Introduction
Idiopathic pulmonary fibrosis (IPF) is a persistent
and destructive interstitial lung disease of unknown etiology with
an estimated median survival time of 3-5 years following diagnosis
(1-3).
It is characterized by abnormal proliferation and remodeling of
fibroblasts and myofibroblasts. The aggregation of myofibroblasts
leads to the deposition of extracellular matrix, an abnormal wound
healing process and remodeling of the lung structure (4). IPF may be an epithelial-driven disease
and the damage and repair of alveolar epithelial cells (AECs) are
important incentives for pulmonary fibrosis (5). Epithelial cells may lose their normal
polarity and shape under various stimuli, which leads to
epithelial-mesenchymal transition (EMT) (6).
Whether EMT occurs during pulmonary fibrosis remains
controversial (7,8). Cell tracking technology is useful for
analyzing the differentiation path of AECs in pulmonary fibrosis
and the origin of fibroblasts. Several studies with lineage tracing
supported the existence of EMT during pulmonary fibrosis. Kim et
al (9) used a SPC-rtTA,
tetO7-CMV-Cre induction expression system to induce the
expression of a lacZ reporter gene in the AECs of transgenic mice.
Triple transgenic mice were then treated with intranasal
instillation of a replication-deficient adenovirus (Ad) expressing
constitutively active TGFβ-1, to establish a pulmonary fibrosis
model; 33% of vimentin-positive cells were X-gal positive (9). Other studies applied β-galactosidase
as the detection marker of AECs and the intratracheal instillation
of bleomycin to establish the pulmonary fibrosis model, revealing
the co-localization of β-galactosidase and mesenchymal markers
(10-14).
Rock et al (15) used the
tamoxifen-induced SPC-CreERT2 (new mutations on the
estrogen receptor that enhances tamoxifen binding ability) system,
labeled ATII cells and performed intratracheal instillation of
bleomycin to establish the pulmonary fibrosis model, which failed
to prove the existence of EMT. Chilosi et al (16) and another study (17) indicated that the markers zinc finger
E-box binding homeobox 1 and β-catenin of the EMT signaling pathway
were co-localized with epithelial markers in the fibrotic region of
human pulmonary fibrosis. As aforementioned, the results of lineage
tracing studies are contradictory and inconsistent with the results
of clinicopathological studies (9-17).
Thus, future studies should comprehensively investigate the
mechanisms of bleomycin-induced pulmonary fibrosis, select a type
of modeling similar to the clinical-pathological process, and use
neutral and efficient labeling and gene reporting systems in
lineage tracing studies to determine whether EMT occurs in
pulmonary fibrosis, which is crucial for the development of
therapeutic strategies for IPF.
In the present study, the classic model of pulmonary
fibrosis was induced by intraperitoneal injection of bleomycin.
Adult
SPC-rtTA+/-/tetO7-CMV-Cre+/-/mTmG+/-
mice were induced by doxycycline and the labeled AECs were detected
by cell membrane-localized enhanced green fluorescence protein
(mEGFP). The co-localization of mEGFP and mesenchymal markers was
detected by multiple immunohistochemical staining to analyze
whether EMT occurred in AECs during pulmonary fibrosis.
Materials and methods
Establishment of transgenic animal
system
SPC-rtTA mice [B6.Cg-Tg(SFTPC-rtTA)5Jaw/J] and
tetO7-CMV-Cre mice
[B6.Cg-Tg(tetO7-cre)1Jaw/J] were purchased from the
Jackson Laboratory. mTmG mice
[B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,
-EGFP)Luo/J] were obtained from Nanjing BioMedical
Research Institute of Nanjing University. All mice were of the
C57BL6 strain. First, SPC-rtTA and tetO7-CMV-Cre mice
were mated to obtain those with both SPC-rtTA and
tetO7-CMV-Cre genes, which were then mated
with mTmG mice to obtain transgenic mice with the genotype
SPC-rtTA+/-/tetO7-CMV-Cre+/-/mTmG+/-.
Male transgenic mice (aged 8-12 weeks) were induced by adding 0.5
mg/ml doxycycline to their drinking water for 15 days and the
expression of Cre in the lungs was examined by multiple IHC in a
number of mice. The remaining mice were raised with normal water
and food for >20 days, after which multiple IHC was performed to
detect the expression of mEGFP and SPC in the lung tissue. Animals
that reached the endpoint of the experiment were then sacrificed by
cervical dislocation to observe the lungs by multiple IHC staining.
The animal care and use complied with the Provisions and General
Recommendation of the Chinese Experimental Animals Administration
Legislation (no. GB14925-2010). The animals were housed at a
temperature of 23±2˚C, relative humidity of 55±15%, and a 12-h
light/dark cycle. Access to standard food and sterile drinking
water was ensured. Animal health and behavior were monitored every
day. The study protocol was approved by the Institutional Animal
Care and Use Committee (IACUC) of China Medical University (CMU)
(IACUC issue no. 14031M).
Bleomycin-induced pulmonary fibrosis
in mice
Adult male transgenic mice aged 8-12 weeks with the
genotype
SPC-rtTA+/-/tetO7-CMV-Cre+/-/mTmG+/-
were induced with doxycycline for 15 days. Mice were then provided
normal water and food for one week. Subsequently, mice were
randomly divided into two groups: Experimental group (n=3;
bleomycin-induced pulmonary fibrosis) and control group (n=3). The
body weight of the mice was 20-25 g. The mice in the experimental
group were intraperitoneally injected with bleomycin (Hanhui; 40
USP/kg in 0.2 ml of 0.9% physiological saline solution) on days 0
and 2, followed by 20 USP/kg on days 4 and 6, and 10 USP/kg on days
9, 12, 15, 18, 21, 24 and 27 to avoid death. The mice in the
control group were injected with 0.2 ml of normal saline. No
animals died during the experimental period. The mice in both
groups were sacrificed by cervical dislocation on day 28.
