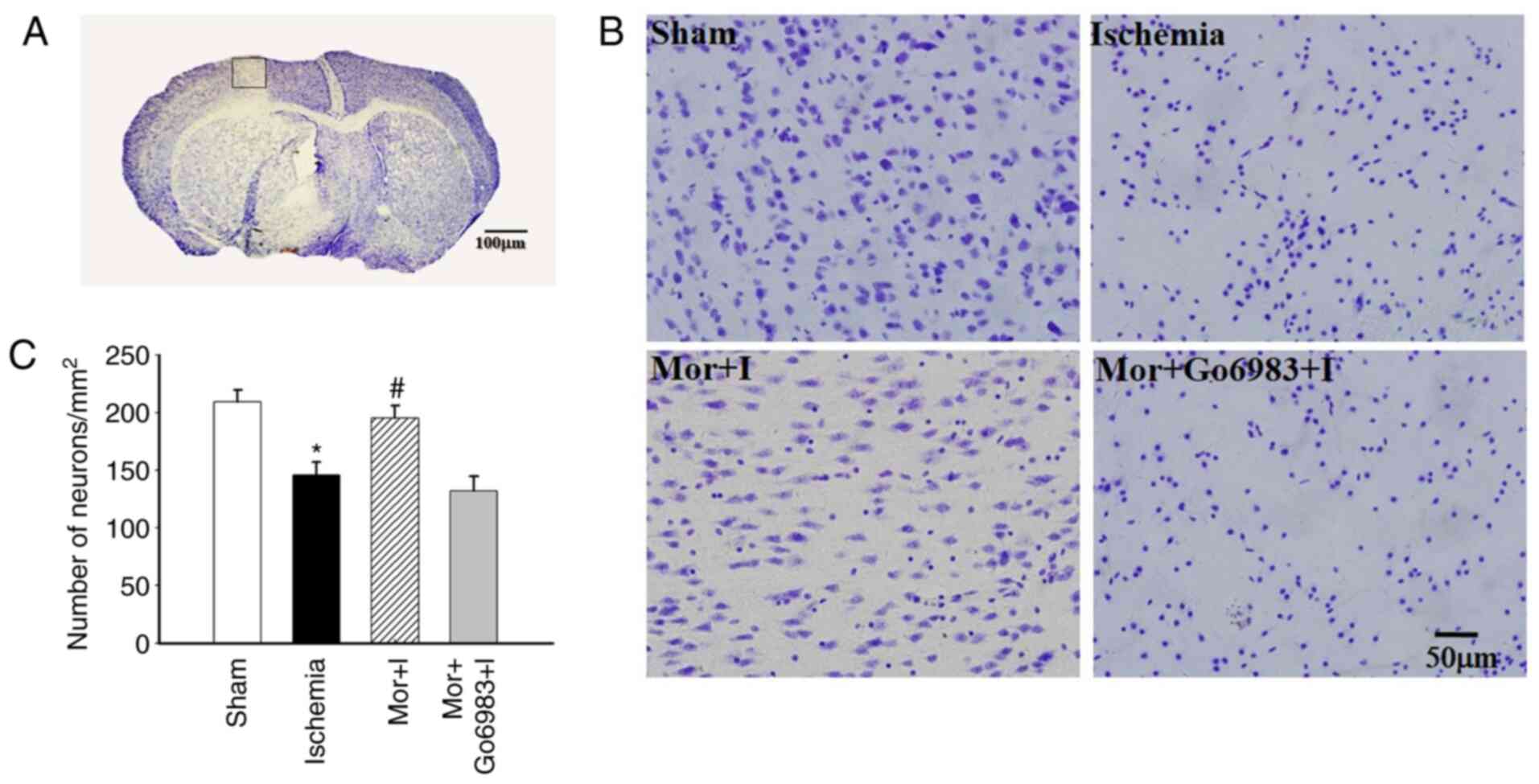
The number of Nissl-stained neural cells in the
peri-ischemic region was significantly decreased in the ischemia
group (149±10.0) compared with the sham group (209±10.4), but was
not significantly different in the MP group (198±9.1). Furthermore,
there was no significant difference in the number of Nissl-stained
neural cells between the ischemia and Mor + I + Go6983 groups
(149±10.0 vs. 138±8.9, respectively; Fig. 2A-C).
