Introduction
Pulmonary artery hypertension (PAH) is a persistent
and fatal disease that is associated with a variety of factors,
including heredity, prolonged hypoxia and inflammation (1). PAH is characterized by a continuous
increase in pulmonary arterial pressure and right-sided heart
failure (1). As a bridge to chronic
pulmonary heart disease, hypoxic pulmonary hypertension is also
associated with high rates of mortality in elderly individuals in
China, which begins with persistent hypoxia (2). A previous study on the mortality of
PAH in Australia showed that patients with pulmonary artery
pressure >60 mmHg had a mortality rate of >40% within 1 year
and a mortality rate of ~80% within 5 years (3). The main histological characteristics
of PAH include the excessive proliferation of pulmonary artery
smooth muscle cells (PASMCs) and the accumulation of PASMCs in the
distal pulmonary arterioles, leading to increased myogenic
arteries, hence reducing the compliance of pulmonary arterioles
(2). Pulmonary vascular remodeling
(PVR), a major cause of PAH irreversibility, occurs due to the
proliferation of PASMCs (3). The
PAH process is accompanied by the activation of mast cells
(4). Mast cells can synthesize and
secrete IL-6(5). IL-6 then
stimulates B cells to secrete autoantibodies (6). The general abundance of autoantibodies
in the circulating blood of PAH rats, coupled with the disability
in targeting the immune intervention to inhibit cell proliferation
of PASMC, suggest that autoantibodies are an important cause of
PASMC apoptosis resistance (7). A
decreased expression of p62 and markedly increased ratio of
LC3-II/LC3-I suggest that autophagy is activated in the lung
tissues of rats during PAH (8). The
activation of autophagy is involved in hypoxia-induced PASMC
proliferation and migration, which is restored to baseline levels
in hypoxia-exposed PASMCs treated with the autophagy inhibitor 3-MA
(9,10). At present, no specific therapeutic
agents that are effective for pulmonary hypertension exist
(11). Only symptom-alleviation
treatments, including endothelin receptor antagonists and
phosphodiesterase inhibitors, in addition to strategies to promote
arterial vasodilation, such as prostanoids and calcium channel
blockers, can be used for clinical treatment (11). However, these treatments
aforementioned can only partially effective in improving the
symptoms and prolong life expectancy (12). Therefore, the present study aimed to
develop more effective PAH treatment strategies and explore the
mechanism underlying the pathological accumulation of PASMCs.
Similarities between the pathophysiology of
malignancies and PAH have been found (13). In particular, focal adhesion kinase
(FAK) and survivin were found to be associated with PAH (14). FAK was originally identified to be
an important therapeutic target protein for diseases involving
abnormal cell proliferation, including liver fibrosis (15) and PAH (16). Operating downstream protein of FAK
(17), survivin is a member of the
inhibitor of apoptosis protein family and promotes the
proliferation of PASMCs through inhibition of the caspase-3 pathway
(18,19). However, the underlying mechanism has
not been fully elucidated. Previous studies have suggested that
angiotensin (Ang) II can increase FAK expression, which is known to
be involved in the development of tumors and is also an important
factor in the development of normal cells such as fibroblasts and
epithelial cells (20). Ang
II-activated FAK could lead to liver fibrosis (21), macrophage accumulation during
atherosclerosis (22,23), proteinuric kidney disease (24) and the growth and extension of
climbing fiber innervation to some extent (25,26).
However, to the best of our knowledge, whether Ang II can also
increase FAK expression in PAH has not been previously
reported.
PAH is often accompanied by the activation of the
renin angiotensin system (RAS) and Ang II (27), leading to the activation of
hypoxia-inducible factor 1 (HIF-1). HIF-1-deficient cells
frequently exhibit increased apoptosis (28). Unfortunately, the activation of
HIF-1 is followed by the activation of Ang II, which in turn
activates RAS, resulting in a vicious circle (29). Within the RAS, ang-converting enzyme
(ACE) converts ang I into vasoconstrictor Ang II, which regulates
salt/water homeostasis and vasoconstriction, modulates blood
pressure and is also implicated in inflammation, endothelial
dysfunction and oxidative stress (30). Therefore, ACE inhibitors (ACEis)
have been widely used to treat diseases such as hypertension
involving the overproduction of Ang II; however, ACEi may cause
side effects such as hypotension (24). The majority of patients with PAH
exhibit reductions in cardiac output, which may cause hypotension
and insufficient organ perfusion (3). Therefore, ACEis treatment may increase
the risk of hypotension and insufficient organ perfusion (31). ACE2 was investigated in the present
study as it decomposes Ang II into Ang (1-7),
which reduces the Ang II concentration, allowing the RAS to
regulate blood pressure, and water and electrolyte balance
(32).
In the present study, it was hypothesized that an
association exists between Ang II and FAK during the occurrence of
PAH. To test this hypothesis, a rat model of PAH was established
with a single administration of monocrotaline and continuous
hypoxia treatment before the application of ACE2 activators or
inhibitors. The severity of PAH in rats was then assessed,
following which changes in the serum levels of FAK in plasma, the
mRNA expression of ACE2, FAK, caspase-3 and survivin, the protein
expression of ACE2, pFAK/FAK, cleaved caspase-3/pro-caspase-3 and
survivin and the expression of FAK around pulmonary arterioles, in
addition to apoptosis, was measured.
Materials and methods
Animals and experimental
intervention
A total of 36 male Sprague-Dawley rats (weight,
160-180 g; age, 8 weeks) were purchased from Changzhou Cavens
Laboratory Animal Co., Ltd. (http://www.cavens.com.cn/). All rats had free access
to food and water and were housed at 22±1˚C with 50±10% relative
humidity at a 12-h light/dark cycle. The animal care was in
accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals (33), and the study was approved by the
Animal Use and Protection Committee of Wuxi People's Hospital
(Wuxi, China). The rats were randomized into six groups
(n=6/group): i) Normal (N) group, subcutaneous injection (iH) and
intraperitoneal injection (ip) with normal saline (NS); ii)
diminazene aceturate (DIZE) group, treatment with iH administration
of 15 mg/kg/day DIZE (Sigma-Aldrich; Merck KGaA), a specific ACE2
activator (34), with an equivalent
volume of NS ip; iii) DX-600 group, treatment with 0.1 µmol/kg/day
ACE2 inhibitor DX-600 (Cayman Chemical Company) (35,36) ip
and an equivalent volume of NS iH; iv) PAH group, continuous
hypoxia with NS ip and iH; v) PAH + DIZE group, continuous hypoxia
with 15 mg/kg/day DIZE iH and NS ip; and vi) PAH + DX-600 group,
continuous hypoxia with 0.1 µmol/kg/day DX-600 ip and NS iH. The
rats were treated for 21 days.
The PAH rat model was established with a single iH
administration of monocrotaline (MCT; cat. no. IC0930; Beijing
Solarbio Science & Technology, Co., Ltd.) and continuous
hypoxia treatment (37). The rats
were then treated with saline, DIZE or DX-600 before beign returned
into the hypoxic chamber. The daily injection time was from 8:00 to
8:30 am in the morning, during which time the rats were in a
normoxic room. During the 28 days of hypoxia, drug administration
was only performed for the first 21 days i.e on day 21 of modeling,
the rats were injected with saline, DIZE or DX-600 for the final
time. No drugs were injected during the last 7 days of modeling.
The successful establishment of modeling was determined by
measuring rat weight, right ventricular systolic pressure (RVSP),
right ventricular hypertrophy index (RVHI) and observing
pathological changes (38). The
rats in the PAH group were fed in a special KY-2F hypoxic tank
(Fig. 1; Jiande Meicheng Analysis
Instrument Factory), whilst rats in the normoxic group were fed in
a normal oxygen environment. The KY-2F oxygen controller monitored
the oxygen concentration in real time.
The PAH groups adapted to a 12% oxygen concentration
for 2 days, and then a continued low flow of nitrogen was
introduced into the tank to control the oxygen concentration to
between 8-9%. On the first day of modeling, MCT 60 mg/kg iH was
administered to the three PAH model groups, while an equivalent
volume of NS iH was administered to the three groups under normal
oxygen conditions. During the modeling period, food and water were
changed for each group, the rats were weighed, drugs were
administered, and the tank was ventilated for 1 h daily. After 1 h,
the rats in the three PAH model groups were placed back into the
hypoxic tank, and the tank was filled with nitrogen. The treatment
lasted for 4 weeks.
Treatment of rats after modeling
The rats were anesthetized with pentobarbital sodium
(40 mg/kg) intraperitoneally and placed flat on the experiment
table. The hair on the chest and neck was removed with a shaver,
the skin was cut through a median and vertical incision and the
thyroid was removed. Cotton threads were passed through the back of
the trachea to expose it fully. Tracheotomy at the middle of the
trachea and tracheal intubation ensured the ventilation of the
rats. After connecting the tracheal tube to the Inspira ventilator
(https://www.dlnaturegene.net/pdlistone/products/11681692.html),
the parameters were set according to the weight of the rats.
Before measuring the RVSP, necessary preparations
were conducted. A sensor was connected to a PowerLab system (AD
Instruments) and zeroed. Then, a plastic catheter with a diameter
of 1 mm was prepared. It was rinsed, and heparin sodium was
injected with a sensor tubing. The internal jugular vein was
located from the right side of the trachea and the catheter placed
into the vein under B-ultrasound (Mindray Medical International,
Co., Ltd.) to ensure its placement in the right ventricle.