Histological examination
After the lungs were removed, the right lung tissue
was immediately fixed in 4% paraformaldehyde and the tissues were
dehydrated and embedded in paraffin. Subsequently, 5-µm paraffin
sections were prepared and the tissues were stained with H&E
(cat. no. WK297; Biolab) and examined under a microscope (Olympus
Corporation). Masson's trichrome staining (cat. no. G1340; Beijing
Solarbio Science & Technology Co., Ltd.) was also performed to
evaluate fibrosis (collagen fibers) following the manufacturer's
standard protocols. The samples were inspected using a BX-51
microscope (Olympus Corporation). ImagePro Plus v. 6.0 (Media
Cybernetics, Inc.) was used to calculate the integrated optical
density and area value for each image, from which the average
optical density value was then calculated.
IHC staining
The paraffin sections (5 µm) of mouse lungs were
deparaffinized and then subjected to antigen retrieval in citrate
buffer (10 mM sodium citrate, pH 6.0) with heating to 100˚C for 10
min. Subsequently, the Immunohistochemical Staining kit (cat. no.
SP-9001; OriGene Technologies, Inc.) was used to complete the
follow-up experiments. The sections were blocked with normal goat
plasma for 20 min at room temperature and then incubated overnight
at 4˚C with the following primary antibodies: Cre recombinase
antigen (rabbit; dilution, 1:100; cat. no. 15036S; Cell Signaling
Technology, Inc.), GFP antigen (rabbit; dilution, 1:200; cat. no.
2956S; Cell Signaling Technology, Inc.), SPC antigen (rabbit;
dilution, 1:50; cat. no. sc-13979; Santa Cruz Biotechnology, Inc.),
α-smooth muscle actin (α-SMA) antigen (rabbit; dilution, 1:100;
cat. no. ab5694; Abcam) and S100 calcium binding protein A4
(S100A4) antigen (rabbit; dilution, 1:100; cat. no. 13018S; Cell
Signaling Technology, Inc.). The sections were incubated for 20 min
with a streptavidin-biotin-peroxidase complex and incubated with
secondary antibody for 20 min at 37˚C. The AEC Chromogenic Reagent
(Boster Biological Technology) and hematoxylin were used for color
development and counterstaining, respectively. The samples were
examined using a BX-51 microscope (Olympus Corporation) at a
magnification of x100 or x200.
Multiple IHC staining
Following micrograph acquisition after the first IHC
staining, the samples were washed with an alcohol gradient (25, 50,
70, 85, 95, 85, 70, 50 and 25%). The antibodies were then stripped
with a buffer containing 65 mM Tris-HCl pH 6.8 (Sigma-Aldrich;
Merck KGaA), 1% SDS (Beijing Solarbio Science & Technology Co.,
Ltd.), 0.113M 2-mercaptoethanol (Sigma-Aldrich; Merck KGaA), 0.1M
NaCl and 2M urea (Sigma-Aldrich; Merck KGaA) in a 56˚C water bath
with agitation twice for 30 min each time. After antibody
stripping, the sections were washed with distilled water for 30 min
(5 min each time) at room temperature. The sections were then
immunostained again, as described earlier.
Reverse-transcription-quantitative
(RT-q)PCR
Total RNA was isolated from the left lobe of each
mouse lung (RNAiso Plus; cat. no. D9108AT; Takara Bio, Inc.) and
transcribed into cDNA according to the kit (cat. no. RR037A; Prime
Script® RT reagent Kit Perfect Real-Time; Takara Bio,
Inc.). Amplification and quantitation were performed on an ABI7500
Real-time PCR System with Takara SYBR® Premix Ex
Taq™ II (cat. no. RR820A; Takara Bio, Inc.). The
following primers were used: α-SMA forward,
5'-CCAACTGGGACGACATGGAA-3' and reverse, 5'-GAGGCATAGAGGGACAGCAC-3';
collagen forward, 5'-ACATGTTCAGCTTTGTGGACC-3' and reverse,
5'-TAGGCCATTGTGTATGCAGC-3'; GAPDH forward,
5'-GGCATTGTGGAAGGGCTCAT-3' and reverse, 5'-GGCAGCACCAGTGGATGCAG-3'.