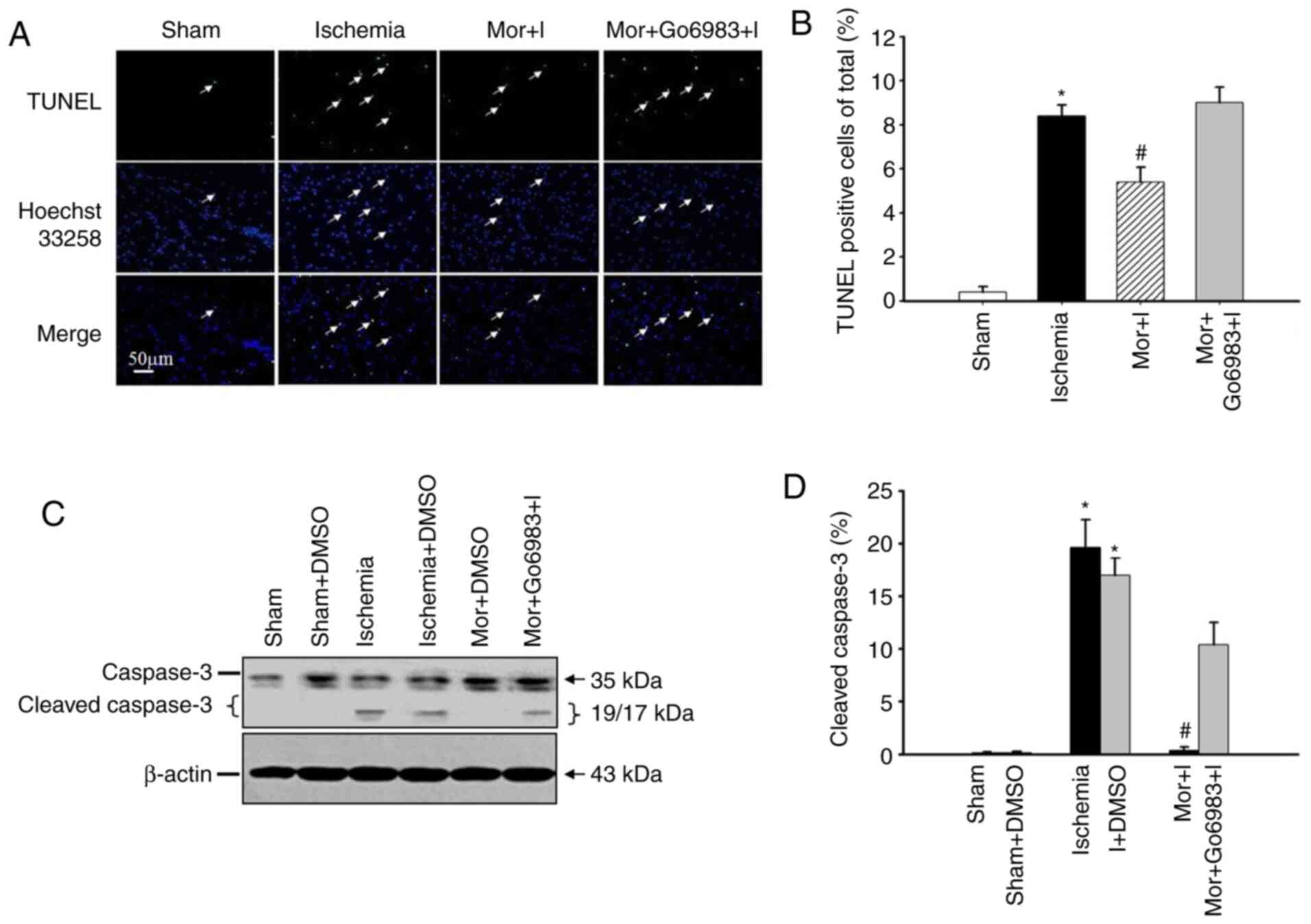
MP inhibits neuronal apoptosis and cleaved
caspase-3 activation, but this inhibition is reversed by the
cPKCγ antagonist. The number of TUNEL-positive neural
cells in the peri-ischemic region was significantly increased in
the ischemia group compared with that in the MP group (ischemia
8.6±1.2% vs. MP 5.6±1.2%), but was not significantly different
between the ischemia and Mor + I + Go6983 groups (ischemia 8.6±1.2%
vs. Mor + I + Go6983 9.2±1.3%; Fig.