Subsequently, the RVSP was recorded. After recording the data, the
catheter was removed and the wound was clamped with forceps to
prevent bleeding.
After cutting the sternum from the middle, the rats
were exsanguinated, and 1.5 ml blood was collected from the right
ventricle and placed in a tube to determine the serum level of
FAK.
The rat heart and lung tissues were washed with
phosphate-buffered saline (PBS), and the excess tissues and trachea
were cut off. The pulmonary artery was separated on ice from the
secondary branch, and the water was blotted with an absorbent
paper. The right apex of the lung was fixed with 4% formaldehyde
for 24 h at room temperature and used for histopathological
examination. The remaining lung was cut into pieces and stored in
liquid nitrogen.
The right and left atrial appendages of the heart
were cut off, and the heart was perfused with PBS from the left
atrial appendage. When the right atrial appendage effluent was
transparent, the right ventricle, the left ventricle and the
ventricular septum were separated. An absorbent paper was used to
dry the three parts, which were then weighed. RVHI was calculated
using the following formula: RVHI=right ventricle/(left ventricle +
ventricular septum).
Quantification of FAK expression
Blood (1.5 ml) was taken from the right ventricle
and placed in a tube. The blood was left to coagulate at room
temperature for 20 min and then centrifuged for 20 min (1,000 x g;
4 ˚C). The supernatant was carefully collected and stored at -80˚C.
FAK expression levels in the serum were determined using an ELISA
kit (cat. no. SBJ-R1023-96T; Nanjing SenBeiJia Biological
Technology, Co., Ltd.; https://www.biomart.cn/infosupply/99217435.html)
according to the manufacturer's instructions. The absorbance values
were measured at 450 nm using a spectrophotometer (Thermo Fisher
Scientific, Inc.).
Reverse transcription-quantitative
(RT-q)PCR
The mRNA expression levels of related factors were
determined to evaluate the effect of DIZE on PAH. The total RNA was
extracted from lung tissue using TRIzol® (Thermo Fisher
Scientific, Inc.). The RNA yield and purity were determined with a
NanoDrop 2000/2000c Spectrophotometer (NanoDrop Technologies;
Thermo Fisher Scientific, Inc.) using the ratio of absorbance at
260 and 280 nm. Reverse transcription was performed with a
PrimeScript™ RT reagent kit (Takara Bio, Inc.) using the
following protocol: 30˚C for 10 min, 42˚C for 30 min and 99˚C for 5
min followed by 5˚C for 5 min. qPCR was then conducted with a
QuantiNova STBR Green PCR kit (Qiagen GmbH). Thermocycling
conditions were as follows: 95˚C for 2 min, followed by 45 cycles
of 94˚C for 15 sec, 55˚C for 15 sec and 68˚C for 30 sec and final
elongation at 72˚C for 5 min. All the primers in this study were
synthesized by Sangon Biotech Co., Ltd. Primer sequences were as
follows: TATA binding protein (TBP) forward,
5'-CCCACCAGCAGTTCAGTAGC-3' and reverse,
5'-GAATTCTGGGTTTGATCATTCTG-3'; FAK forward,
5'-CACCTGATGGAAGAGCGGCTAATC-3' and reverse,
5'-GGATCGGTCAAGGTTGGCAGTG-3'; caspase-3 forward,
5'-GTACAGAGCTGGACTGCGGTATTG-3' and reverse,
5'-AGTCGGCCTCCACTGGTATCTTC-3'; survivin forward,
5'-TGGCGGAGGCTGGCTTCATC-3' and reverse,
5'-CGGTCAGTTCTTCCACCTGCTTC-3'; and ACE2 forward,
5'-AAGCCACCTTACGAGCCTCCTG-3' and reverse,
5'-AACAATGCCAACCACTACCGTTCC-3'. The 2-ΔΔCq method (with
TBP as an internal reference primer) was used to quantify mRNA
expression (39).
Western blot analysis
The lung tissues were homogenized using RIPA buffer
(cat. no. P0013C; Beyotime Institute of Biotechnology) and protease
inhibitor cocktail (cat. no. HY-K0010; MedChem Express). The
protein concentrations were determined using a bicinchoninic acid
assay kit (cat. no. P0012S; Beyotime Institute of Biotechnology).
The samples were centrifuged at 3,000 x g at 4˚C for 30 min to
obtain the supernatants. Sample volumes containing 60 µg protein
were separated via 12% SDS-PAGE and transferred onto PVDF membranes
(Biosharp Life Sciences). After blocking nonspecific binding with 5%
non-fat milk at 25˚C for 1 h, the membranes were incubated with the
following primary antibodies at 4˚C overnight: Mouse anti-b-tubulin
(1:1,000; cat. no. 2146; Cell Signaling Technology, Inc.); rabbit
anti-ACE2 (1:1,000; cat. no. ab108252; Abcam), rabbit anti-FAK
(1:1,000; cat. no. ab40794; Abcam), rabbit anti-phosphorylated
(p)-FAK (Y397; 1:1,000; cat. no. ab81298; Abcam), rabbit
anti-caspase-3 (pro-caspase 3, 34 kDa; cleaved caspase 3, 17 kDa;
1:500; cat. no. ab13847; Abcam) and rabbit anti-survivin (1:500;
cat. no. ab134170; Abcam). After washing with Tris Buffered Saline
with 0.1% Tween-20 (TBST, cat. no. 9997s; Cell Signaling
Technology, Inc.), the membranes were incubated with diluted
HRP-conjugated goat anti-mouse IgG (1:1,000; cat. no. 91196s; Cell
Signaling Technology, Inc.) or goat anti-rabbit IgG (1:3,000, cat.
no. 7074s; Cell Signaling Technology, Inc.) for 2 h at 25˚C and
developed with enhanced chemiluminescence reagents (EMD Millipore).
The optical densities were analyzed using Image Lab (version 4.0;
Bio-Rad Laboratories, Inc.). β-tubulin was used as a loading
control.
Hematoxylin-eosin (HE) staining
The right pulmonary apex tissue was fixed with 4%
paraformaldehyde for 24 h at 25˚C, dehydrated using an ascending
ethanol gradient, embedded in paraffin wax, embedded and cut into
sections with a thickness of 4 µm. The sections were dewaxed using
xylene, rehydrated with a descending graded ethanol series, stained
with H&E (0.5% hematoxylin for 5 min followed by 0.5% eosin for
1 min, both at 25˚C), hydrated with a graded alcohol series and
dehydrated with xylene. The images were acquired using an inverted
light microscope (magnification, x400; model, Leica DMi8; Leica
Microsystems GmbH) under white light. Pulmonary arterioles with a
diameter of ~50 µm were observed using Image-Pro Plus (version 6.0;
Media Cybernetics, Inc.).
In total, 20-40 small pulmonary arteries with a
diameter of ~50 µm, were taken from each group. According to the
morphology of the arteries, they were divided into non-muscularized
arteries (internal acinar arteries without muscularization of the
vessel wall), partially muscularized arteries (the muscularization
of the vessel wall ≤75% of the vascular radius) and
fully-muscularized arteries (the muscularization of the vessel wall
>75% of the vascular radius) (40). The extent of muscularization of each
group was calculated according to the ratio. In addition, 25
pulmonary arterioles with a diameter of ~50 µm were selected in
each group, before Image-Pro Plus (version 6.0; Media Cybernetics,
Inc.) was used to measure the inner and outer diameters of the
lumen. From these, the ratio of the pulmonary artery media
thickness to the outer diameter was calculated to deduce the media
fraction thickness of each group.
TUNEL staining
The paraffin-embedded sections, prepared using
protocols aforementioned, were incubated at 60˚C for 1 h and
stained using a One-Step TUNEL Apoptosis Assay Kit (cat. no.
KGA7073; Nanjing KeyGen Biotech Co., Ltd.) according to the
manufacturer's protocol. First, tissue slides were treated with
proteinase K (100 µl) for 30 min at 37˚C. After rinsing with PBS,
the tissue slides were treated with a TdT enzyme reaction solution
(50 µl; a mixture of 45 µl equilibration buffer, 1 µl
biotin-11-dUTP and 4 µl TdT enzyme) for 60 min at 37˚C. The samples
were incubated in a humidity chamber (plastic box with PBS).
Sections were incubated with streptavidin-fluorescein (5 µl) and
labeling buffer (45 µl) for 30 min at 37˚C. Sections were then
incubated with 5 µg/ml DAPI for 10 min at 25˚C. After rinsing with
PBS, the sections were treated with glycerin PBS seal tablets
(glycerin: PBS=6:4; cat. no. ST1353; Beyotime Institute of
Biotechnology). The apoptotic cells showing green fluorescence were
observed under an inverted fluorescence microscope (magnification,
x400) and photographed using Image-Pro Plus (version 6.0; Media
Cybernetics, Inc.). In total, five random fields were selected for
each sample, which were analyzed using ImageJ software v1.0
(National Institutes of Health). The positive points of DAPI were
the total number of cells, whilst the positive points of FITC were
the apoptotic positive cells. Finally, the proportion of apoptotic
cells calculated using the formula FITC/DAPI x100%.
