Introduction
Tuberous sclerosis complex (TSC) is an autosomal
dominant disorder with multisystemic involvement, usually due to a
pathogenic mutation in the tuberous sclerosis 1 (TSC1) or
TSC2 genes (1). A positive
genetic test is considered sufficient for the diagnosis. However,
10 to 25% of patients do not exhibit the mentioned mutations upon
genetic testing (2). The clinical
diagnostic criteria include eleven major features and six minor
features from different organ systems and a definite diagnosis
requires either two major criteria or one major and at least two
minor criteria (2). Infantile
spasms or focal seizures usually initially occur during the first
two years of life in over 80% of the patients, with good control of
seizures under antiepileptic drugs (AEDs) in more than half of
patients (3). The cerebral
malformations considered among the diagnostic criteria, also
standing at the origin of the epileptic seizures, include
subependymal nodules, subependymal giant cell astrocytoma, and
cortical dysplasia (2,3).
The dermatological features of TSC make up the
largest part of the diagnostic criteria and are present in almost
all patients (2). A simple physical
examination, with close attention paid to skin lesions, can suggest
the diagnosis even soon after birth, making possible the diagnosis
of TSC before the appearance of neurological signs or developmental
delay (4). This allows swift
interventions and treatment. A large trial (EPISTOP) demonstrated
that preventive treatment of infants with TSC with vigabatrin
reduced the risk and severity of epilepsy (5). The effects of this early intervention
have a much larger impact on the chances of these infants to have
normal development and a long-term quality of life. Previous
studies have demonstrated that in children with TSC epilepsy
remission is significantly correlated with better cognitive
outcomes (6-8).
Cerebral cavernous malformations (CCMs) are
capillary-venous malformations with irregular structure found in
the brain and in the spinal cord. They can be asymptomatic or can
cause variable neurological manifestations, most often seizures
(9,10). CCMs usually become evident between
the second and fifth decades of life; half of cases are discovered
incidentally and around 80% of cases are sporadic (9). When multiple CCMs are identified or in
the event of a single CCM in a person with a positive family
history, genetic testing for the pathogenic genes
KRIT1/CCM1, MGC4607/CCM2 and
PDCD10/CCM3 is indicated (10,11).
Apart from the neurological features, further supporting evidence
for the suspicion of familial CCM are vascular ocular and
dermatological lesions, which have been described in different
families (11,12).
Case report
We present the case of a 31-year-old female,
diagnosed with TSC from the age of 18, after a first generalized
seizure. She was admitted to our clinic in 2019 for increased
seizure frequency. Personal history included chronic kidney disease
and right nephrectomy due to renal angiomyolipoma. Treatment on
admission consisted of lamotrigine 250 mg/day and everolimus 7.5
mg/day. Everolimus treatment was started in 2018 with favorable
effects on renal function and a reduction in seizure frequency.
Genetic testing for TSC1 and TSC2 mutations was
negative. The patient provided informed consent for publication and
data usage concerning the case report.
The patient described two types of seizures: i)
seizures with motor automatisms of the right leg continuing with
bilateral leg clonias, without loss of awareness and duration of up
to 1 min; and ii) focal to bilateral tonic-clonic seizures,
observed by the family during the night, and without any other
details available from the patient, with a duration of up to 2-3
min and postictal sleepiness and confusion. On admission, the
patient reported 2-3 focal seizures/day from a previous frequency
of 2-4 seizures/month.
Neurological examination revealed an oriented
patient, with normal speech, no signs of neck stiffness, diplopia
and horizontal nystagmus on right lateral gaze, hemihypoesthesia of
right face and limbs, no motor deficits or ataxia, brisk deep
tendon reflexes and distal legs paresthesia. General examination
showed an overweight patient with no fever, multiple facial and
gingival angiofibroma, right latero-lumbar scar post nephrectomy
with normal cardiovascular and respiratory functions. We also noted
the presence of renal angiomyolipomas, as documented on abdominal
ultrasound.
VideoEEG identified frequent epileptiform discharges
and slowing of the background rhythm predominantly over the frontal
paramedian derivations, with slightly higher amplitudes and spread
in the right hemisphere.