Quantification was performed by the 2-∆∆Ct method
(18) with GAPDH for
normalization.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Comparisons between groups were performed by using
unpaired the independent t-test. All analyses were performed using
GraphPad Prism 5 (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Labeled type II AECs and their
descendants may be detected by multiple IHC staining
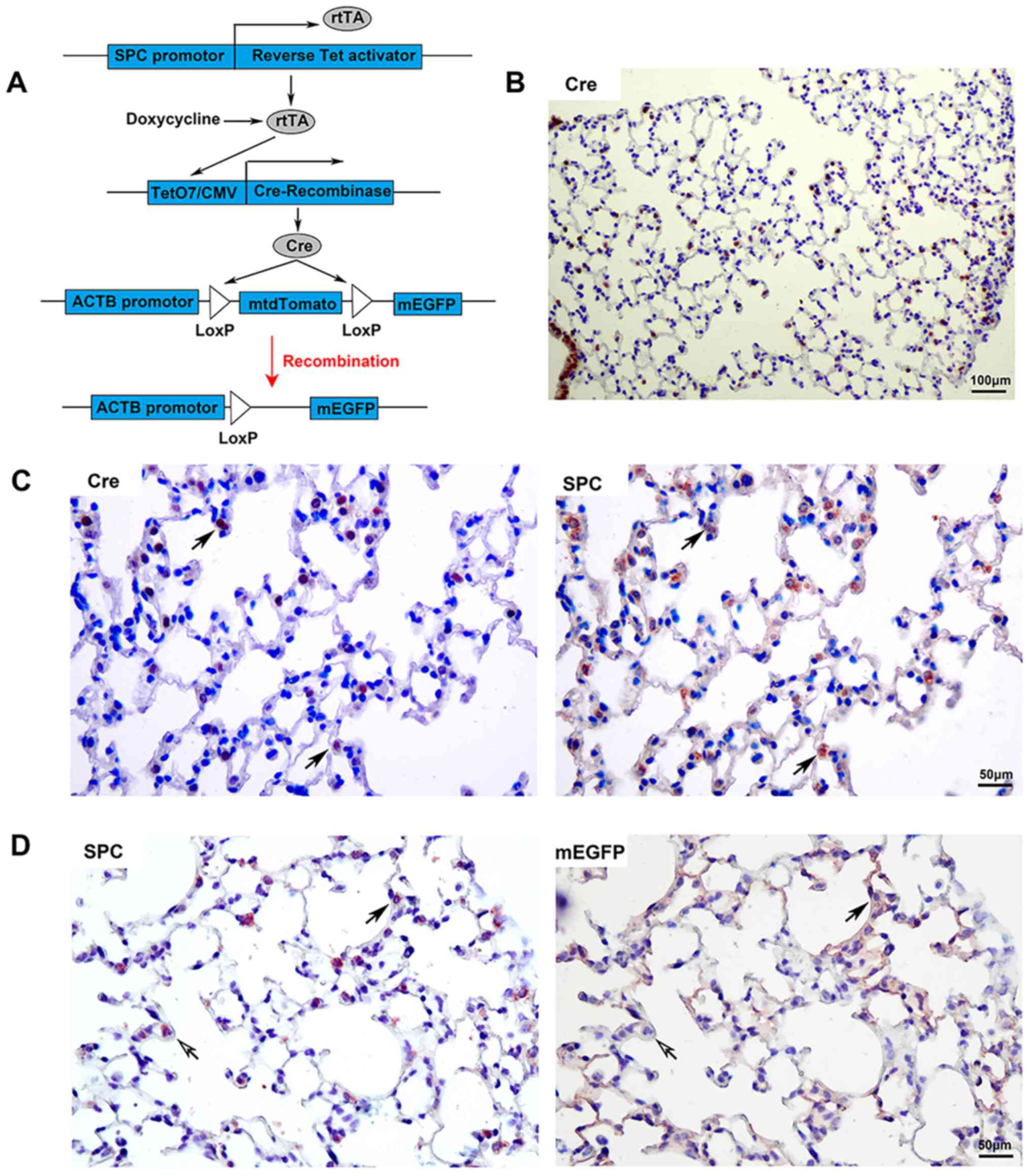
The transgenic mouse system involves three
transgenic components. It expressed rtTA under the control of
recombinant SPC promoter; in the presence of inducer doxycycline,
the transcription factor rtTA was able to activate the expression
of Cre recombinase. The mtdTomato sequence flanked by Loxp sites
was removed by site-specific recombinase Cre. Thus, mEGFP was
constitutively and irreversibly expressed under the control of the
ACTB promotor (Fig. 1A). Adult
transgenic mice with the genotype
SPC-rtTA+/-/tetO7-Cre+/-/mTmG+/-
were induced with doxycycline for 15 days. Cre was determined to be
localized in the airway and peripheral lung tissues of
doxycycline-induced knockout transgenic mice (Fig. 1B). Co-localization of Cre and SPC
was observed in the peripheral lung tissue of transgenic mice
(Fig. 1C); the proportion of
Cre+SPC+/SPC+ was 63.27±7.51%.
Male transgenic mice were induced with doxycycline,
and the multiple IHC staining technique was used to detect the
expression of SPC and EGFP (Fig.
1D). mEGFP was detected in the peripheral lung tissue of
transgenic mice. Certain mEGFP+ cells were type II AECs
(with expression of SPC, which is a type II AEC marker); other
mEGFP+ cells spread over the surface were type I AECs
(without expression of SPC). The expression of mEGFP was caused by
gene recombination when their parent cells were type II AECs. There
were also a small number of SPC+ cells that did not
express mEGFP in lung tissue, which suggests that gene knockout
efficiency was <100%. The right panel of Fig. 1D indicates that the membrane bonded
with EGFP that was detected, while there was no SPC stain in the
cytoplasm, which indicated that the anti-SPC antibody used in the
first round of IHC staining was fully stripped after IHC staining.
Furthermore, it did not interfere with the second round of IHC
staining to detect mEGFP. All of these results demonstrated that
multiple IHC staining may be used for cell lineage tracing.