Immunohistochemistry (IHC)
staining
IHC staining was conducted to evaluate the
immunoreactivity of FAK. The paraffin-embedded sections, prepared
using protocols aforementioned, were dewaxed with xylene, hydrated
with a descending graded ethanol series. The tissue sections were
then blocked using an enhanced endogenous peroxidase blocking
buffer (cat. no. P0100B; Beyotime Institute of Biotechnology) at
25˚C for 10 min. After washing twice with PBS, the sections were
heated to 96-98˚C for 15 min inducing epitope retrieval with a
citrate antigen retrieval solution (cat. no. P0081; Beyotime
Institute of Biotechnology). The sections were rinsed with PBS and
then blocked with 10% goat serum (cat. no. C0265; Beyotime
Institute of Biotechnology) at 37˚C for 30 min. The sections were
then probed with rabbit anti-FAK (1:250; cat. no. ab40794; Abcam)
at 4˚C for 12 h followed by HRP-conjugated goat anti-rabbit
secondary antibodies at 37˚C for 1 h (1:5,000; cat. no. ab205718;
Abcam). Fixed tissues were stained with DAB (cat. no. P0203;
Beyotime Institute of Biotechnology) at 25˚C for 30 min. After
washing thoroughly with distilled water, the sections were stained
with hematoxylin at 25˚C for 10 min and then differentiated with an
acidic ethanol differentiation solution at 25˚C for 5 sec (cat. no.
C0107; Beijing Solarbio Science & Technology, Co., Ltd.). After
further rinsing with distilled water, the sections were hydrated
with a graded alcohol series and dehydrated with xylene. They were
observed under x400 magnification with an inverted light microscope
under white light and the images were collected using Image-Pro
Plus (version 6.0; Media Cybernetics, Inc.). FAK expression was
indicated as yellowish-brown particles.
Statistical analysis
The data are presented as the mean ± standard error
of mean. Statistical analysis was performed using SPSS version 22.0
(IBM Corp.). Comparison among groups was performed by one-way
analysis of variance. Post hoc analyses were performed using
Tukey's multiple comparisons test. P<0.05 was considered to
indicate a statistically significant difference.
Results
ACE2 alleviates PAH-induced
morphological changes
The hypoxic treatment lasted for 4 weeks. The rats
in each group were weighed and treated according to the procedure.
The preoperative body weight of rats was significantly higher in
the N group compared with that in the PAH group (Fig. 2B). By contrast, the RVSP and RVHI
values were lower in the N group compared with in the PAH group
(Fig. 2A and C). The H&E staining results indicated
that the pulmonary arteries in the N group had large and thin walls
with a regular arrangement of the endothelium and smooth muscle
cells (Fig. 2D). The pulmonary
arterioles in the PAH group were similar in external diameter, but
had decreased internal diameter, increased wall asymmetry, stenosis
of the lumen, decreased number of nonmuscular arteries and
increased number of muscular arteries compared with the N group
(Fig. 2D). In addition, reduced
distal pulmonary artery muscularization and media fraction
thickness were observed in the N group, compared with the PAH group
(Fig. 2D). Compared with the N
group, the rats in the PAH group showed significant weight loss,
increased RVSP and RVHI and thickened pulmonary arterioles.
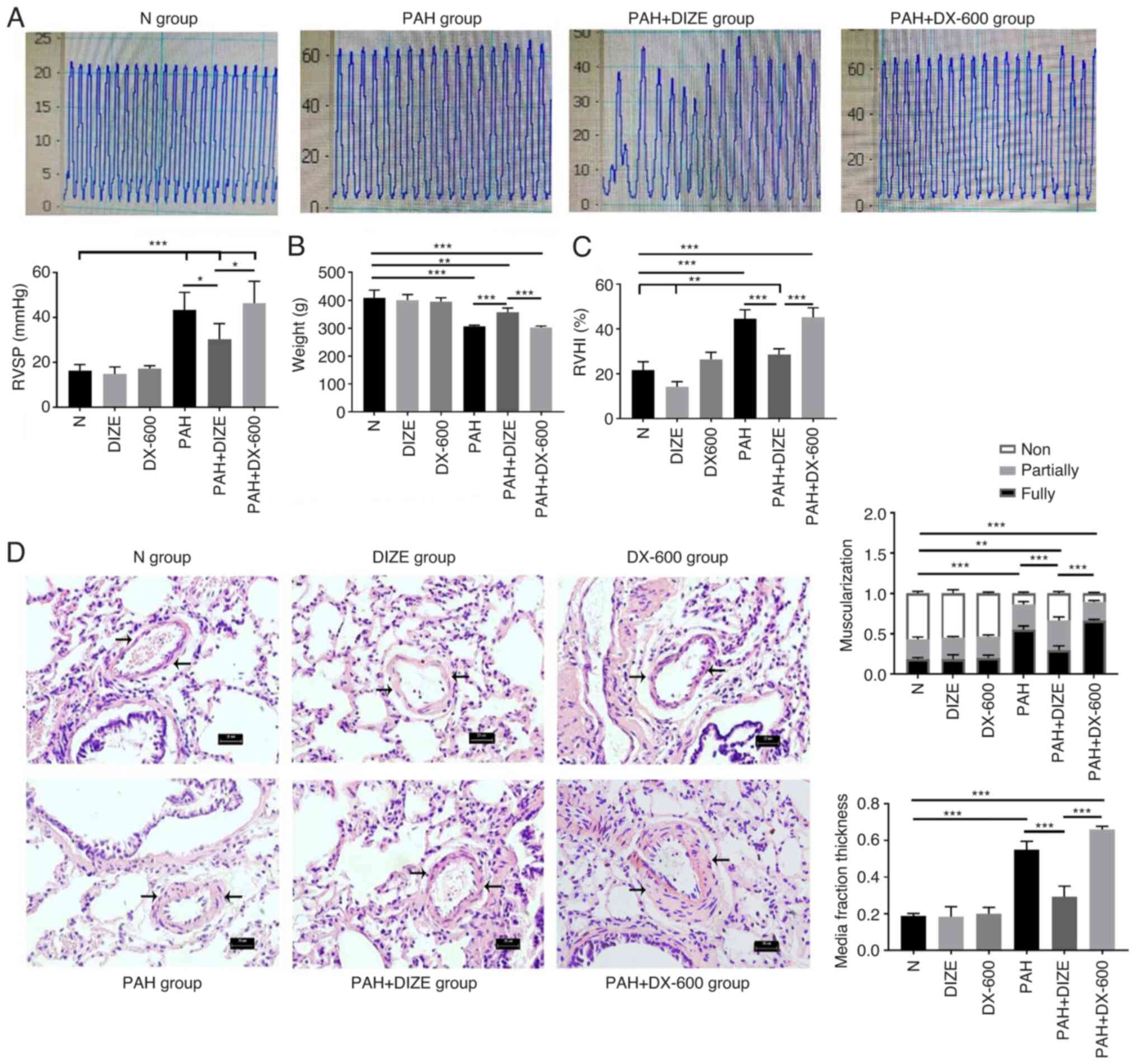 | Figure 2Effect of DIZE on normal rats and in
a hypoxia-induced PAH rat model. Rats were randomly divided into N,
DIZE, DX-600, PAH, PAH + DIZE and PAH + DX-600 groups (n=6/group).
The DIZE, PAH + DIZE, DX-600 and PAH + DX-600 groups were injected
with DIZE or DX-600 for the first 3 weeks of hypoxia. (A)
Representative trace of RVSP in the N, PAH, PAH + DIZE and
PAH+DX-600 groups, and quantification of RVSP in all groups. (B)
Weight, (C) RVHI and (D) morphology of the pulmonary arterioles in
the N, DIZE, DX-600, PAH, PAH + DIZE and PAH + DX-600 groups
(magnification, x400). In addition, percentage of muscularization
and assessment of medial thickness in all groups were conducted.
*P<0.05, **P<0.01,
***P<0.001. RVSP, right ventricular systolic
pressure; RVHI, right ventricular hypertrophy index; N, normoxic;
DIZE, diminazene aceturate; PAH, pulmonary artery hypertension. |
Compared with the PAH + DIZE group, the rats in the
PAH and PAH + DX-600 groups had significantly reduced weight
(Fig. 2B). DIZE caused no
significant effect on the RVSP of rats in the normoxia groups (data
not shown), but a reduction in RVSP was noted in the PAH + DIZE
group compared with the PAH group (Fig.
2A). HE staining and micrograph examination showed that the
thickening of pulmonary arterioles was less severe in the PAH +
DIZE group compared with in the PAH group, whilst the thickening in
the PAH + DX-600 group was more severe (Fig. 2D). These results showed the
muscularization of the distal pulmonary artery in the PAH + DIZE
group was less than that in the PAH group and the PAH + DX-600
group (Fig. 2D). The media fraction
thickness of the distal pulmonary artery was also compared in each
group, which found that the proportion of the media fraction in the
PAH group and the PAH+DX-600 group was significantly higher than
that in the PAH + DIZE group (Fig.
2D). These findings suggested that PAH modeling was successful.
Additionally, DIZE caused no significant difference in weight,
RVSP, RVHI or pathology between normoxic groups. However, DIZE
treatment in rats with PAH led to increased weight, decreased RVSP
and RVHI, and thinner pulmonary arterioles compared with PAH
alone.
DIZE reduces FAK expression in the PAH
rat model
Blood samples were collected, and hemorrhagic serum
was separated to observe the effect of DIZE on FAK expression in
rats using ELISA (Fig. 3). No
significant difference was found in FAK expression between the N
and DIZE groups; both groups exhibited reduced levels compared with
the DX-600 group. FAK levels in the PAH group were significantly
higher compared with that in the N group and the DIZE group.