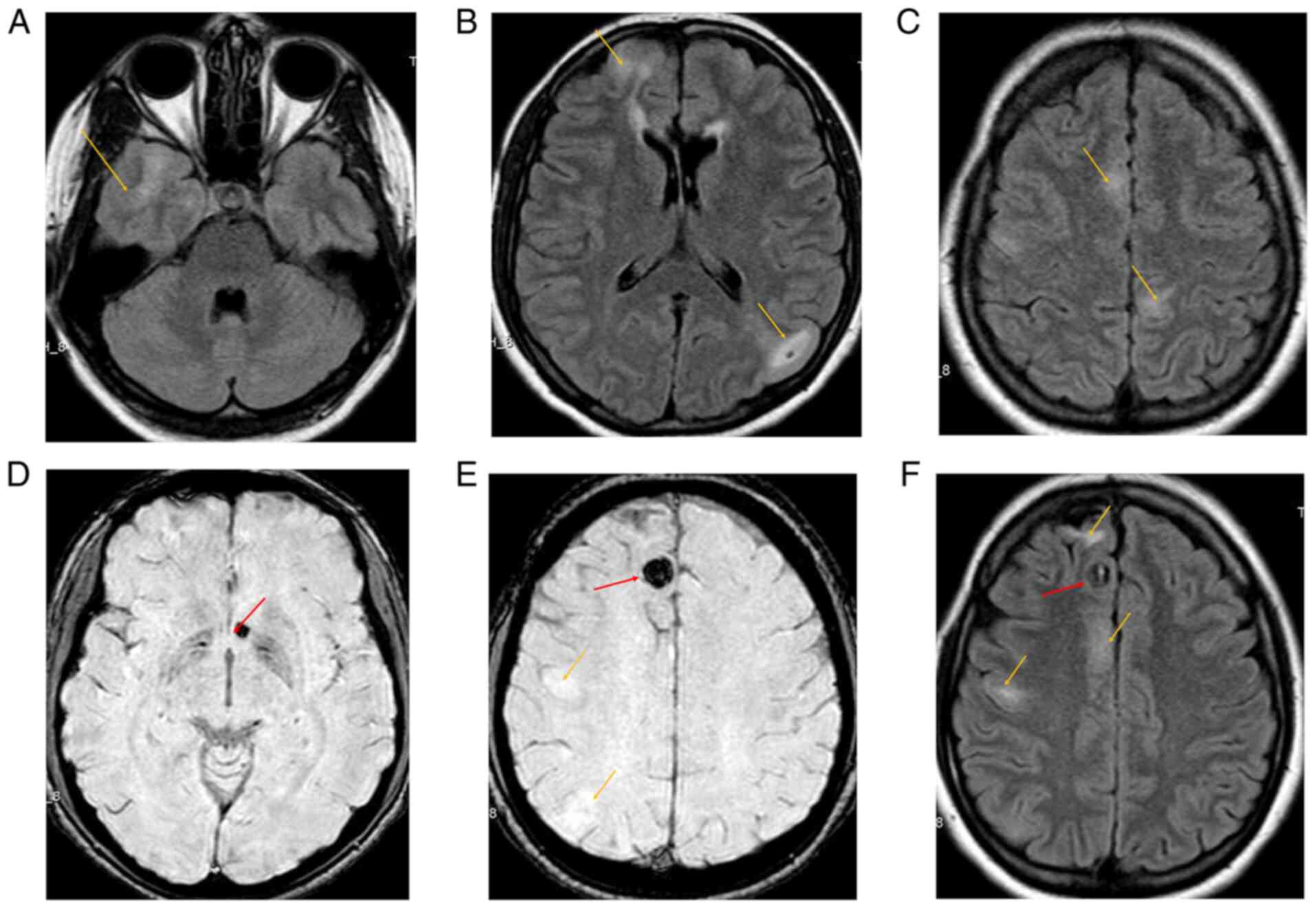
Cerebral magnetic resonance imaging (MRI) and
angioMRI were performed, revealing numerous hyperintense
cortico-subcortical lesions fronto-parietal bilaterally, suggestive
of cortical tubers. Additionally, round-oval shaped hypointense
lesions were visible on susceptibility weighted imaging (SWI)
sequences, located right frontal parasagittal and left
juxtaventricular, which were suggestive of cavernomas with
hemosiderin deposition (Fig.
1).
The patient was administered lamotrigine 400 mg/day
and everolimus 7.5 mg/day and experienced a marked improvement in
seizure frequency (1/week). Genetic testing for familial
cavernomatosis was proposed, but, unfortunately, she refused this
testing due to costs. The patient expressed an intent to have
children in the upcoming years, therefore the lamotrigine dose was
gradually reduced to 200 mg/day over the next 6 months.