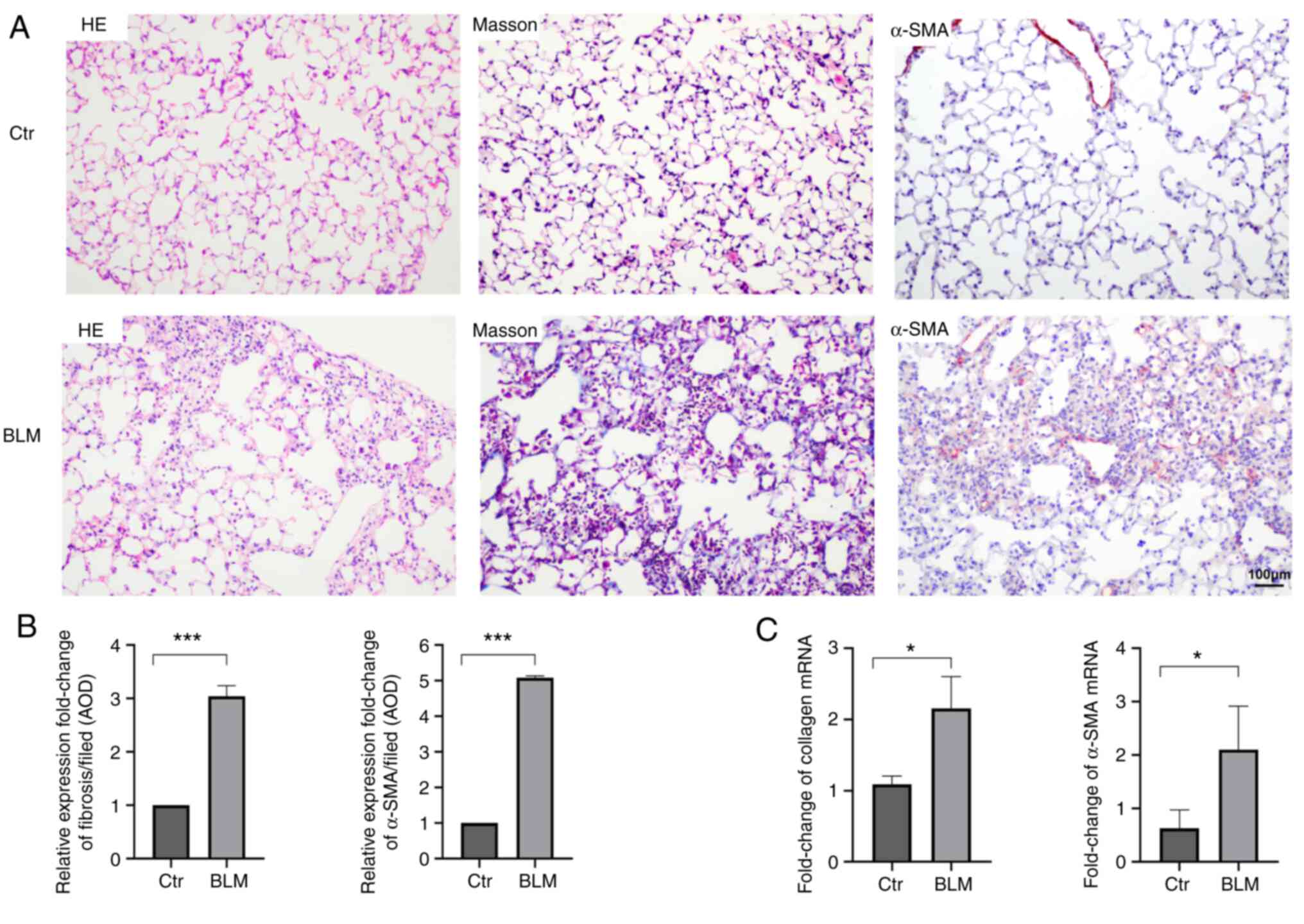
Bleomycin-induced pulmonary fibrosis
in transgenic mice
After induction and modeling, the mice in the
experimental group exhibited decreased appetite, weight loss
(Fig. S1), unkempt and dull fur
and shortness of breath. On day 28, histopathological examination
indicated inflammatory-cell infiltration in the alveolar septum and
destruction of the normal alveolar structure in the experimental
group. Masson staining indicated that the alveolar structure was
destroyed, the normal structure disappeared, and obvious collagen
fiber deposition occurred in the experimental group compared with
the control group. Furthermore, the expression of α-SMA and
collagen mRNA in the lung tissue was significantly higher in the
experimental group than in the control group (P<0.05), which
indicated that myogenic fibroblasts and fibrotic foci were formed
(Fig. 2).
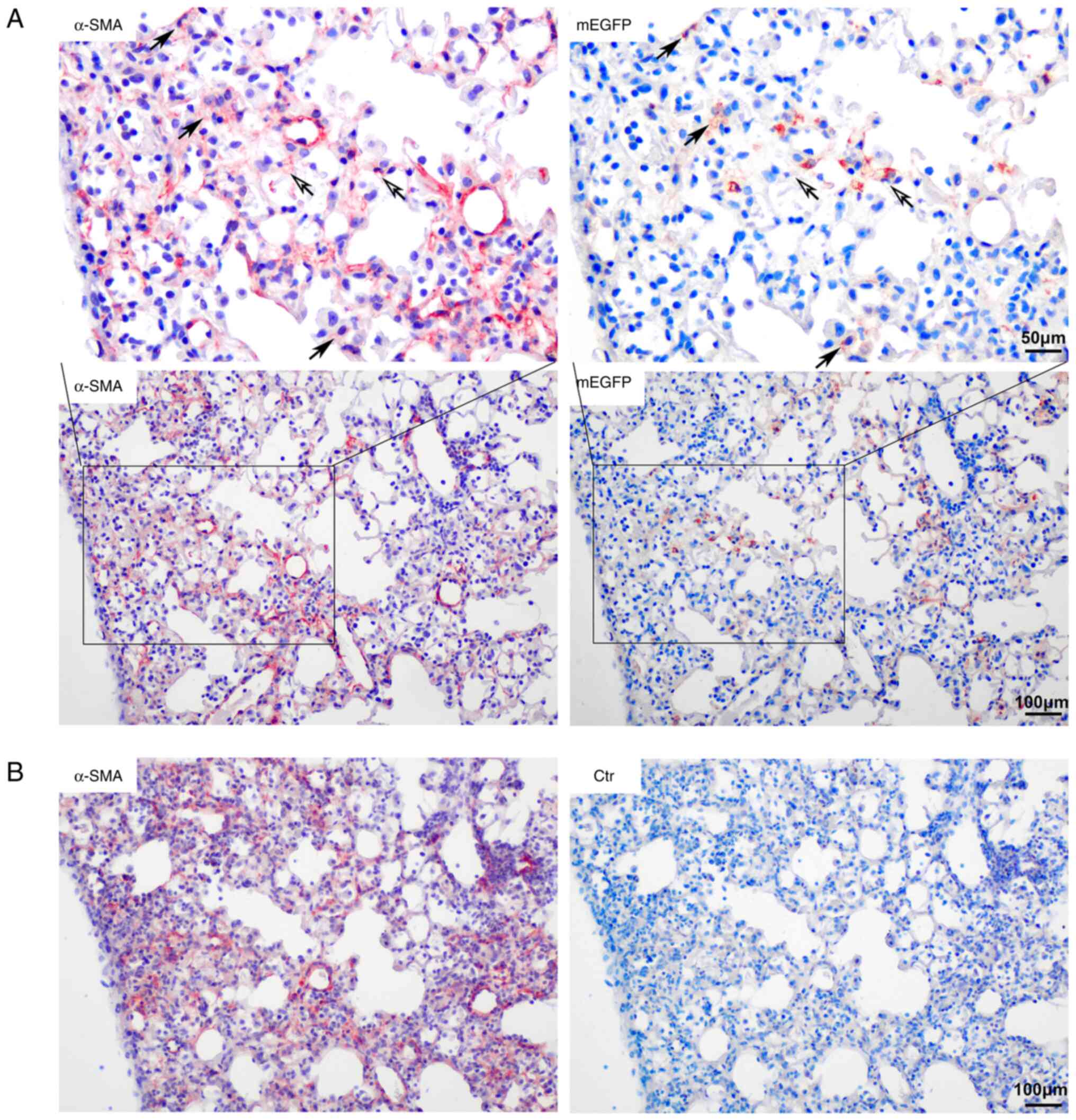