Furthermore, FAK expression was significantly lower in the PAH +
DIZE group compared with those in the PAH and PAH + DX-600 groups
(Fig. 3). These findings suggested
that ACE2 could reduce FAK expression in the PAH group, but the
effect was not as marked in the normoxic group.
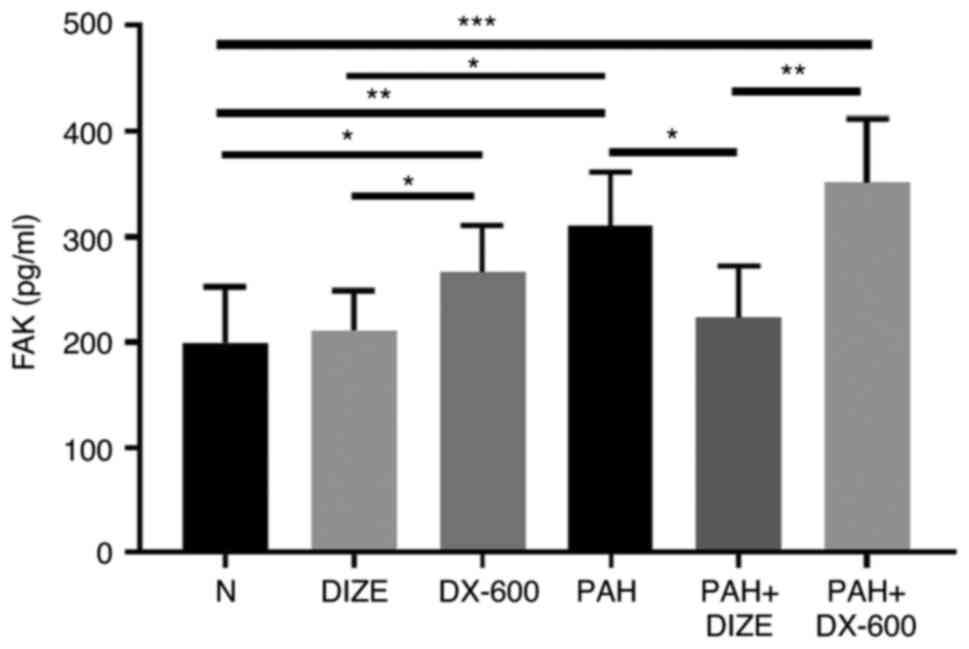 | Figure 3Effect of DIZE on FAK expression in
normal rats and a hypoxia-induced PAH rat model. Rats were randomly
divided into the N, DIZE, DX-600, PAH, PAH + DIZE and PAH + DX-600
groups (n=6/group). The DIZE, PAH + DIZE, DX-600 and PAH + DX-600
groups were injected with DIZE or DX-600 for the first 3 weeks of
hypoxia. *P<0.05, **P<0.01,
***P<0.001. FAK, focal adhesion kinase; N, normoxic;
DIZE, diminazene aceturate; PAH, pulmonary artery hypertension. |
DIZE inhibits the expression of FAK
and its downstream proteins in the PAH rat model
The mRNA expression levels of ACE2 and caspase-3
decreased, and the mRNA expression levels of FAK and survivin
increased, in the PAH group compared with the N group (Fig. 4A-D). These results indicated that
PAH was accompanied by inhibition of ACE2 and activation of FAK.
Correspondingly, the addition of DIZE increased the mRNA expression
levels of ACE2, and decreased the mRNA expression levels of FAK and
survivin, in PAH animals compared with the PAH only group. However,
significant differences in the mRNA expression of caspase-3 between
the PAH group and the PAH + DIZE group were not observed, nor
between the PAH group and the PAH + DX-600 group (Fig. 4A-D).
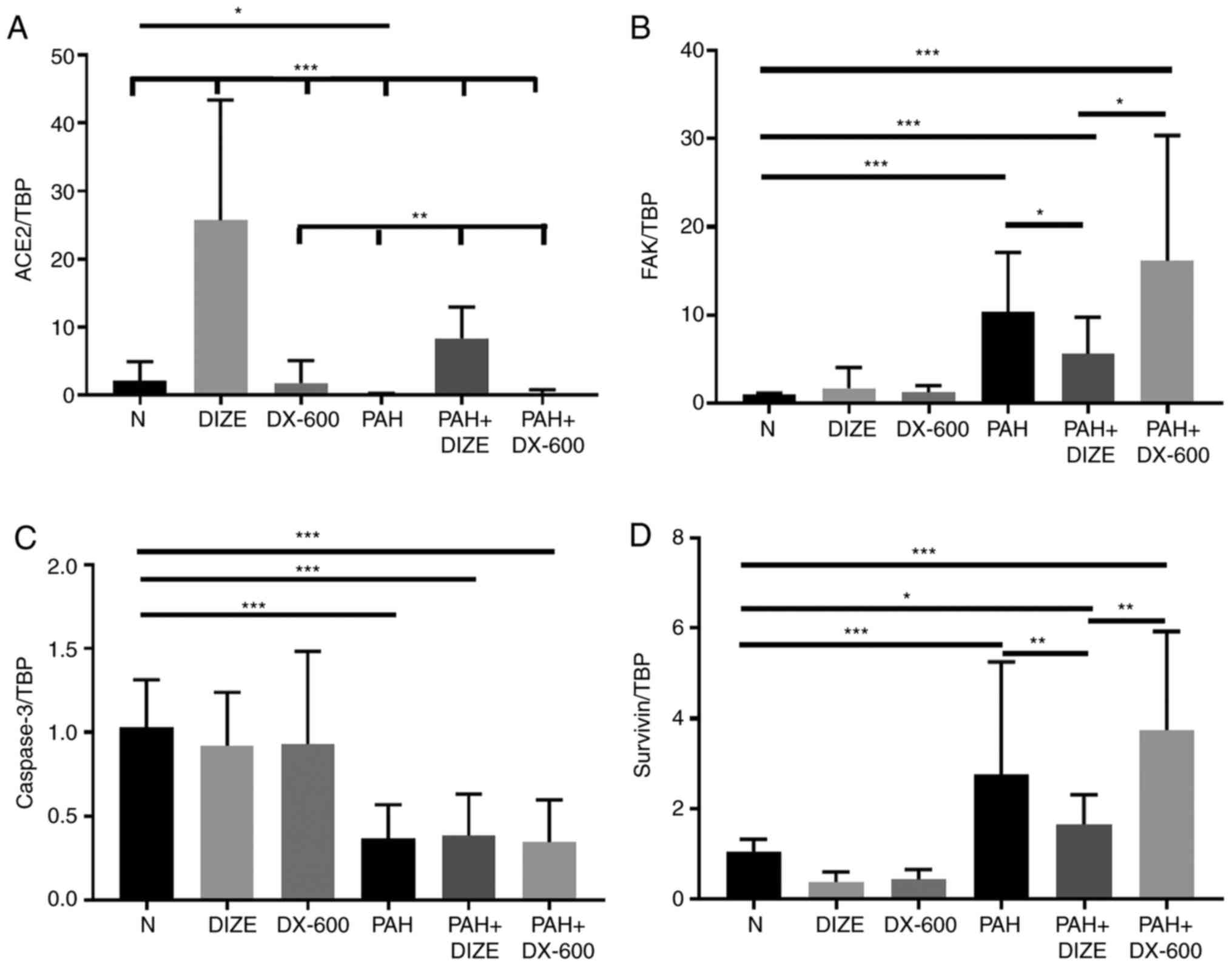 | Figure 4Effect of DIZE on mRNA expression in
the rat PAH model. Rats were randomly divided into N, DIZE, DX-600,
PAH, PAH + DIZE and PAH + DX-600 groups (n=6/group). The DIZE, PAH
+ DIZE, DX-600 and PAH + DX-600 groups were injected with DIZE or
DX-600 for the first 3 weeks of hypoxia. mRNA expression of (A)
ACE2, (B) FAK, (C) caspase-3 and (D) survivin.
*P<0.05, **P<0.01,
***P<0.001. TBP, TATA-binding protein; ACE2,
angiotensin-converting enzyme 2; FAK, focal adhesion kinase; N,
normoxic; DIZE, diminazene aceturate; PAH, pulmonary artery
hypertension. |
The protein expression levels of the aforementioned
factors were observed using western blot analysis to verify whether
they were affected by the addition of DIZE. The results showed that
the trend was similar to that of mRNA results. Compared with the
PAH group, the expression of ACE2 in the N group and the DIZE group
was significantly higher. However, there was no significant
difference in ACE2 expression between the DX-600 group and the PAH
group. Compared with the PAH + DIZE group, the expression of ACE2
in the PAH group and the PAH + DX-600 group was significantly
decreased (Fig. 5A). The p-FAK/FAK
ratio in the DX-600 group was significantly increased compared with
the N group and the DIZE group, whilst this ratio in the PAH + DIZE
group was significantly reduced compared with the PAH group and the
PAH + DX-600 group (Fig. 5B).
Compared with the N group, the cleaved caspase-3/pro-caspase-3
ratio of the three PAH groups increased, whilst the ratio in the
PAH + DIZE group was significantly increased compared with the
other two PAH groups (Fig. 5C). The
expression of survivin in PAH group was significantly higher than
that in N group, DIZE group, DX-600 group and PAH + DIZE group, but
there was no significant difference between the PAH group and
PAH+DX-600 group. The expression of survivin in the PAH + DIZE
group was significantly lower than that in the PAH group and the
PAH + DX-600 group (Fig. 5D).
