Introduction
Asthma, a chronic inflammatory disease that is
associated with type-2 cytokines such as IL-4 and IL5, accelerates
airway mucus overproduction, eosinophilia, immunoglobulin E (IgE)
synthesis and bronchial hyperresponsiveness (1). Migration of airway smooth muscle cells
(ASMCs) increases airway smooth muscle mass, to promote airway
remodelling, which is a key characteristic of asthma pathogenesis
(2). Asthma can be well controlled
by an optimized inhaled drug dose and continuous treatment
(3). However, asthma often develops
into a lifelong condition with a resulting economic burden and may
even lead to early permanent disability or premature death
(4). It would therefore be
beneficial to explore new approaches in the treatment of
asthma.
It has been demonstrated that long noncoding RNAs
(lncRNAs) exert functions in various cellular processes such as
proliferation and migration by regulating DNA, RNA and protein
expression levels (5-9).
Long noncoding RNA taurine-upregulated gene1 (TUG1) takes part in a
host of respiratory diseases. Silencing of TUG1 has been indicated
to regulate LIM domain kinase 2b through histone methyl transferase
enhancer of zeste homolog 2, inhibiting cell growth in small cell
lung cancer (6). TUG1 was also
indicated to promote lung adenocarcinoma cell viability and
suppress cell apoptosis (7). TUG1
facilitates pulmonary vascular remodelling through targeting the
microRNA (miR)-374c/forkhead box c1 axis in hypoxic pulmonary
hypertension (8). The results of
recent research also suggest that TUG1 increases the ASMC growth in
asthma by regulating the miR-590-5p-mediated FGF1(9). Therefore, it is hypothesized that TUG1
may play a role in asthma progression.
As biological molecules, miRNAs take part in the
development of respiratory diseases including asthma (10,11).
miR-217 is expressed at low levels in asthma, and its upregulation
suppresses ASMC proliferation and migration by inhibiting zinc
finger E-box homeobox 1 expression (12). miR-142 reduces proliferation and
induces apoptosis in the ASMCs of asthmatic rats during airway
remodelling (13). miR-138 has been
demonstrated to attenuate the proliferation of ASMCs from asthmatic
donors (14). In previous studies
it was suggested that miR-138 is a target of TUG1. Yan et al
(15) demonstrated that TUG1
contributed to the development of colorectal cancer via a reduction
in miR-138-5p expression and Zhu et al (16) reported that TUG1 promoted cervical
cancer cell growth and metastasis by suppressing miR-138
expression. However, the specific regulatory relationship between
TUG1 and miR-138-5p in asthma remains unclear.
E2F3, a member of the E2F family, participates in
the transcriptional activation of genes that regulate the
proliferation of both tumour and primary cells (17). Researchers have suggested that E2F3
is associated with the development of respiratory diseases. Cooper
et al (18) indicated that
small cell lung cancer shows enhanced expression of nuclear
staining for E2F3 and Al Ahmed and Nada (19) reported that E2F3 expression
increased in lung cancer patients and that overexpression of E2F3
was positively related to metastatic lung cancer. Silencing of
lncRNA PVT1 attenuated the viability and migration of ASMCs in
asthma by mediating the miR-203a/E2F3 axis (20). However, to the best of our
knowledge, the potential regulatory mechanism of TUG1 on the
miR-138-5p/ E2F3 axis in asthma has not previously been
evaluated.
In the present study, a rat asthma model and a
platelet-derived growth factor-BB (PDGF-BB)-treated ASMC cell model
were applied. Whether TUG1 controlled the viability and migration
of ASMCs via miR-138-5p and E2F3 in asthma was then
investigated.
Materials and methods
Animals
Sprague Dawley rats (female; 60-80 g; 4-weeks-old)
were purchased from the Institute of Zoology, Chinese Academy of
Sciences. Rats were fed standard chow and water and maintained
under a 12-h light/dark cycle (25˚C; 50% humidity). This study was
performed with the approval of the Animal Ethics Committee of Hunan
Children's Hospital (Changsha, China).