3A and B).
Compared with the sham group, the protein expression
level of cleaved caspase-3 in the peri-ischemic region was
significantly increased in the ischemia group (ischemia 19.6±2.7%
vs. sham 0.1±0.1%). Moreover, compared with the ischemia group, the
protein expression level of cleaved caspase-3 in the peri-ischemic
region was significantly decreased in the MP group (ischemia
19.6±2.7% vs. MP 1.4±0.3%). However, there was no significant
difference in the cleaved caspase-3 protein expression between the
ischemia and Mor + I + Go6983 groups (ischemia 19.6±2.7% vs. Mor +
I + Go6983 10.4±2.1%; Fig. 3C and
D).
In vitro experiment
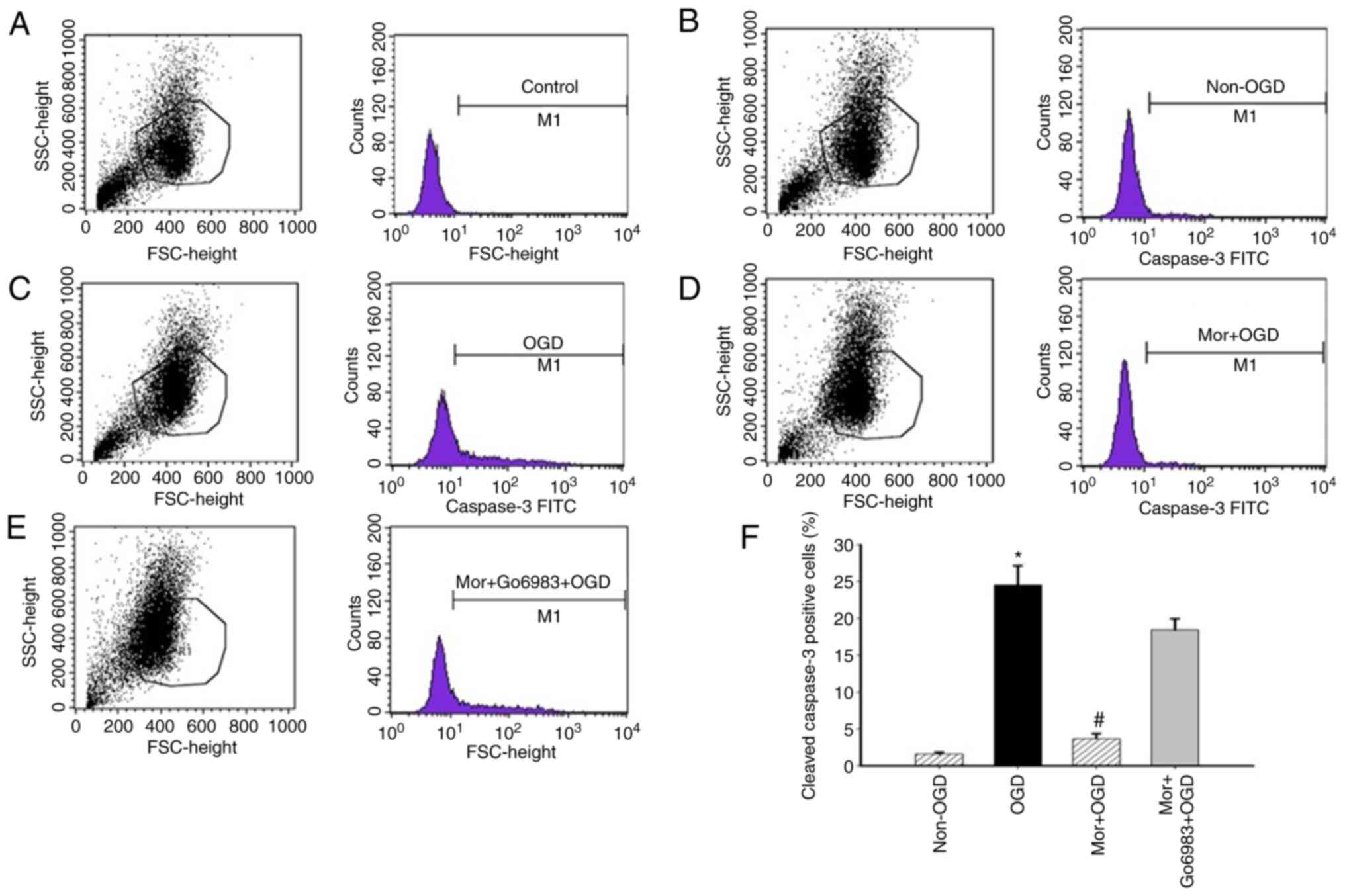
MP reduces the number of cleaved
caspase-3-positive cells after OGD, and the cPKCγ antagonist
prevents this MP-induced reduction. In the flow cytometry
analysis, the percentage of cleaved caspase-3-positive cells was
significantly increased in OGD N2a cells compared with N2a cells
under normoxic conditions (OGD 24.5±2.6% vs. normoxic 1.6±0.2%).