Discussion
After performing a thorough literature search and to
the best of our knowledge, we conclude that this is the first
reported case with an association between TSC with negative genetic
testing and multiple CCMs in a patient with epileptic seizures.
Most TSC patients have mutations in the TSC1
or TSC2 genes. In a review by Henske et al, multiple
studies suggest that the majority of patients have a TSC2
mutation. The mutations result in a loss of inhibition of the mTOR
pathway and consequent overexpression of the mTOR complex, with
dysregulation of normal cell functioning (1). However, a significant percentage of
patients with clinically diagnosed TSC do not exhibit these
mutations, as is the case of our patient, and are thought to have
mosaicism or another, yet unidentified, pathomechanism (1,2).
Further studies are needed to complete the list of pathogenic
mutations which result in TSC, as well as to establish the risk of
transmission to the next generation, especially in cases similar to
our patient.
One particularity of our case consisted of the late
diagnosis of TSC, at the age of 18 years, after a generalized
epileptic seizure. After performing a more thorough history, the
patient reported a few episodes that suggested epileptic auras
(13) or short focal seizures
during childhood, which were ignored at that moment and considered
isolated events, not requiring medical attention. Previous studies
have highlighted the difficulties in the recognition of subtle
focal seizures, but commonly, a diagnosis is established within the
first years of life (3,13-15).
The main dermatological criteria for TSC diagnosis
include: hypomelanotic macules, angiofibromas, fibrous cephalic
plaque, shagreen patch, ‘confetti’ skin lesions, intraoral and
ungual fibromas (16). These must
be carefully differentiated from similar skin lesions, which can be
found in other, associated or independent dermatological
conditions, such as vitiligo, lichen sclerosus, lichen planus,
single or multiple cutaneous tumors, and localized scleroderma
(17-19).
At least one type of skin lesions is found in more than 90% of TSC
patients, irrespective of age, the most commonly observed after
birth being hypomelanotic macules (20). Our patient exhibited multiple facial
and intraoral angiofibromas, which usually appear between ages 2
and 5 years, but in rare cases can also appear later in life,
during adulthood (2). The lack of
recognition of these typical lesions for TSC until adulthood in our
patient underlines the importance of education among general
medical practitioners and neurologists on rare inherited
neuro-cutaneous disorders, or at least a dermatology referral when
suspicious lesions are observed.
Another particularity of the case is the presence of
multiple CCMs in addition to the typical cortical tubers of TSC.
CCMs usually occur as single lesions, predominantly in the brain
white matter and the spinal cord. In patients with multiple
lesions, approximately 70% have a familial form (9,11),
when one of the pathogenic variants of the genes KRIT1,
CCM2 or PDCD10 is inherited. There is a recognized
higher risk of bleeding, with the possible emergence of seizures,
when multiple CCMs are present (21,22),
patient age is younger than 45 years and the presence of a
developmental venous anomaly (23).
In cavernoma-related epilepsy, hemosiderin deposits
adjacent to cortical tissue and repeated microhemorrhages are
thought to induce hyperexcitability due to free radicals and lipid
peroxides generated by iron ions (9,24).
Considering the clinical data and investigations, we hypothesize
that the increase in seizure frequency was secondary to a bleeding
in the frontal paramedian cavernoma, which resolved in a short time
and required only AED dose adjustments.
Periodical neurological consults in the long run
were recommended, especially in the event of a pregnancy,
considering a higher risk of recurrent bleeding, as well as a
possible increase in seizure frequency, secondary to hemodynamic
and hormonal changes related to pregnancy (3,6,9,21,22,25).
Familial CCMs have been described to be associated
with a wide array of skin lesions: hyperkeratotic cutaneous
capillary-venous malformations (HCCVM), angiokeratoma, cherry
angiomas, cutaneous bluish nodules, multiple small bluish papules,
capillary vascular anomalies, nodular venous malformations, and
port-wine stains (11,12,26).
The emergence of these lesions varies from birth to late adulthood
(11,12,26).
The exact prevalence of dermatological vascular lesions associated
with familial CCM is unknown, but one study indicated that 6 of 13
members of an affected family had peculiar skin lesions (11). Even though our patient seemingly had
only facial and intraoral angiofibromas, it is possible that some
of the facial lesions were actually angiokeratoma, which is
associated with a few cases of familial CCM (27,28).
Moreover, considering the variable age of the emergence of the
dermatological lesions, periodic dermatological consultations were
recommended to our patient.