Asthmatic rat model establishment
A total of 20 rats were randomly divided into two
groups (n=10): The asthma group and the control group. In the
asthma group, rats were intraperitoneally injected with 1 ml 10%
ovalbumin antigenic suspension (OAS, containing 100 mg ovalbumin
and 100 mg aluminium hydroxide (both MilliporeSigma) at 1, 7 and 15
days. On day 21, the rats were further stimulated by a 7-day
ultrasonic atomizing inhalation of 2% OAS for 30 min daily. Similar
procedures were performed on the rats in the control group, but the
OAS was replaced with normal saline.
Airway resistance measurement
Inspiratory and expiratory resistances were detected
using a rodent ventilator (model 7025; Ugo Basile SRL).
Methacholine (MilliporeSigma) at an initial dose of 0.0625
(dissolved in 0.9% sodium chloride) was intravenously injected into
the rats. To obtain a response curve of lung resistance, the dose
was increased two-fold with each injection, up to 1 mg/kg. The
injection interval was 5 min. Before the next methacholine
injection, 50 µl methacholine was administered over 4 sec through
the return of resistance curves to the pre-methacholine level.
Following methacholine administration, the response was detected
directly as the peak elevated above the baseline, with the use of
AniRes2005 software (Beijing Bestlab High-Tech Co., Ltd.).
PDGF-BB-induced asthma model in
ASMCs
Rats were anesthetized with 50 mg/kg pentobarbital
sodium and euthanized by cervical dislocation. ASMCs were isolated
from the airway of rats in the control group as previously
described (21). The ASMCs were
centrifuged (500 x g for 15 min; 4˚C) and cultured in Dulbecco's
modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc.) with 10% foetal bovine serum (FBS; Invitrogen;
Thermo Fisher Scientific, Inc.) at 37˚C and 5% CO2. The
ASMCs were randomly divided into two groups: The PDGF-BB group
(ASMCs were stimulated with 25 ng/ml PDGF-BB for 24 h) and the
control group (ASMCs without treatment). The PDGF-BB-induced ASMCs
acted as the asthmatic model at the cellular level (22).
Enzyme-linked immunosorbent assay
(ELISA)
After measuring airway reactivity, serum was
obtained by lethal cardiac puncture of anesthetized rats. The level
of serum IgE in the rats was measured with ELISA using a rat IgE
ELISA kit (cat. no. RAB0799MSDS; Sigma Aldrich) according to the
manufacturer's guidelines.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissues and cells using
the TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and was reverse-transcribed into cDNA using the
PrimeScript RT reagent kit (Takara Bio, Inc.). PCR reaction was
performed on an ABI 7500HT Fast Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) under the following
conditions: 95˚C for 5 min, 40 cycles of 72˚C for 34 sec and 60˚C
for 20 sec. Relative expression was calculated using the
2-ΔΔCq method (23).
GAPDH, U6 and β-actin were used for the normalization of TUG1,
miR-138-5p and E2F3, respectively. The primer sequences are shown
in Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Name of primer | Sequences,
5'-3' |
|---|
| TUG1-F |
GGACACAATTCGCCACGACTT |
| TUG1-R |
GCGCAGTCCCAGATTCCA |
| GAPDH-F |
AGAAGGCTGGGGCTCATTTG |
| GAPDH-R |
AGGGGCCATCCACAGTCTTC |
| miR-138-5p-F |
GCTTAAGGCACGCGG |
| miR-138-5p-R |
GTGCAGGGTCCGAGG |
| U6-F |
CGCTTCGGCAGCACATATAC |
| U6-R |
AAATATGGAACGCTTCACGA |
| E2F3-F |
ACAAACAACCAAGACCACAATG |
| E2F3-R |
GGGAGGCAGTAAGTTCACAAAC |
| β-actin-F |
CATGTACGTTGCTATCCAGGC |
| β-actin-R |
CTCCTTAATGTCACGCACGAT |
Cell transfection
The short hairpin (sh)-TUG1
(5'-GTCTGCATTGAGGATATAG-3'), sh-negative control (NC;
5'-CCTCTAGGTAAGCATAATTTT-3'), miR-138-5p mimics
(5'-AGCUGGUGUUGUGAAUCAGGCCG-3'), miR-NC
(5'-UUGUACUACACAAAAGUACUG-3'), pcDNA3.1 TUG1 (pcDNA-TUG1) and
sh-E2F3 (5'-CATTGAGGTTTACTTATGT-3') were synthesized by Shanghai
GenePharma, Co., Ltd. PDGF-BB-induced ASMCs grown to 85% confluence
were transfected or co-transfected with 50 nM of the above-listed
agents using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) for 4 h at 37˚C. The ASMCs in the control
group did not receive any transfection. Subsequently, 48 h after
transfection, ASMCs were harvested to perform further
experiments.