Furthermore, the percentage of cleaved caspase-3-positive N2a cells
was 3.7±0.7% in the MP treatment group and 18.4±1.5% in the Go6983
group. It was found that MP treatment significantly decreased the
number of cleaved caspase-3-positive OGD N2a cells (OGD 24.5±2.6%
vs. Mor + OGD 3.7±0.7%). However, pretreatment with Go6983 in OGD
N2a cells before MP inhibited the reduction in the number of
cleaved caspase-3-positive cells induced by MP (Mor + OGD 3.7±0.7%
vs. Mor + Go6983 + OGD 18.4±1.5%; Fig.
4A-F).
Discussion
Stroke is a severe perioperative complication in
elderly patients receiving surgery and represents a major
socioeconomic burden. The traditional treatment methods, involving
thrombolytic agents, tissue plasminogen activator, anticoagulation
therapy and even carotid endarterectomy, can only be used for the
therapy of ischemic strokes that have already occurred, and the
time window is very narrow (30).
Moreover, there is a lack of effective methods for the prevention
and therapy of perioperative stroke. Sedative anesthetics, such as
barbiturates, propofol and dexmedetomidine, have been reported to
provide neuroprotection in the perioperative period (34). However, they are not routinely used
prior to surgery due to their poor effect on decreasing patient
anxiety, and their inhibitory effects on the circulation and
cardiac function of the patients. Therefore, safe and more
effective alternative strategies are urgently required for the
prevention of stroke in the preoperative period.
As a potent opioid analgesic, Mor is used to
ameliorate preoperative patient anxiety and perioperative pain
(35). Previous studies have
reported MP-induced protection in numerous organs (36,37),
particularly in the brain (38).
The main findings of the present study were that MP could
significantly improve the neurological outcomes of MCAO model mice
and that cPKCγ was involved in MP-induced neuroprotection.
At the onset of an acute ischemic stroke, lack of
oxygen and other nutrients triggers a series of events causing
electrophysiological, metabolic and molecular disorders, leading to
irreversible brain tissue damage, which can manifest as
neurological deficits with regards to behavior and cell loss with
regards to morphology. In the MCAO model, animals develop
neurological deficits following recovery from anesthesia (20). Thus, the degree of neurological
deficits was evaluated in the present study, according to the NDSS
classification described by Rodriguez et al (31), who suggest that the peri-infarct
region (penumbra) becomes apparent and peak brain edema occurs at 6
h after MCAO. As this model has a high success rate of inducing
cerebral ischemia in the cortex, it can make the results of
experiment more uniform (32). In
the present study, consistent MCAO-induced neurological deficits
were observed, and application of MP at 24 h before MCAO
significantly improved the neurological outcomes of MCAO model
animals. This result was in line with the findings from a previous
study by Zhao et al (39),
in which application of Mor at 24 h before MCAO modeling decreased
the cerebral infarct volume and improved neurological outcomes at
24 h after MCAO modeling in adult rats. Given that improved
neurological outcomes were obtained at 6 and 24 h after MCAO
modeling, in the present study as well by Zhao et al
(39), it was suggested that
MP-induced neuroprotection should last >1 day. In addition, our
previous study reported that naloxone, a non-selective opioid
receptor antagonist, could block MP-induced neuroprotection
(40). Similarly, this finding was
confirmed by Arabian et al (41). All these results indicate that
opioid receptors are important for the protective effect of MP
against cerebral ischemic injury.