In conclusion, this is the first reported
association of tuberous sclerosis with negative genetic testing and
multiple cerebral cavernomas in a patient with epilepsy. The
collaboration between medical specialists, especially in the fields
of neurology and dermatology, is vital in neuro-cutaneous diseases,
as certain features relevant to a correct diagnosis may pass
unnoticed and may result in a delay in treatment of the affection.
On the long term, periodical consultations of both types of
specialists are recommended, considering that the emergence of
specific lesions or subtle neurological signs can also occur later
in life. Multidisciplinary management and whole genome sequencing
of such complex cases could lead to a better understanding of the
intricacies of inherited neuro-cutaneous disorders and improve the
quality of life of these patients.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Any further information regarding this case report
can be obtained from the corresponding author upon reasonable
request.
Authors' contributions
AAA performed the analysis and interpretation of the
and drafted the manuscript. BRT performed the data collection and
assisted in the drafting of the manuscript. IGL played a major role
in the acquisition of the data. ICL, ALT, AOD performed the
revision of the manuscript for content regarding the literature
findings and the patient data. All authors have read and agreed to
the published version of the manuscript.
Ethics approval and consent to
participate
Patient written consent was obtained. No ethics
committee approval was needed as the article uses only readily
available data that the patient agreed to be used (anonymously) for
publication. No experimental intervention was performed, and the
patient was investigated and treated according to the usual
hospital standards.
Patient consent for publication
Written consent was obtained for publication of the
case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Henske EP, Jóźwiak S, Kingswood JC,
Sampson JR and Thiele EA: Tuberous sclerosis complex. Nat Rev Dis
Primers. 2(16035)2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Northrup H and Krueger DA: International
Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis
complex diagnostic criteria update: Recommendations of the 2012
international tuberous sclerosis complex consensus conference.
Pediatr Neurol. 49:243–254. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nabbout R, Belousova E, Benedik MP, Carter
T, Cottin V, Curatolo P, Dahlin M, D´Amato L, D'Augères GB, de
Vries PJ, et al: Epilepsy in tuberous sclerosis complex: Findings
from the TOSCA study. Epilepsia Open. 4:73–84. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Davis PE, Filip-Dhima R, Sideridis G,
Peters JM, Au KS, Northrup H, Bebin EM, Wu JY, Krueger D and Sahin
M: Tuberous Sclerosis Complex Autism Center of Excellence Research
Network. Presentation and diagnosis of tuberous sclerosis complex
in infants. Pediatrics. 140(e20164040)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
De Ridder J, Lavanga M, Verhelle B,
Vervisch J, Lemmens K, Kotulska K, Moavero R, Curatolo P, Weschke
B, Riney K, et al: Prediction of neurodevelopment in infants with
tuberous sclerosis complex using early EEG characteristics. Front
Neurol. 11(582891)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chu-Shore CJ, Major P, Camposano S,
Muzykewicz D and Thiele EA: The natural history of epilepsy in
tuberous sclerosis complex. Epilepsia. 51:1236–1241.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Curatolo P and Moavero R: Can we change
the course of epilepsy in tuberous sclerosis complex? Epilepsia.
51:1330–1331. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Curatolo P, Jóźwiak S and Nabbout R: TSC
Consensus Meeting for SEGA and Epilepsy Management. Management of
epilepsy associated with tuberous sclerosis complex (TSC): Clinical
recommendations. Eur J Paediatr Neurol. 16:582–586. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Rosenow F, Alonso-Vanegas MA, Baumgartner
C, Blümcke I, Carreño M, Gizewski ER, Hamer HM, Knake S, Kahane P,
Lüders H, et al: Cavernoma-related epilepsy: Review and
recommendations for management-report of the surgical task force of
the ILAE commission on therapeutic strategies. Epilepsia.
54:2025–2035. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Morrison L and Akers A: Cerebral cavernous
malformation, familial. In: GeneReviews® [Internet].
Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G and
Amemiya A (eds). University of Washington, Seattle, WA, 1993.
|
|
11
|
de Vos IJ, Vreeburg M, Koek GH and van
Steensel MA: Review of familial cerebral cavernous malformations
and report of seven additional families. Am J Med Genet A.
173:338–351. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Toll A, Parera E, Giménez-Arnau AM, Pou A,
Lloreta J, Limaye N, Vikkula M and Pujol RM: Cutaneous venous
malformations in familial cerebral cavernomatosis caused by KRIT1
gene mutations. Dermatology. 218:307–313. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Russo A, Arbune A, Bansal L, Mindruta I,
Gobbi G and Duchowny M: The localizing value of epileptic auras:
Pitfalls in semiology and involved networks. Epileptic Disord.