Co-expression of α-SMA and mEGFP in
bleomycin-induced pulmonary fibrotic lesions
A small number of cells in fibrotic foci in mice
with bleomycin-induced pulmonary fibrosis were type II AECs.
Certain α-SMA+ cells expressed mEGFP simultaneously in
the center of the lesion area and certain mEGFP+ cells
on the alveolar wall also exhibited α-SMA staining signals. Most
mEGFP+ cells had no α-SMA expression. The number of
α-SMA/mEGFP double-positive cells in the fibrotic tissue was low,
as 1.94±0.08% of α-SMA-positive cells were mEGFP-positive. Most of
the cells in the fibrotic foci did not express mEGFP and they were
no descendants of type II AECs (Fig.
3).
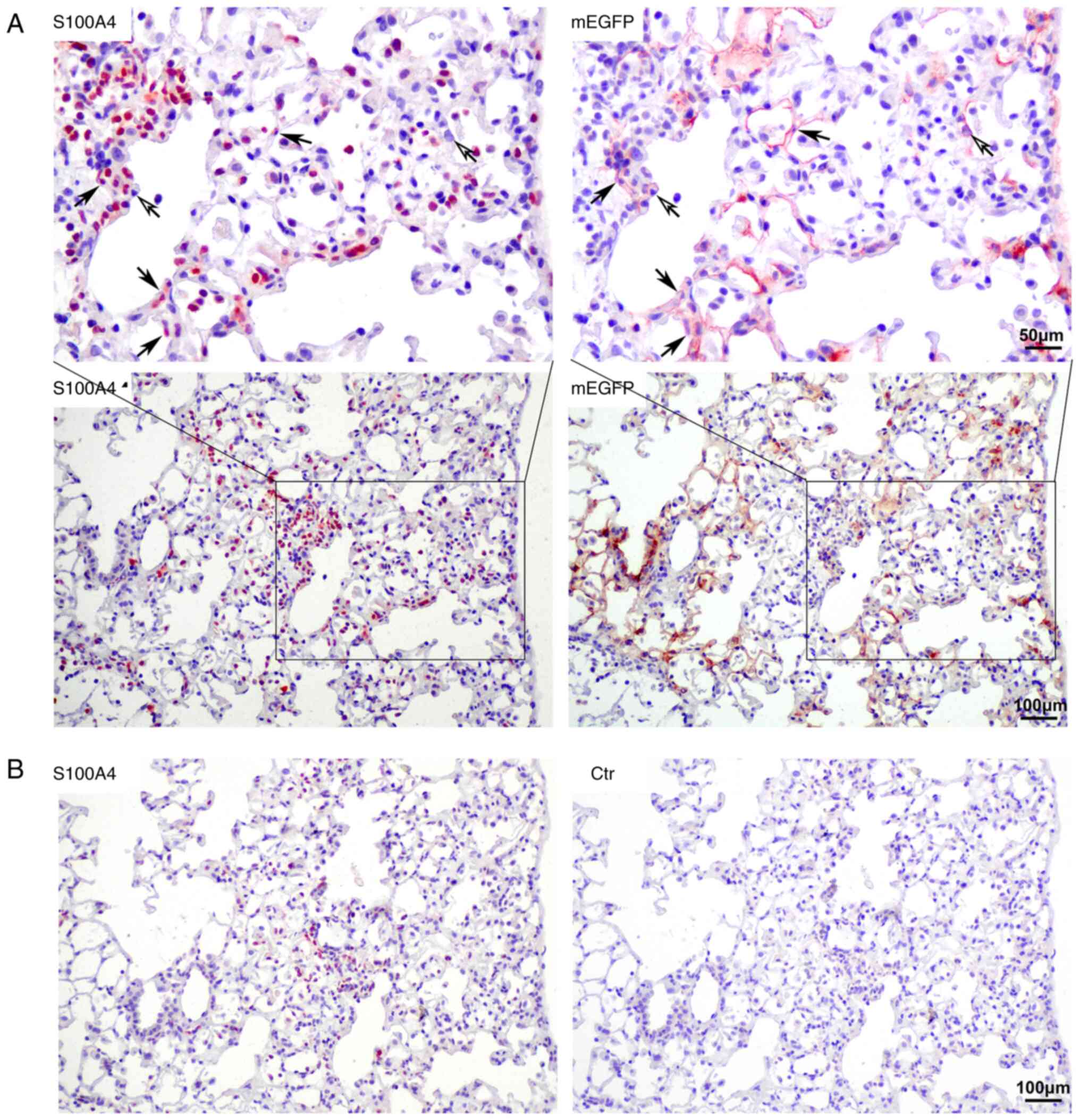
Cells co-expressing S100A4 and mEGFP
in bleomycin-induced pulmonary fibrotic lesions
Numerous S100A4-positive cells were observed in the
pulmonary fibrotic lesion areas in the lungs of the model mice.