It has been reported that the protective effects of
acute and chronic opioid treatment in cerebral ischemic injury are
mediated via different signaling pathways (42). For example, chronic MP is mediated
by the signaling pathways involving protein kinase A (PKA) and Gs
proteins, while acute MP is mediated via PKC and Gi proteins
(41). Our previous study revealed
that PKC activation was significantly inhibited in the OGD-treated
hippocampal sections (20).
Moreover, in a mouse model of MCAO-induced ischemic stroke, Zhang
et al (20) confirmed that
cPKCγ knockout significantly increased the cerebral infarct volume
after 1 h MCAO/72 h reperfusion, as detected using TTC staining. It
has also been shown that, in primary cultured cortical neurons,
cPKCγ knockout can aggravate OGD-induced cell death and
morphological damage of neurites, while cPKCγ restoration can
alleviate ischemic injury (43). In
the present experiment, when MCAO model mice were treated with
intracerebroventricular injection of the cPKCγ antagonist prior to
MP, the MP-induced neuroprotective effect was eliminated, as shown
by the deteriorated neurological outcomes, expanded cerebral
infarct volume and increased neural cell loss. Collectively, these
findings suggest that PKC serves an important role in the
protection of MP against cerebral ischemic injury in MCAO model
mice; however, the involvement of other signaling molecules, such
as PKA, cannot be excluded.
To further determine the detailed mechanisms
underlying MP-mediated neuroprotection, neuronal apoptosis was also
evaluated using TUNEL staining in the current study. The present
results indicated that MP could significantly inhibit neuronal
apoptosis and caspase-3 activation, but a cPKCγ antagonist could
notably diminish the inhibitory effect of MP on MCAO-induced
neuronal apoptosis and caspase-3 activation. Moreover, in the
OGD-model N2a cells, the flow cytometry results revealed that MP
decreased the number of OGD-induced caspase-3-positive cells, while
the use of a cPKCγ antagonist eliminated the inhibitory effect of
MP on the activation of OGD-induced caspase-3-positive cells. Thus,
it was suggested that MP may protect against cerebral ischemic
injury via a cPKCγ-dependent anti-apoptosis pathway.
The available evidence indicates that
mitochondrial-related mechanisms may be involved in Mor treatment
and pretreatment (39). However,
oxidative stress can disrupt the mitochondrial membrane potential,
which promotes apoptosis-inducing factor release and activates the
caspase cascade. Furthermore, caspases serve as important drug
targets in ischemic organ injury (44). In the activation of the caspase
cascade, caspase-3 plays a key role by acting as the final executor
of the apoptosis pathway (45).
Scientific reports have shown the effect of µ-opioid agonists on
the release of apoptosis-inducing factor and cytochrome c,
as well as on caspase-3 activation, as the final executor of the
apoptosis pathway, using immunoblotting in toxin- and drug-treated
neuronal cells (45,46). The present study demonstrated that
MP could significantly inhibit neuronal apoptosis and caspase-3
activation. Moreover, it was found that, in OGD-induced N2a cells,
MP decreased the number of OGD-induced caspase-3-positive cells.
Thus, it was suggested that such opioid effects may be mediated by
anti-apoptotic activities via the reduction of the suppression of
caspase-3 activation. Additional implicated mechanisms, such as
apoptosis-inducing factor release and ion balance, must be further
elucidated.
There were certain limitations to the present study.
First, only one dose of Mor was tested. Thus, it remains unknown
whether the protective effect of MP against cerebral ischemic
injury was dose-dependent. Second, only the protective effect of MP
at 6 h after cerebral ischemia was observed. Therefore, the time
window of MP protection against cerebral ischemic injury must be
determined. Third, this experiment was only focused on the
involvement of the cPKCγ-mediated anti-apoptosis pathway in the
protective role of MP against cerebral ischemic injury. The
available evidence indicates that other mechanisms, such as
mitochondrial ATP-sensitive potassium channels, PI3K and
extracellular signal-regulated kinase pathways, autophagy,
inflammation and oxidative stress, may contribute to opioid-induced
preconditioning (11,47). The findings of the present study did
not provide definitive evidence regarding the exact roles of these
pathways and their interactions with the cPKCγ-mediated
anti-apoptosis pathway in the protective effect of MP against
cerebral ischemic injury. To address these aforementioned issues,
further experiments are required.