21:519–528. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Canevini MP, Kotulska-Jozwiak K, Curatolo
P, La Briola F, Peron A, Słowińska M, Strzelecka J, Vignoli A and
Jóźwiak S: Current concepts on epilepsy management in tuberous
sclerosis complex. Am J Med Genet C Semin Med Genet. 178:299–308.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ebrahimi-Fakhari D, Mann LL, Poryo M, Graf
N, Von Kries R, Heinrich B, Ebrahimi-Fakhari D, Flotats-Bastardas
M, Gortner L, Zemlin M and Meyer S: Incidence of tuberous sclerosis
and age at first diagnosis: New data and emerging trends from a
national, prospective surveillance study. Orphanet J Rare Dis.
13(117)2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Teng JM, Cowen EW, Wataya-Kaneda M,
Gosnell ES, Witman PM, Hebert AA, Mlynarczyk G, Soltani K and
Darling TN: Dermatologic and dental aspects of the 2012
international tuberous sclerosis complex consensus statements. JAMA
Dermatol. 150:1095–1101. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mihăilă B, Dinică RM, Tatu AL and Buzia
OD: New insights in vitiligo treatments using bioactive compounds
from piper nigrum. Exp Ther Med. 17:1039–1044. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Tatu AL and Nwabudike LC: Male genital
lichen sclerosus-a permanent therapeutic challenge. J Am Acad
Dermatol. 79 (Suppl 1)(AB185)2018.
|
|
19
|
Tatu AL and Nwabudike LC: The treatment
options of male genital lichen sclerosus et atrophicus: Treatments
of genital lichen sclerosus. In: Proceedings of the 14th national
congress of urogynecology and the national conference of the
Romanian association for the study of pain, Eforie, pp262-264,
2017.
|
|
20
|
Portocarrero LKL, Quental KN, Samorano LP,
Oliveira ZNP and Rivitti-Machado MCDM: Tuberous sclerosis complex:
Review based on new diagnostic criteria. An Bras Dermatol.
93:323–331. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Josephson CB, Leach JP, Duncan R, Roberts
RC, Counsell CE and Al-Shahi Salman R: Scottish Audit of
Intracranial Vascular Malformations (SAIVMs) steering committee and
collaborators. Seizure risk from cavernous or arteriovenous
malformations: Prospective population-based study. Neurology.
76:1548–1554. 2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Mouchtouris N, Chalouhi N, Chitale A,
Starke RM, Tjoumakaris SI, Rosenwasser RH and Jabbour PM:
Management of cerebral cavernous malformations: From diagnosis to
treatment. ScientificWorldJournal. 2015(808314)2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kashefiolasl S, Bruder M, Brawanski N,
Herrmann E, Seifert V, Tritt S and Konczalla J: A benchmark
approach to hemorrhage risk management of cavernous malformations.
Neurology. 90:e856–e863. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Moran NF, Fish DR, Kitchen N, Shorvon S,
Kendall BE and Stevens JM: Supratentorial cavernous haemangiomas
and epilepsy: A review of the literature and case series. J Neurol
Neurosurg Psychiatry. 66:561–568. 1999.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Arbune AA, Craiu D, Cuciureanu ID, Iulian
C, Pavel LL, Berbece IS, Grigore CA and Dulamea A: Challenges of
valproate treatment during pregnancy: Pros and cons. Rev Chim.
71:456–459. 2020.
|
|
26
|
Campione E, Diluvio L, Terrinoni A, Di
Stefani A, Orlandi A, Chimenti S and Bianchi L: Progressive
late-onset of cutaneous angiomatosis as possible sign of cerebral
cavernous malformations. Dermatol Online J. 19(2)2013.PubMed/NCBI
|
|
27
|
Whitworth WR, Hick RW, Nelson KC and
Sidhu-Malik NK: Cerebral cavernous malformations associated with
cutaneous angiokeratomas and hemangiomas. Cutis. 96:329–332.
2015.PubMed/NCBI
|
|
28
|
Errichetti E, Piccirillo A and Ricciuti F
and Ricciuti F: An unusual case of multiple angiokeratomas arising
on a medium-sized congenital melanocytic nevus. Indian J Dermatol
Venereol Leprol. 78:374–375. 2012.PubMed/NCBI View Article : Google Scholar
|