S100A4 is also considered to be an important marker of fibrosis
(19). Multiple IHC staining
indicated that only a small number of S100A4-positive cells
originated from type II AECs and 9.68±2.06% of S100A4-positive
cells were mEGFP-positive. These were S100A4 and mEGFP
double-positive cells and they were mainly distributed on the
alveolar wall. Thus, the origin of most of the S100A4+
cells was not related to type II AECs (Fig. 4).
Discussion
In the present study, the classic intraperitoneal
administration of bleomycin was used to establish pulmonary
fibrosis in mice (20).
Lineage-tracing studies with neutral and efficient labeling and
gene reporting systems were performed to clarify whether EMT
existed in pulmonary fibrosis. The results indicated that EMT
contributes to the pathogenesis of pulmonary fibrosis, but it is
not the major causative factor of this condition.
Bleomycin may be injected using intratracheal
administration or systemically (intraperitoneally, intravenously or
subcutaneously) (21). The
intratracheal injection has been widely applied in lineage tracing
studies (10-14).
When injected through the trachea into the lungs, bleomycin
directly damages AECs. Fibrosis induced by this method has been
reported to be self-limiting after 28 days. Previous studies also
suggested that this routine may be inconsistent with the
pathological changes in human IPF (20-24).
When bleomycin is systemically injected, the initial site of injury
is the pulmonary vascular endothelium; after endothelial cell
injury, the drug targets alveolar epithelium. When bleomycin is
intraperitoneally delivered, the dose may be easily defined and the
method is simple to perform; it may cause repeated tissue damage
and repair, followed by irreversible pulmonary fibrosis, which is
considered more clinically relevant than intratracheal
administration (20,21,25).
In the present study, intraperitoneal administration of bleomycin,
which is closer to the clinicopathological changes of IPF, was used
to establish pulmonary fibrosis in mice.
Existing lineage tracing experiments, cell labeling
methods and reporter gene systems are somewhat limited. First, the
inducers used in some research may have a potential effect on
fibrosis (15). Tamoxifen, an
inducer of the CreERT2 system, is a selective estrogen
receptor modulator, which is also known for its inhibitory effect
on the production of TGF-β. Its anti-fibrotic effect has been
examined in certain fibrotic diseases (26). There is an interval between the end
of induction and the beginning of the modeling that serves to avoid
side effects of tamoxifen. However, this interval was different
between studies on pulmonary fibrosis (15,27).
Furthermore, proper labeling time is important for accurately
controlling the labeling range. Certain studies were initiated from
the mouse embryonic stage, which may lead to a wider range of
labeling (28). In addition, a
suitable reporter gene should be used to mark target cells.
Previous studies have indicated that β-galactosidase is expressed
in both fibroblasts and epithelial cells of human lung tissue
affected by IPF and bleomycin-induced pulmonary fibrosis (29,30).
X-gal staining should be avoided in pulmonary fibrosis cell tracing
studies. In the present study, the
SPC-rtTA+/-/tetO7-CMV-Cre+/-/mTmG+/-
system was used and induced with doxycycline from adulthood. The
inducer doxycycline and the reporter gene product mEGFP in this
system are neutral and do not interfere with pulmonary fibrosis;
thus, more objective results may be observed.
In the present study, the labeling method of type II
AECs was improved to obtain more objective tracking results. The
SPC-rtTA+/-/tetO7-CMV-Cre+/-/mTmG+/-
system was used and induced with doxycycline from adulthood. The
results indicated that only type II epithelial cells expressed Cre
in the peripheral lung tissue. The labeling efficiency in AEC II of
transgenic mice was 63.27±7.51% by IHC co-localization with Cre and
SPC. At the beginning of bleomycin modeling, the labeled cells were
type II epithelial cells, while there was only a small number of
newly differentiated type I epithelial cells. By using mEGFP as a
reporter gene, the outline of the cell was marked without this
being disturbed by cell senescence. Multiplex IHC staining
techniques are useful for examining changes in the expression of
target molecules and the basic histological changes in the lesion
while avoiding background fluorescence interference and sensitivity
to low-abundance antigens (31,32).
The present study reproduced the process of differentiation of type
II AECs in bleomycin-induced pulmonary fibrosis, which is more
clinically relevant than intratracheal administration.
The expression of mEGFP was detected in the fibrotic
region, suggesting the existence of progeny cells of epithelial
cells. Multiplex IHC staining indicated that certain cells were
α-SMA+/mEGFP+ or
S100A4+/mEGFP+ double-positive in
bleomycin-induced pulmonary fibrosis. This confirms that EMT may be
one of the origins of myofibroblasts. However, only a small
proportion of fibroblasts was identified as originating from type
II epithelial cells. This suggests that the origin of cells in
fibrotic lesions is complex. Fibroblasts may have other sources,
including the proliferation of lung fibroblasts, transformation of
vascular endothelial cells and pericytes, as well as
differentiation of bone marrow progenitor cells (33,34).