In conclusion, the present study demonstrated that
MP may protect against cerebral ischemic injury via a
cPKCγ-mediated anti-apoptosis pathway.
Acknowledgements
The authors would like to thank Dr Yi-Qiong Liu
(College of Life Sciences, PKU-IDG/McGovern Institute for Brain
Research, Peking University, Beijing, China) for providing helpful
suggestions.
Funding
Funding: The present study was supported by the National Science
Foundation of China (grant nos. 81671044, 81970344, 314711475 and
31671205). The funders had no part in experimental design, data
collection or analysis, decision to publish, or preparation of the
manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors participated in the design of the
studies, analysis of the data and review of the manuscript. XYZ
performed the immunohistochemistry, western blotting and behavioral
tests. FSX performed statistical analysis and generated figures.
XYZ and FSX wrote the manuscript. GYW and JFL interpreted the data
and revised the manuscript. TZL and CXP designed the experiments.
XYZ, FSX and GYW confirm the authenticity of the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocol was designed in accordance
with the Animal Protection Law of the People's Republic of China
and was approved by the Animal Ethics Committee of Beijing Tongren
Hospital Affiliated with Capital Medical University (approval nos.
AEEI-2014-114, AEEI-2018-141 and AEEI-2019-115).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dabrowska S, Andrzejewska A, Lukomska B
and Janowski M: Neuroinflammation as a target for treatment of
stroke using mesenchymal stem cells and extracellular vesicles. J
Neuroinflammation. 16(178)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mergenthaler P, Dirnagl U and Meisel A:
Pathophysiology of stroke: Lessons from animal models. Metab Brain
Dis. 19:151–167. 2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Moncayo J, de Freitas GR, Bogousslavsky J,
Altieri M and van Melle G: Do transient ischemic attacks have a
neuroprotective effect? Neurology. 54:2089–2094. 2000.PubMed/NCBI View Article : Google Scholar
|
|
4
|
de Lau LM, den Hertog HM, van den Herik EG
and Koudstaal PJ: Predicting and preventing stroke after transient
ischemic attack. Expert Rev Neurother. 9:1159–1170. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Rancurel G: Transient ischemic attacks in
the elderly: New definition and diagnostic difficulties. Psychol
Neuropsychiatr Vieil. 3:17–26. 2005.PubMed/NCBI(In French).
|
|
6
|
Archer DP, Walker AM, McCann SK, Moser JJ
and Appireddy RM: Anesthetic neuroprotection in experimental stroke
in rodents: A systematic review and meta-analysis. Anesthesiology.
126:653–665. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang J, Haddad GG and Xia Y: delta-, but
not mu- and kappa-, opioid receptor activation protects neocortical
neurons from glutamate-induced excitotoxic injury. Brain Res.
885:143–153. 2000.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang J, Gibney GT, Zhao P and Xia Y:
Neuroprotective role of delta-opioid receptors in cortical neurons.
Am J Physiol Cell Physiol. 282:C1225–C1234. 2002.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Endoh H, Taga K, Yamakura T, Sato K,
Watanabe I, Fukuda S and Shimoji K: Effects of naloxone and
morphine on acute hypoxic survival in mice. Crit Care Med.
27:1929–1933. 1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Endoh H, Honda T, Ohashi S and Shimoji K:
Naloxone improves arterial blood pressure and hypoxic ventilatory
depression, but not survival, of rats during acute hypoxia. Crit
Care Med. 29:623–627. 2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Peart JN, Gross ER and Gross GJ:
Opioid-induced preconditioning: Recent advances and future
perspectives. Vascul Pharmacol. 42:211–218. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mellor H and Parker PJ: The extended
protein kinase C superfamily. Biochem J. 332:281–292.