Therefore, it was concluded that DIZE promoted the expression of
ACE2 and cleaved caspase-3/pro-caspase-3, whilst inhibiting the
levels of p-FAK/FAK and survivin in rats with PAH (Fig. 5A-D).
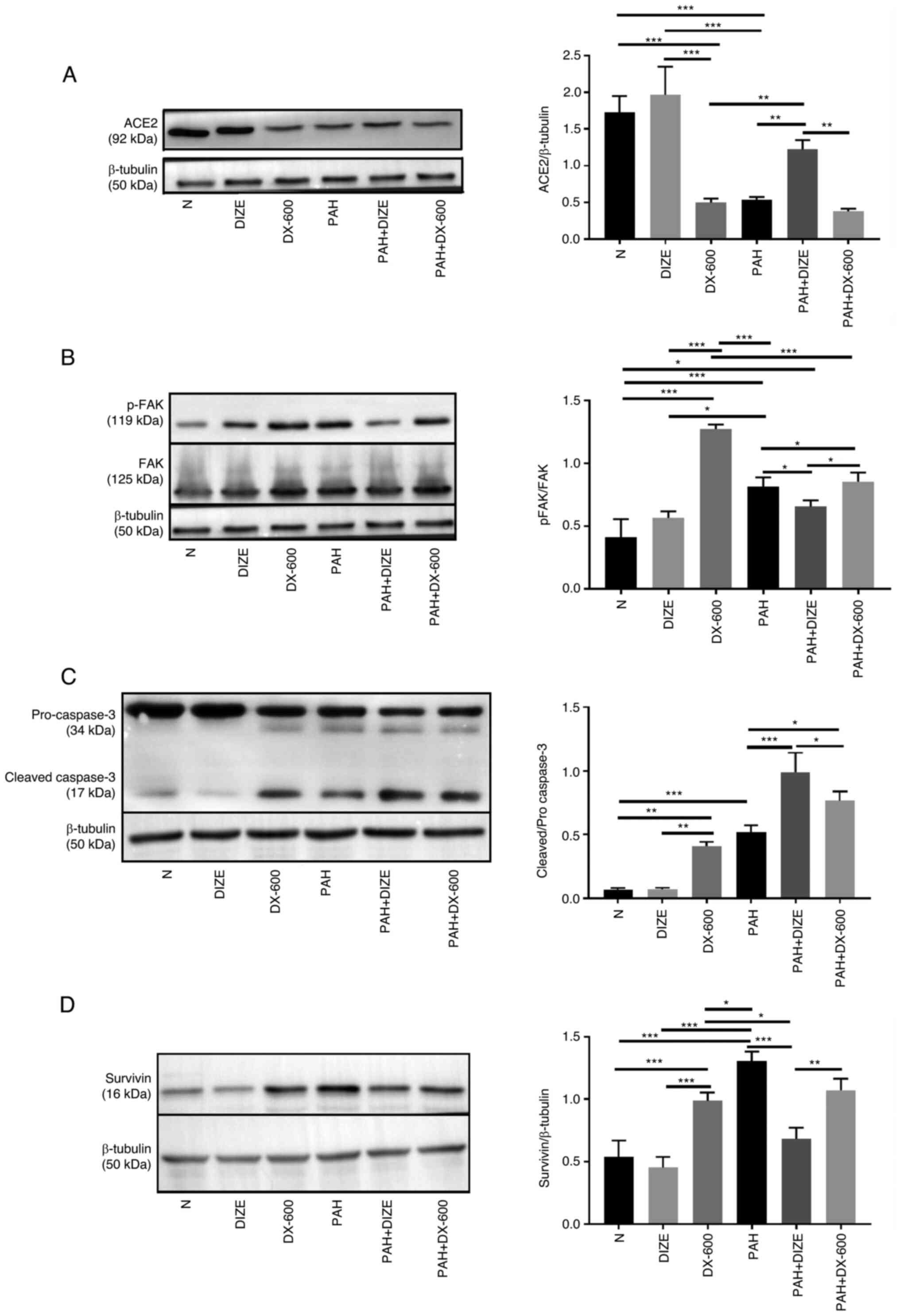 | Figure 5Effect of DIZE on protein expression
in the rat PAH model. Rats were randomly divided into N, DIZE,
DX-600, PAH, PAH + DIZE and PAH + DX-600 groups (n=6/group). The
DIZE, PAH + DIZE, DX-600 and PAH + DX-600 groups were injected with
DIZE or DX-600 for the first 3 weeks of hypoxia. Representative
western blots and quantification of the expression of (A) ACE2, (B)
p-FAK/FAK (C), cleaved caspase-3/pro-caspase-3 and (D) survivin in
the rat model. *P<0.05, **P<0.01,
***P<0.001. ACE2, angiotensin-converting enzyme 2;
FAK, focal adhesion kinase; N, normoxic; DIZE, diminazene
aceturate; PAH, pulmonary artery hypertension; p,
phosphorylated. |
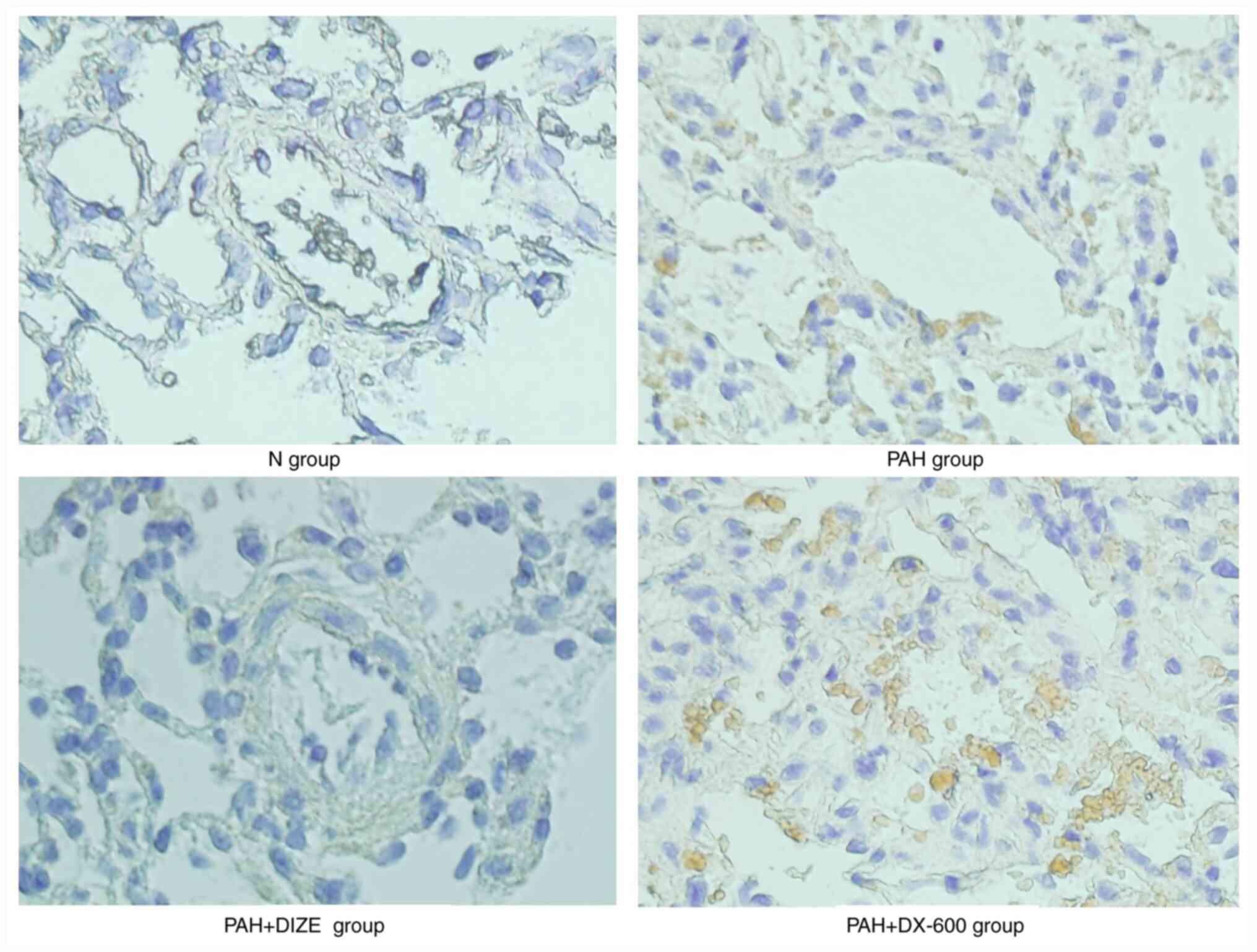
IHC staining was performed to further observe FAK
expression around the pulmonary arterioles. Analysis of the three
groups that underwent normoxia were not significantly different
under the microscope (data not shown). FAK expression was notably
increased in the PAH group compared with the N group. Additionally,
FAK expression was markedly lower in the PAH + DIZE group compared
with in the PAH group, while its expression level was markedly
higher in the PAH + DX-600 group compared with in the
aforementioned three groups (Fig.
6).
These findings suggested that PAH was accompanied by
inhibition of ACE2 and activation of FAK. However, ACE2 activation
in PAH inhibited FAK expression.