On the other hand, EMT may not be the only direct phenotypic
transformation of epithelial cells into fibroblasts. Yamaguchi
et al (35) reported that
fibroblastic foci were covered with alveolar epithelium and certain
AECs also expressed vimentin, implying partial EMT, and those cells
had the functions of both epithelial cells and mesenchymal cells,
such as adhesion and migration. Whether epithelial cells with
mesenchymal markers may release profibrotic factors or exhibit a
partial EMT phenotype requires further investigation. It was
noticed that the distribution of
α-SMA+/mEGFP+ or
S100A4+/mEGFP+ double-positive cells in
fibrotic lesion was not exactly the same. In patients with IPF and
bleomycin-induced pulmonary fibrosis, S100A4 may be secreted by
fibroblasts and promote pulmonary fibrosis through fibroblast
activation (36,37). How S100A4 participates in abnormal
epithelial-mesenchymal interactions requires further
investigation.
Of note, the present study had several limitations.
First, the sample size of mice used in the experiment was
relatively small. In the present study, mice with a uniform genetic
background and uniform modeling methods were strictly selected,
which met the minimal requirement for sufficient statistical power.
Furthermore, α-SMA+/mEGFP+ or
S100A4+/mEGFP+ double-positive cells appeared
more in alveoli with obvious fibrosis and less in regions with mild
fibrosis or even normal fibrosis. In future studies, the proportion
of positive cells will be observed according to different stages of
pulmonary fibrosis, which may be of great significance. Finally,
the present results were all from animal experiments, which require
to be confirmed in clinical patient samples if possible.
In conclusion, the present study identified
descendants of epithelial cells in the fibrotic region. A small
number of mEGFP-positive cells had co-localization of α-SMA and
S100A4, which indicated that EMT contributed to the pathogenesis of
pulmonary fibrosis; however, it was not the major causative factor
of pulmonary fibrosis.
Supplementary Material
Weight changes in BLM-induced mice
compared with the Ctr group. **P<0.01 and
***P<0.001. Ctr, control; BLM, bleomycin.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the National Natural Science
Foundation of China (grant no. 31271231).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CC designed the experiments, completed and revised
the writing of the manuscript. WT performed the main experimental
work and was involved in drafting the manuscript. YW contributed to
data acquisition and analysis. YC made contributions to the
conception and design of the study, and manuscript revision. All
authors read and approved the manuscript. YW and YC confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
All animal experiments were performed according to
the Guidelines for Animal Care of CMU were and approved by the
IACUC of CMU (IACUC issue no. 14031M). All applicable
international, national and institutional guidelines for the care
and use of animals were followed.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Barratt SL, Creamer A, Hayton C and
Chaudhuri N: Idiopathic pulmonary fibrosis (IPF): An overview. J
Clin Med. 7(201)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Raghu G, Remy-Jardin M, Myers JL, Richeldi
L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F,
et al: Diagnosis of idiopathic pulmonary fibrosis. An official
ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care
Med. 198:e44–e68. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Richeldi L, Collard HR and Jones MG:
Idiopathic pulmonary fibrosis. Lancet. 389:1941–1952.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Moeller A, Gilpin SE, Ask K, Cox G, Cook
D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, et
al: Circulating fibrocytes are an indicator of poor prognosis in
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
179:588–594. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Selman M and Pardo A: The leading role of
epithelial cells in the pathogenesis of idiopathic pulmonary
fibrosis. Cell Signal. 66(109482)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Salton F, Ruaro B, Confalonieri P and
Confalonieri M: Epithelial-mesenchymal transition: A major
pathogenic driver in idiopathic pulmonary fibrosis? Medicina
(Kaunas). 56(608)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Salton F, Volpe MC and Confalonieri M:
Epithelial-mesenchymal transition in the pathogenesis of idiopathic
pulmonary fibrosis. Medicina (Kaunas). 55(83)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kim KK, Kugler MC, Wolters PJ, Robillard
L, Galvez MG, Brumwell AN, Sheppard D and Chapman HA: Alveolar
epithelial cell mesenchymal transition develops in vivo during
pulmonary fibrosis and is regulated by the extracellular matrix.
Proc Natl Acad Sci USA. 103:13180–13185. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
DeMaio L, Buckley ST, Krishnaveni MS,
Flodby P, Dubourd M, Banfalvi A, Xing Y, Ehrhardt C, Minoo P, Zhou
B, et al: Ligand-independent transforming growth factor-β type I
receptor signalling mediates type I collagen-induced
epithelial-mesenchymal transition. J Pathol. 226:633–644.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kim KK, Wei Y, Szekeres C, Kugler MC,
Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg
JA and Chapman HA: Epithelial cell alpha3beta1 integrin links
beta-catenin and Smad signaling to promote myofibroblast formation
and pulmonary fibrosis. J Clin Invest. 119:213–224. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Tanjore H, Xu XC, Polosukhin VV, Degryse
AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS
and Lawson WE: Contribution of epithelial-derived fibroblasts to
bleomycin-induced lung fibrosis. Am J Respir Crit Care Med.
180:657–665. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Degryse AL, Tanjore H, Xu XC, Polosukhin
VV, Jones BR, McMahon FB, Gleaves LA, Blackwell TS and Lawson WE:
Repetitive intratracheal bleomycin models several features of
idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol.