1998.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kaleli HN, Ozer E, Kaya VO and Kutlu O:
Protein kinase C isozymes and autophagy during neurodegenerative
disease progression. Cells. 9(553)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bright R and Mochly-Rosen D: The role of
protein kinase C in cerebral ischemic and reperfusion injury.
Stroke. 36:2781–2790. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Perez-Pinzon MA, Dave KR and Raval AP:
Role of reactive oxygen species and protein kinase C in ischemic
tolerance in the brain. Antioxid Redox Signal. 7:1150–1157.
2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kurkinen K, Busto R, Goldsteins G,
Koistinaho J and Pérez-Pinzón MA: Isoform-specific membrane
translocation of protein kinase C after ischemic preconditioning.
Neurochem Res. 26:1139–1144. 2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Niu C, Li J, Cui X, Han S, Zu P, Li H and
Xu Q: Changes in cPKC isoform-specific membrane translocation and
protein expression in the brain of hypoxic preconditioned mice.
Neurosci Lett. 384:1–6. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sun MK, Hongpaisan J, Nelson TJ and Alkon
DL: Poststroke neuronal rescue and synaptogenesis mediated in vivo
by protein kinase C in adult brains. Proc Natl Acad Sci USA.
105:13620–13625. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Voronkov AV and Mamleev AV: Endothelial
dysfunction and Protein kinase C activity development interrelation
at ischemic injury of a brain. Patol Fiziol Eksp Ter. 60:134–142.
2016.PubMed/NCBI
|
|
20
|
Zhang N, Yin Y, Han S, Jiang J, Yang W, Bu
X and Li J: Hypoxic preconditioning induced neuroprotection against
cerebral ischemic injuries and its cPKCγ-mediated molecular
mechanism. Neurochem Int. 58:684–692. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wolf R, Koch T, Schulz S, Klutzny M,
Schröder H, Raulf E, Bühling F and Höllt V: Replacement of
threonine 394 by alanine facilitates internalization and
resensitization of the rat mu opioid receptor. Mol Pharmacol.
55:263–268. 1999.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Mann A, Illing S, Miess E and Schulz S:
Different mechanisms of homologous and heterologous μ-opioid
receptor phosphorylation. Br J Pharmacol. 172:311–316.
2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vanderschuren LJ, De Vries TJ, Wardeh G,
Hogenboom FA and Schoffelmeer AN: A single exposure to morphine
induces long-lasting behavioural and neurochemical sensitization in
rats. Eur J Neurosci. 14:1533–1538. 2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ferguson SM, Thomas MJ and Robinson TE:
Morphine-induced c-fos mRNA expression in striatofugal circuits:
Modulation by dose, environmental context, and drug history.
Neuropsychopharmacology. 29:1664–1674. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cunningham CL, Bakner L, Schuette LM and
Young EA: Morphine and ethanol pretreatment effects on expression
and extinction of ethanol-induced conditioned place preference and
aversion in mice. Psychopharmacology (Berl). 238:55–66.
2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Blanchard OL and Smoliga JM: Translating
dosages from animal models to human clinical trials--revisiting
body surface area scaling. FASEB J. 29:1629–1634. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sacchetti B and Bielavska E:
Chelerythrine, a specific PKC inhibitor, blocks acquisition but not
consolidation and retrieval of conditioned taste aversion in rat.
Brain Res. 799:84–90. 1998.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gschwendt M, Dieterich S, Rennecke J,
Kittstein W, Mueller HJ and Johannes FJ: . Inhibition of protein
kinase C mu by various inhibitors. Differentiation from protein
kinase c isoenzymes. FEBS Lett. 392:77–80. 1996.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Muñoz A, Nakazaki M, Goodman JC, Barrios
R, Onetti CG, Bryan J and Aguilar-Bryan L: Ischemic preconditioning
in the hippocampus of a knockout mouse lacking SUR1-based K(ATP)
channels. Stroke. 34:164–170. 2003.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rabinstein AA: Update on treatment of
acute ischemic stroke. Continuum (Minneap Minn). 26:268–286.