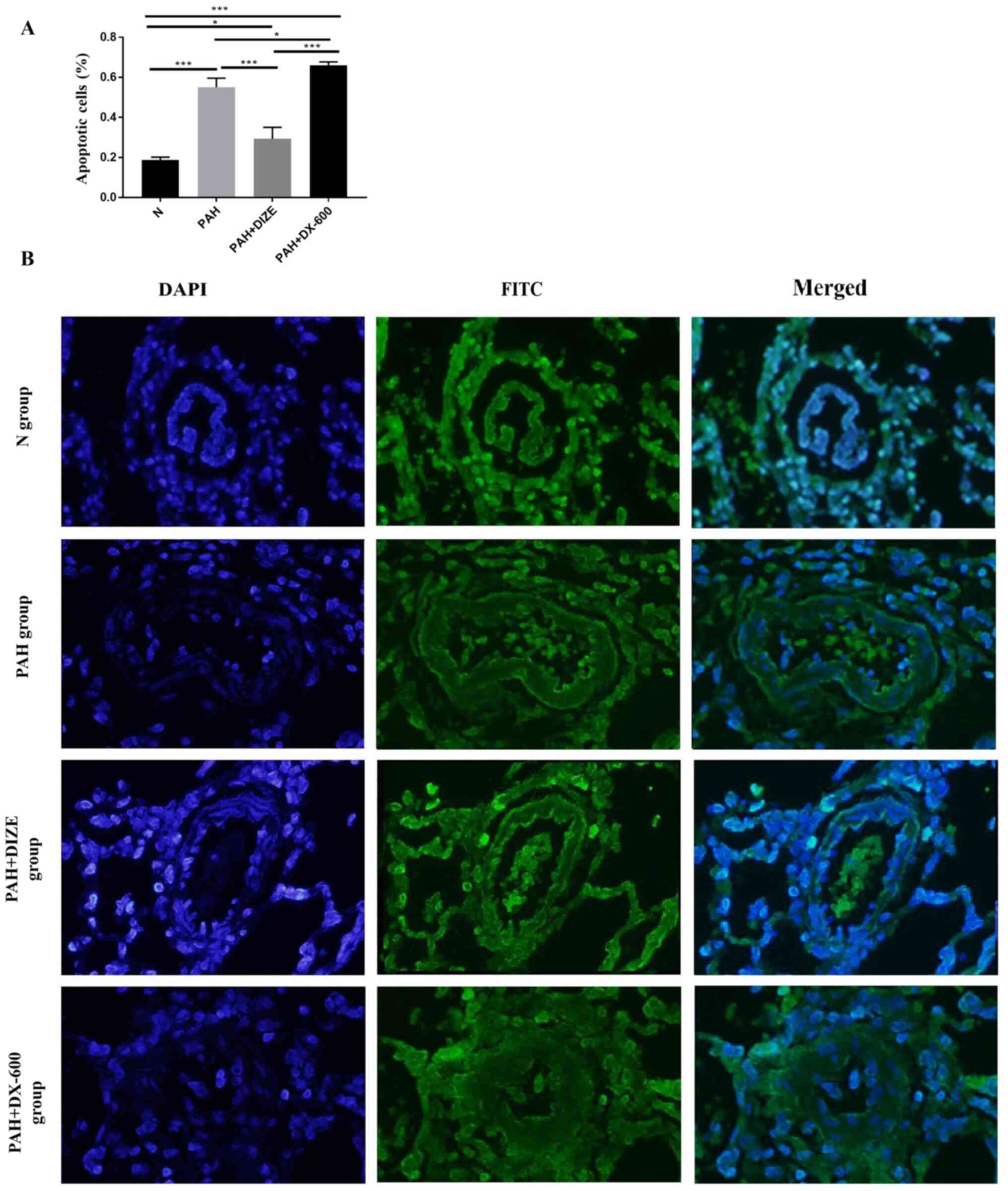
DIZE increases apoptosis in PAH
rats
TUNEL staining was performed to assess the effect of
DIZE on rats with PAH. The apoptosis level appeared reduced in the
PAH group compared with the N group. Conversely, the apoptosis
level was higher in the PAH + DIZE group compared with the PAH
group and the PAH + DX-600 group (Fig.
7A and B). These findings
suggested that DIZE promoted apoptosis around the pulmonary
arterioles.
Discussion
In the present study, it was identified that ACE2
was downregulated during PAH development in a PAH rat model of MCT
treatment combined with continuous hypoxia, and that ACE2
upregulation helped promote apoptosis and led to relief from PAH.
In addition, ACE2 activation inhibited p-FAK/FAK and survivin
expression in a PAH rat model, and promoted cleaved
caspase-3/pro-caspase-3 expression, thus promoting apoptosis around
the pulmonary arterioles and reducing the circumference of the
arterioles. However, compared with the PAH + DIZE group, the
expression of p-FAK/FAK and survivin in the PAH + DX-600 group was
increased, whilst the expression of cleaved caspase-3/pro-caspase-3
was decreased, which was also accompanied by the thickening of
pulmonary arterioles and the decrease of cell apoptosis around
pulmonary arterioles. Taken together, these findings suggested that
ACE2 promoted apoptosis in rats with PAH by inhibiting the
FAK/survivin pathway and promoting caspase-3 expression.
The present study showed that rats with PAH showed a
decrease in ACE2 expression compared with controls, which was
consistent with previous reports (34,41).
Research into ACE2 has examined its role in a number of conditions,
including the treatment of myocardial infarction, myocardial
fibrosis (42), heart failure
(43) and liver fibrosis (44). ACE2 expression is also reportedly
reduced under hypoxic conditions (45). ACE2 activation has been shown to
attenuate the devastating effects of Ang II in the cardiovascular
system by affecting macrophage function (46) and increasing Ang (1-7)
generation (47). ACE2, a novel ACE
homolog, binds to the Mas receptor and metabolizes ang I into
inactive Ang (1-9),
as well as Ang II into Ang (1-7).
Ang (1-7)
can also be generated directly from ang (1-9)
via cleavage by ACE (42). It has
been reported that Ang (1-7)
activation promotes nitric oxide release, increases superoxide
dismutase-2 expression, reduces the production of inflammatory
factors and reduces plasma oxidative stress in right heart
hypertrophy and cardiac fibrosis (48,49).
Perivascular pulmonary arterial and systemic
inflammation is reported to contribute to the initiation and
maintenance of pulmonary vascular hypertension, while oxidative
stress and inflammation, in an interactive manner, play a major
role in the development of PVR (50). This is consistent with the
experimental results of the present study, which showed that ACE2
activation improved PAH and reduced right ventricular hypertrophy.
PAH, whether idiopathic or related to underlying diseases, such as
human immunodeficiency virus infection, results from complex vessel
remodeling involving both PASMC proliferation and inflammation
(51). Early application of ACE2
can also reduce PVR and promote apoptosis, but the specific
mechanism is unknown (41). The
proliferation of PASMCs is the pathological basis of PVR, which, in
turn, is a major factor in the continuous progression and
irreversibility of PAH (52). To
investigate the association between ACE2 and PASMC apoptosis, the
expression of apoptosis-related proteins was further examined in
the present study. The results suggested that ACE2 may promote
apoptosis via caspase-3 activation and FAK inhibition. Previous
studies demonstrated that ACE2 improved acute lung injury (ALI) by
inhibiting apoptosis (53,54). In ALI, neutrophils release
proinflammatory and pro-apoptotic cytokines to harm neighboring
cells and weaken the alveolar-capillary barrier; ACE2 reduces the
severity of ALI by hydrolyzing Ang II and the ALI-induced apoptosis
of pulmonary endothelial cells (54). However, in proliferative lung
diseases such as PAH, ACE2 can promote apoptosis by hydrolyzing Ang
II (55). It is currently believed
that the inhibition of ACE2 activity causes the imbalance between
Ang II and Ang (1-7).
As a bridge between Ang II and Ang (1-7),
inhibited ACE2 will cause an increase in Ang II and a decrease in
Ang (1-7)
(56). Although research on Ang II
is still in its infancy, increased Ang II levels has been shown to
be an important risk factor in breast cancer, COVID-19 and PAH
(56,57). The therapeutic effect of ACE2 varies
with the diseases caused by Ang II. Therefore, it is hypothesized
that Ang II-mediated pulmonary vascular inflammation promotes the
proliferation of PASMCs in PAH and that ACE2 inhibits it. In
addition, ACE2 activation has been indicated to increase caspase-3
expression through the ACE2-Ang (1-7)-Mas
axis in cultured human umbilical vein endothelial cells (58), in line with the experimental results
of the present study.
To understand how ACE2 activates caspase-3 in PAH,
the present study focused on signaling pathways associated with
tumor progression, since there are similarities with PAH in terms
of aberrant cell proliferation (59). As Ang II has been widely shown to
promote FAK activation during the development of liver fibrosis and
kidney disease, it is hypothesized that this association may also
exist in the development of PAH and the hydrolysis of Ang II by
ACE2 may inhibit the activation of FAK (21,23,24).