299:L442–L452. 2010.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Degryse AL, Tanjore H, Xu XC, Polosukhin
VV, Jones BR, Boomershine CS, Ortiz C, Sherrill TP, McMahon FB,
Gleaves LA, et al: TGFβ signaling in lung epithelium regulates
bleomycin-induced alveolar injury and fibroblast recruitment. Am J
Physiol Lung Cell Mol Physiol. 300:L887–L897. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Rock JR, Barkauskas CE, Cronce MJ, Xue Y,
Harris JR, Liang J, Noble PW and Hogan BL: Multiple stromal
populations contribute to pulmonary fibrosis without evidence for
epithelial to mesenchymal transition. Proc Natl Acad Sci USA.
108:E1475–E1483. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chilosi M, Caliò A, Rossi A, Gilioli E,
Pedica F, Montagna L, Pedron S, Confalonieri M, Doglioni C, Ziesche
R, et al: Epithelial to mesenchymal transition-related proteins
ZEB1, β-catenin, and β-tubulin-III in idiopathic pulmonary
fibrosis. Mod Pathol. 30:26–38. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Harada T, Nabeshima K, Hamasaki M, Uesugi
N, Watanabe K and Iwasaki H: Epithelial-mesenchymal transition in
human lungs with usual interstitial pneumonia: Quantitative
immunohistochemistry. Pathol Int. 60:14–21. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Boye K and Maelandsmo GM: S100A4 and
metastasis: A small actor playing many roles. Am J Pathol.
176:528–535. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
B Moore B, Lawson WE, Oury TD, Sisson TH,
Raghavendran K and Hogaboam CM: Animal models of fibrotic lung
disease. Am J Respir Cell Mol Biol. 49:167–179. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Mouratis MA and Aidinis V: Modeling
pulmonary fibrosis with bleomycin. Curr Opin Pulm Med. 17:355–361.
2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Moore BB and Hogaboam CM: Murine models of
pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol.
294:L152–L160. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Degryse AL and Lawson WE: Progress toward
improving animal models for idiopathic pulmonary fibrosis. Am J Med
Sci. 341:444–449. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lawson WE, Polosukhin VV, Stathopoulos GT,
Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, et
al: Increased and prolonged pulmonary fibrosis in surfactant
protein C-deficient mice following intratracheal bleomycin. Am J
Pathol. 167:1267–1277. 2005.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Adamson IY and Bowden DH: The pathogenesis
of bloemycin-induced pulmonary fibrosis in mice. Am J Pathol.
77:185–197. 1974.PubMed/NCBI
|
|
26
|
Yoldas O, Karaca T, Bilgin BC, Yilmaz OH,
Simsek GG, Alici IO, Uzdogan A, Karaca N, Akin T, Yoldas S and
Akbiyik F: Tamoxifen citrate: A glimmer of hope for silicosis. J
Surg Res. 193:429–434. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
El Agha E, Moiseenko A, Kheirollahi V, De
Langhe S, Crnkovic S, Kwapiszewska G, Szibor M, Kosanovic D,
Schwind F, Schermuly RT, et al: Two-way conversion between
lipogenic and myogenic fibroblastic phenotypes marks the
progression and resolution of lung fibrosis. Cell Stem Cell.
20:261–273.e3. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Perl AK, Wert SE, Nagy A, Lobe CG and
Whitsett JA: Early restriction of peripheral and proximal cell
lineages during formation of the lung. Proc Natl Acad Sci USA.
99:10482–10487. 2002.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Schafer MJ, White TA, Iijima K, Haak AJ,
Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y,
et al: Cellular senescence mediates fibrotic pulmonary disease. Nat
Commun. 8(14532)2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chen X, Xu H, Hou J, Wang H, Zheng Y, Li
H, Cai H, Han X and Dai J: Epithelial cell senescence induces
pulmonary fibrosis through Nanog-mediated fibroblast activation.
Aging (Albany NY). 12:242–259. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Pirici D, Mogoanta L, Kumar-Singh S,
Pirici I, Margaritescu C, Simionescu C and Stanescu R: Antibody
elution method for multiple immunohistochemistry on primary
antibodies raised in the same species and of the same subtype. J
Histochem Cytochem. 57:567–575. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bolognesi MM, Manzoni M, Scalia CR,
Zannella S, Bosisio FM, Faretta M and Cattoretti G: Multiplex
staining by sequential immunostaining and antibody removal on
routine tissue sections. J Histochem Cytochem. 65:431–444.
2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Borensztajn K, Crestani B and Kolb M:
Idiopathic pulmonary fibrosis: From epithelial injury to
biomarkers-insights from the bench side. Respiration. 86:441–452.
2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bagnato G and Harari S: Cellular
interactions in the pathogenesis of interstitial lung diseases. Eur
Respir Rev. 24:102–114. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yamaguchi M, Hirai S, Tanaka Y, Sumi T,
Miyajima M, Mishina T, Yamada G, Otsuka M, Hasegawa T, Kojima T, et
al: Fibroblastic foci, covered with alveolar epithelia exhibiting
epithelial-mesenchymal transition, destroy alveolar septa by
disrupting blood flow in idiopathic pulmonary fibrosis. Lab Invest.
97:232–242. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xia H, Gilbertsen A, Herrera J, Racila E,
Smith K, Peterson M, Griffin T, Benyumov A, Yang L, Bitterman PB
and Henke CA: Calcium-binding protein S100A4 confers mesenchymal
progenitor cell fibrogenicity in idiopathic pulmonary fibrosis. J
Clin Invest. 127:2586–2597. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lee JU, Chang HS, Shim EY, Park JS, Koh
ES, Shin HK, Park JS and Park CS: The S100 calcium-binding protein
A4 level is elevated in the lungs of patients with idiopathic
pulmonary fibrosis. Respir Med. 171(105945)2020.PubMed/NCBI View Article : Google Scholar
|