2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Rodriguez R, Santiago-Mejia J, Gomez C and
San-Juan ER: A simplified procedure for the quantitative
measurement of neurological deficits after forebrain ischemia in
mice. J Neurosci Methods. 147:22–28. 2005.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wexler EJ, Peters EE, Gonzales A, Gonzales
ML, Slee AM and Kerr JS: An objective procedure for ischemic area
evaluation of the stroke intraluminal thread model in the mouse and
rat. J Neurosci Methods. 113:51–58. 2002.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ashwal S, Tone B, Tian HR, Cole DJ and
Pearce WJ: Core and penumbral nitric oxide synthase activity during
cerebral ischemia and reperfusion. Stroke. 29:1037–1047.
1998.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zwerus R and Absalom A: Update on
anesthetic neuroprotection. Curr Opin Anaesthesiol. 28:424–430.
2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Thomazeau J, Rouquette A, Martinez V,
Rabuel C, Prince N, Laplanche JL, Nizard R, Bergmann JF, Perrot S
and Lloret-Linares C: Acute pain factors predictive of
post-operative pain and opioid requirement in multimodal analgesia
following knee replacement. Eur J Pain. 20:822–832. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Murry CE, Richard VJ, Reimer KA and
Jennings RB: Ischemic preconditioning slows energy metabolism and
delays ultrastructural damage during a sustained ischemic episode.
Circ Res. 66:913–931. 1990.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Schultz JE, Rose E, Yao Z and Gross GJ:
Evidence for involvement of opioid receptors in ischemic
preconditioning in rat hearts. Am J Physiol. 268:H2157–H2161.
1995.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gao CJ, Niu L, Ren PC, Wang W, Zhu C, Li
YQ, Chai W and Sun XD: Hypoxic preconditioning attenuates global
cerebral ischemic injury following asphyxial cardiac arrest through
regulation of delta opioid receptor system. Neuroscience.
202:352–362. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhao P, Huang Y and Zuo Z: Opioid
preconditioning induces opioid receptor-dependent delayed
neuroprotection against ischemia in rats. J Neuropathol Exp Neurol.
65:945–952. 2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liu Y, Li J, Yang J, Ji F, Bu X, Zhang N
and Zhang B: Inhibition of PKCgamma membrane translocation mediated
morphine preconditioning-induced neuroprotection against
oxygen-glucose deprivation in the hippocampus slices of mice.
Neurosci Lett. 444:87–91. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Arabian M, Aboutaleb N, Soleimani M,
Mehrjerdi FZ, Ajami M and Pazoki-Toroudi H: Role of morphine
preconditioning and nitric oxide following brain ischemia
reperfusion injury in mice. Iran J Basic Med Sci. 18:14–21.
2015.PubMed/NCBI
|
|
42
|
Peart JN and Gross GJ: Cardioprotective
effects of acute and chronic opioid treatment are mediated via
different signaling pathways. Am J Physiol Heart Circ Physiol.
291:H1746–H1753. 2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhang N, Zhu H, Han S, Sui L and Li J:
cPKCγ alleviates ischemic injury through modulating synapsin Ia/b
phosphorylation in neurons of mice. Brain Res Bull. 142:156–162.
2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Faubel S and Edelstein CL: Caspases as
drug targets in ischemic organ injury. Curr Drug Targets Immune
Endocr Metabol Disord. 5:269–287. 2005.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Eftekhar-Vaghefi S, Esmaeili-Mahani S,
Elyasi L and Abbasnejad M: Involvement of Mu opioid receptor
signaling in the protective effect of opioid against
6-hydroxydopamine-induced SH-SY5Y human neuroblastoma cells
apoptosis. Basic Clin Neurosci. 6:171–178. 2015.PubMed/NCBI
|
|
46
|
Arabian M, Aboutaleb N, Soleimani M, Ajami
M, Habibey R, Rezaei Y and Pazoki-Toroudi H: Preconditioning with
morphine protects hippocampal CA1 neurons from ischemia-reperfusion
injury via activation of the mTOR pathway. Can J Physiol Pharmacol.
96:80–87. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lu S, Liao L, Zhang B, Yan W, Chen L, Yan
H, Guo L, Lu S, Xiong K and Yan J: Antioxidant cascades confer
neuroprotection in ethanol, morphine, and methamphetamine
preconditioning. Neurochem Int. 131(104540)2019.PubMed/NCBI View Article : Google Scholar
|