This hypothesis is consistent with the experimental results of the
present study, which indicated that ACE2 inhibited FAK expression
and phosphorylation and expression of its downstream factors. The
results of the present study also indicated that FAK inhibition
increased caspase-3 expression. Pro-caspase-3 is predominant in
normal cells, and caspase-3 can be cleaved after activation of
apoptosis signals to promote cell apoptosis (60). This is consistent with the
experimental results in the PAH model groups, where there was an
increase in the ratio of cleaved caspase-3/pro-caspase-3, with the
highest level seen in the PAH + DIZE group. Increased levels of FAK
expression are common in patients with liver cancer and cirrhosis
and may not be improved after liver transplantation, increasing
patient mortality (61). A study
reported that after FAK inhibition, caspase-3 expression increased
and the p38 pathway was mediated to inhibit compensatory cell
proliferation in liver cancer (62). Although the sequence of changes in
the expression of FAK and caspase-3 cannot be clarified, the
decrease in FAK expression and the increase in caspase-3 expression
were simultaneously observed, which was previusly found in
myocardial ischemia (63),
triple-negative breast cancer (64)
and human non-small cell lung cancer (65).
In conclusion, ACE2 activation may reduce the
progression of PAH through the inhibition of FAK expression.
Further experiments will be conducted to explore this pathway in
cultured PASMCs, to confirm that ACE2 inhibits FAK and reduces PAH
by inhibiting PASMC proliferation. Further mechanistic studies may
be of great benefit for the development of clinical guidance and
improved patient outcomes.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by grants from the Wuxi
Medical Innovation Team (grant no. CXTD001) and the Major Natural
Science Research Project of Jiangsu Higher Education Institutions
(grant no. 19KJA560004).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RW, ZW, JX and SG contributed to the study design,
data acquisition, analysis, statistical analysis and manuscript
preparation. RW and JW contributed to analysis and interpretation
of data, and revised the manuscript critically for important
intellectual content. All authors read and approved the final
manuscript. RW, ZW, JX and SG confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Wuxi People's Hospital Affiliated
to Nanjing Medical University (Wuxi, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang L, Zhu X, Zhao LP, Wang M, Liu X,
Chen Y, Chen J and Xu W: Effect of beraprost on pulmonary
hypertension due to left ventricular systolic dysfunction. Medicine
(Baltimore). 98(e14965)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mammoto T, Muyleart M, Konduri GG and
Mammoto A: Twist1 in hypoxia-induced pulmonary hypertension through
transforming growth factor-β-smad signaling. Am J Respir Cell Mol
Biol. 58:194–207. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Agrawal V, Byrd BF III and Brittain EL:
Echocardiographic evaluation of diastolic function in the setting
of pulmonary hypertension. Pulmonary Circulation.
9(2045894019826043)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bartelds B, van Loon RLE, Mohaupt S,
Wijnberg H, Dickinson MG, Boersma B, Takens J, van Albada M and
Berger RMF: Mast cell inhibition improves pulmonary vascular
remodeling in pulmonary hypertension. Chest. 141:651–660.
2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sayed BA, Christy A, Quirion MR and Brown
MA: The master switch: The role of mast cells in autoimmunity and
tolerance. Annu Rev Immunol. 26:705–739. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tkaczyk C, Frandji P, Botros HG, Poncet P,
Lapeyre J, Peronet R, David B and Mécheri S: Mouse bone
marrow-derived mast cells and mast cell lines constitutively
produce B cell growth and differentiation activities. J Immunol.
157:1720–1728. 1996.PubMed/NCBI
|
|
7
|
Breitling S, Hui Z, Zabini D, Hu Y,
Hoffmann J, Goldenberg NM, Tabuchi A, Buelow R, Dos Santos C and
Kuebler WM: The mast cell-B cell axis in lung vascular remodeling
and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol.
312:L710–L721. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mao M, Yu X, Ge X, Gu R, Li Q, Song S,
Zheng X, Shen T, Li X, Fu Y, et al: Acetylated cyclophilin A is a
major mediator in hypoxia-induced autophagy and pulmonary vascular
angiogenesis. J Hypertens. 35:798–809. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Rui C, Jiang M, Bo L, Zhong W, Wang Z,
Yuan W and Yan J: The role of autophagy in pulmonary hypertension:
A double-edge sword. Apoptosis. 23:459–469. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Overgaard J: Hypoxic radiosensitization:
Adored and ignored. J Clin Oncol. 25:4066–4074. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Barnes H, Brown Z, Burns A and Williams T:
Phosphodiesterase 5 inhibitors for pulmonary hypertension. Cochrane
Database Syst Rev. 31(CD012621)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
West CM, Wearing OH, Rhem RG and Scott GR:
Pulmonary hypertension is attenuated and ventilation-perfusion
matching is maintained during chronic hypoxia in deer mice native
to high altitude. Am J Physiol Regul Integr Comp Physiol.
320:R800–R811. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Pullamsetti SS, Kojonazarov B, Storn S,
Gall H, Salazar Y, Wolf J, Weigert A, El-Nikhely N, Ghofrani HA,
Krombach GA, et al: Lung cancer-associated pulmonary hypertension:
Role of microenvironmental inflammation based on tumor cell-immune
cell cross-talk. Sci Transl Med. 9(eaai9048)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li XN, Xu JJ, Wu JB, Ji L, Yuan CH and
Wang ZP: Curcumin exerts protective effect on PC12 cells against
lidocaine-induced cytotoxicity by suppressing the formation of
NLRP3 inflammasome. Eur Rev Med Pharmacol Sci. 24:7092–7100.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhao XK, Yu L, Cheng ML, Che P, Lu YY,
Zhang Q, Mu M, Li H, Zhu LL, Zhu JJ, et al: Focal adhesion kinase
regulates hepatic stellate cell activation and liver fibrosis. Sci
Rep. 7:4032–4044. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Paulin R, Meloche J, Courboulin A, Lambert
C, Haromy A, Courchesne A, Bonnet P, Provencher S, Michelakis ED
and Bonnet S: Targeting cell motility in pulmonary arterial
hypertension. Eur Respir J. 43:531–544. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lin C, Li X, Luo Q, Yang H, Li L, Zhou Q,
Li Y, Tang H and Wu L: RELM-β promotes human pulmonary artery
smooth muscle cell proliferation via FAK-stimulated surviving. Exp
Cell Res. 351:43–50. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ferreira-Pinto MJ, Silva AF,
Nogueira-Ferreira R, Padrao AI, Moreira-Goncalves D, Carneiro F,
Costa R, Ribeiro L, Leite-Moreira AF and Henriques-Coelho T:
Survivin role in pulmonary arterial hypertension. Eur Heart J. 34
(Suppl 1)(P302)2013.
|
|
19
|
Fan Z, Liu B, Zhang S, Liu H, Li Y, Wang
D, Liu Y, Li J, Wang N, Liu Y and Zhang B: YM155, a selective
survivin inhibitor, reverses chronic hypoxic pulmonary hypertension
in rats via upregulating voltage-gated potassium channels. Clin Exp
Hypertens. 37:381–387. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mitra SK and Schlaepfer DD:
Integrin-regulated FAK-Src signaling in normal and cancer cells.
Curr Opin Cell Biol. 18:516–523. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Clarke NE, Fisher MJ, Porter KE, Lambert
DW and Turner AJ: Angiotensin converting enzyme (ACE) and ACE2 bind
integrins and ACE2 regulates integrin signalling. PLoS One.
7(e34747)2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen Y, Cao J, Zhao Q, Luo H, Wang Y and
Dai W: Silencing MR-1 attenuates atherosclerosis in
ApoE-/- mice induced by angiotensin II through
FAK-Akt-mTOR-NF-kappaB signaling pathway. Korean J Physiol
Pharmacol. 22:127–134. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Xu XP, He HL, Hu SL, Han JB, Huang LL, Xu
JY, Xie JF, Liu AR, Yang Y and Qiu HB: Ang II-AT2R increases
mesenchymal stem cell migration by signaling through the FAK and
RhoA/Cdc42 pathways in vitro. Stem Cell Res Ther.
8(164)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hall G, Wu G and Winn M: 112 Ang II
Induces FAK activation and podocyte migration via a TRPC6-dependent
mechanism. Am J Kidney Dis. 57(B44)2011.
|
|
25
|
Seguin LR, Villarreal RS and Ciuffo GM:
AT2receptors recruit c-Src, SHP-1 and FAK upon
activation by Ang II in PND15 rat hindbrain. Neurochem Int.
60:199–207. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Watanabe F, Miyazaki T, Takeuchi T, Fukaya
M, Nomura T, Noguchi S, Mori H, Sakimura K, Watanabe M and Mishina
M: Effects of FAK ablation on cerebellar foliation, Bergmann glia
positioning and climbing fiber territory on Purkinje cells. Eur J
Neurosci. 27:836–854. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
George AJ, Thomas WG and Hannan RD: The
renin-angiotensin system and cancer: Old dog, new tricks. Nat Rev
Cancer. 10:745–759. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Jiang Y, Zhou Y, Peng G, Liu N, Tian H,
Pan D, Liu L, Yang X, Li C, Li W, et al: Topotecan prevents
hypoxia-induced pulmonary arterial hypertension and inhibits
hypoxia-inducible factor-1α and TRPC channels. Int J Biochem Cell
Biol. 104:161–170. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Imanishi M, Tomita S, Ishizawa K, Kihira
Y, Ueno M, Izawa-Ishizawa Y, Ikeda Y, Yamano N, Tsuchiya K and
Tamaki T: Smooth muscle cell-specific Hif-1α deficiency suppresses
angiotensin II-induced vascular remodelling in mice. Cardiovasc
Res. 102:460–468. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kittana N: Angiotensin-converting enzyme
2-Angiotensin 1-7/1-9 system: Novel promising targets for heart
failure treatment. Fundam Clin Pharmacol. 32:14–25. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhou X, Zhang P, Liang T, Chen Y, Liu D
and Yu H: Relationship between circulating levels of
angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis and
coronary heart disease. Heart Vessels. 35:153–161. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
de Man FS, Tu L, Handoko ML, Rain S,
Ruiter G, François C, Schalij I, Dorfmüller P, Simonneau G, Fadel
E, et al: Dysregulated renin-angiotensin-aldosterone system
contributes to pulmonary arterial hypertension. Am J Respir Crit
Care Med. 186:780–789. 2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the care and use of laboratory animals. 8th
edition. The National Academies Press, Washington, DC, 2011.
|
|
34
|
Rigatto K, Casali KR, Shenoy V, Katovich
MJ and Raizada MK: Diminazene aceturate improves autonomic
modulation in pulmonary hypertension Eur J. Pharmacol. 713:89–93.
2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yanhong Z, Lina M and Shanshan C:
Mechanism of small intestine injury in non-steroidal
anti-inflammatory drugs rats mediated by RAS-p38MAPK signal
pathway. In: Proceedings of the 4th International Conference on
Digestive Diseases of the World Federation of Chinese Medicine
Societies, Zhengzhou, pp308-311, 2013.
|
|
36
|
Fraga-Silva RA, Sorg BS, Wankhede M,
Dedeugd C, Jun JY, Baker MB, Li Y, Castellano RK, Katovich MJ,
Raizada MK and Ferreira AJ: ACE2 activation promotes antithrombotic
activity. Mol Med. 16:210–215. 2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li Y, Wang Y, Li Y, Qian Z, Zhu L and Yang
D: Osthole attenuates pulmonary arterial hypertension in
monocrotaline-treated rats. Mol Med Rep. 16:2823–2829.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kwon MY, Hwang N, Park YJ, Perrella MA and
Chung SW: NOD2 deficiency exacerbates hypoxia-induced pulmonary
hypertension and enhances pulmonary vascular smooth muscle cell
proliferation. Oncotarget. 9:12671–12681. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jones R, Jacobson M and Steudel W:
Alpha-smooth-muscle actin and microvascular precursor smooth-muscle
cells in pulmonary hypertension. Am J Respir Cell Mol Biol.
20:582–594. 1999.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hemnes AR, Rathinasabapathy A, Austin EA,
Brittain EL, Carrier EJ, Chen X, Fessel JP, Fike CD, Fong P,
Fortune N, et al: A potential therapeutic role for
angiotensin-converting enzyme 2 in human pulmonary arterial
hypertension. Eur Respir J. 51(1702638)2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang J, He W, Guo L, Zhang Y, Li H, Han S
and Shen D: The ACE2-Ang (1-7)-Mas receptor axis attenuates cardiac
remodeling and fibrosis in post-myocardial infarction. Mol Med Rep.
16:1973–1981. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Patel VB, Lezutekong JN, Chen X and Oudit
GY: Recombinant human ACE2 and the angiotensin 1-7 axis as
potential new therapies for heart failure. Can J Cardiol.
33:943–946. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Hu Q, Hu Z, Chen Q, Huang Y, Mao Z, Xu F
and Zhou X: BML-111 equilibrated ACE-AngII-AT1R and
ACE2-Ang-(1-7)-Mas axis to protect hepatic fibrosis in rats.
Prostaglandins Other Lipid Mediat. 131:75–82. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shenoy V, Qi Y, Katovich MJ and Raizada
MK: ACE2, a promising therapeutic target for pulmonary
hypertension. Curr Opin Pharmacol. 11:150–155. 2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hammer A, Yang G, Friedrich J, Kovacs A,
Lee DH, Grave K, Jörg S, Alenina N, Grosch J, Winkler J, et al:
Role of the receptor Mas in macrophage-mediated inflammation in
vivo. Proc Natl Acad Sci USA. 113:14109–14114. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yang G, Chu PL, Rump LC, Le TH and
Stegbauer J: ACE2 and the homolog collectrin in the modulation of
nitric oxide and oxidative stress in blood pressure homeostasis and
vascular injury. Antioxid Redox Signal. 26:645–659. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhang ZZ, Cheng YW, Jin HY, Chang Q, Shang
QH, Xu YL, Chen LX, Xu R, Song B and Zhong JC: The sirtuin 6
prevents angiotensin II-mediated myocardial fibrosis and injury by
targeting AMPK-ACE2 signaling. Oncotarget. 8:72302–72314.
2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zhang J, Dong J, Martin M, He M, Gongol B,
Marin TL, Chen L, Shi X, Yin Y, Shang F, et al: AMP-activated
protein kinase phosphorylation of angiotensin-converting enzyme 2
in endothelium mitigates pulmonary hypertension. Am J Respir Crit
Care Med. 198:509–520. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Sun XQ, Abbate A and Bogaard HJ: Role of
cardiac inflammation in right ventricular failure. Cardiovasc Res.
113:1441–1452. 2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Amsellem V, Lipskaia L, Abid S, Poupel L,
Houssaini A, Quarck R, Marcos E, Mouraret N, Parpaleix A, Bobe R,
et al: CCR5 as a treatment target in pulmonary arterial
hypertension. Circulation. 130:880–891. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Bello-Klein A, Mancardi D, Araujo AS,
Schenkel PC, Turck P and de Lima Seolin BG: Role of redox
homeostasis and inflammation in the pathogenesis of pulmonary
arterial hypertension. Curr Med Chem. 25:1340–1351. 2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Fang Y, Gao F, Hao J and Liu Z:
microRNA-1246 mediates lipopolysaccharide-induced pulmonary
endothelial cell apoptosis and acute lung injury by targeting
angiotensin-converting enzyme 2. Am J Transl Res. 9:1287–1296.
2017.PubMed/NCBI
|
|
54
|
Wang L, Li Y, Qin H, Xing D, Su J and Hu
Z: Crosstalk between ACE2 and PLGF regulates vascular permeability
during acute lung injury. Am J Transl Res. 8:1246–1252.
2016.PubMed/NCBI
|
|
55
|
Li G, Liu Y, Zhu Y, Liu A, Xu Y, Li X, Li
Z, Su J and Sun L: ACE2 activation confers endothelial protection
and attenuates neointimal lesions in prevention of severe pulmonary
arterial hypertension in rats. Lung. 191:327–336. 2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Bujak-Gizycka B, Madej J, Bystrowska B,
Toton-Zuranska J, Kus K, Kolton-Wroz M, Jawien J and Olszanecki R:
Angiotensin 1-7 formation in breast tissue is attenuated in breast
cancer-a study on the metabolism of angiotensinogen in breast
cancer cell lines. J Physiol Pharmacol. 70:503–514. 2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Amirfakhryan H and Safari F: Outbreak of
SARS-CoV2: Pathogenesis of infection and cardiovascular
involvement. Hellenic J Cardiol. 62:13–23. 2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Zhang X, Pan Y and Jin HM: Aldosterone
induced endothelial cell apoptosis via modulation of ACE2-Ang
(1-7)-Mas receptor axis. J Shanghai Jiao Univ (Med Sci). 33:6–11.
2013.
|
|
59
|
Gao ML, Chen L, Li YF, Xue XC, Chen L,
Wang LN, Shah W and Kong Y: Synergistic increase of oxidative
stress and tumor markers in PAH-exposed workers. Asian Pac J Cancer
Prev. 15:7105–7112. 2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Putt KS, Chen GW, Pearson JM, Sandhorst
JS, Hoagland MS, Kwon JT, Hwang SK, Jin H, Churchwell MI, Cho MH,
et al: Small-molecule activation of procaspase-3 to caspase-3 as a
personalized anticancer strategy. Nat Chem Biol. 2:543–550.
2006.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Fujii T, Koshikawa K, Nomoto S, Okochi O,
Kaneko T, Inoue S, Yatabe Y, Takeda S and Nakao A: Focal adhesion
kinase is overexpressed in hepatocellular carcinoma and can be
served as an independent prognostic factor. J Hepatol. 41:104–111.
2004.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Shang N, Bank T, Ding X, Breslin P, Li J,
Shi B and Qiu W: Caspase-3 suppresses diethylnitrosamine-induced
hepatocyte death, compensatory proliferation and
hepatocarcinogenesis through inhibiting p38 activation. Cell Death
Dis. 9(558)2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kanazawa H, Imoto K, Okada M and Yamawaki
H: Canstatin inhibits hypoxia-induced apoptosis through activation
of integrin/focal adhesion kinase/Akt signaling pathway in H9c2
cardiomyoblasts. PLoS One. 12(e0173051)2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Oh E, Sung D, Cho Y, Kim JY, Lee N, Kim
YJ, Cho TM and Seo JH: Abstract 5463: Disulfiram suppresses
metastasis via induction of anoikis and calpain activation in
triple-negative breast cancer. Cancer Res. 77 (Suppl
13)(S5463)2017.
|
|
65
|
Cai F, Chen M, Zha D, Zhang P, Zhang X,
Cao N, Wang J, He Y, Fan X, Zhang W, et al: Curcumol potentiates
celecoxib-induced growth inhibition and apoptosis in human
non-small cell lung cancer. Oncotarget. 8:115526–115545.
2017.PubMed/NCBI View Article : Google Scholar
|