MTT assay
ASMCs and PDGF-BB-treated ASMCs were seeded into
96-well plates (2x103 cells/well) and cultured with 5%
CO2 at 37˚C for 72 h. Cell viability was then measured
using an MTT cell proliferation assay kit (Sigma-Aldrich; Merck
KGaA), according to the manufacturer's guidelines. In brief, the
stock MTT dye solution (5 mg/ml; Sigma-Aldrich; Merck KGaA) was
added to each well after the plates were incubated at 37˚C with 5%
CO2. Following incubation for 2 h, the supernatant was
removed and 100 µl dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA)
was added to each well. The viability [optical density (OD450)] was
analyzed by a Multiskan Spectrum microplate reader (Thermo Fisher
Scientific, Inc.).
Transwell assay
Transwell assay was performed to assess the level of
cell migration using Transwell chambers (8-µM pore size; Corning
Inc.). The lower chamber was filled with 0.6 ml DMEM containing 1%
FBS. The ASMCs and PDGF-BB-treated ASMCs (1x105) were
suspended in 0.1 ml serum-free DMEM and placed in the upper
chamber. After culture for 24 h, the ASMCs and PDGF-BB-treated
ASMCs were fixed with 4% paraformaldehyde at 37˚C for 1 h and
stained with 0.5% crystal violet for 10 min at 37˚C. Cell migration
was assessed by counting the cells under a light microscope
(magnification, x400; Olympus Corporation) in five randomly
selected views.
Dual-luciferase reporter assay
The potential binding sites between TUG1 and
miR-138-5p or miR-138-5p and E2F3 were predicted by Starbase
(version 2.0; http://starbase.sysu.edu.cn) or TargetScan (release
7.2; http://www.targetscan.org/vert_72/), respectively.
TUG1 and E2F3 with WT or MUT miR-138-5p-binding sites were
generated and subcloned into the psiCHECK-2 vectors (Promega
Corporation). PDGF-BB-treated ASMCs grew until 85% confluence and
were co-transfected with the relevant luciferase vectors (80 ng)
and miR-NC or miR-138-5p mimics (50 nM) using
Lipofectamine® 3000 at 37˚C for 4 h. At 48 h
post-incubation, luciferase activity was determined by the
dual-luciferase assay system (Promega Corporation). The activity of
firefly luciferase was normalized to that of Renilla
luciferase.
Lentivirus production and
infection
A shRNA-targeted TUG1 and its corresponding
scrambled NC were designed and synthesized by Shanghai GenePharma,
Co., Ltd. They were inserted into the pGLVU6/Puro vector (Shanghai
GenePharma Co., Ltd.) to construct sh-TUG1 and sh-NC. The vectors
containing sh-TUG1 or sh-NC together with pMD2.G and psPAX2
plasmids (Shaanxi YouBio Technology Co., Ltd.) were then
co-transfected into 293T cells using Lipofectamine 3000. Three days
post transfection, cell supernatants containing sh-TUG1 or sh-NC
lentiviruses were collected. Sh-TUG1 or sh-NC lentiviruses were
extracted and purified from cell supernatants. Rats received a tail
intravenous injection of 100 µl (5x107 TU/ml) sh-TUG1
(n=5, sh-TUG1 group) or sh-NC (n=5, sh-NC group) lentivirus every
24 h for three days. At 30 min after the last injection of
lentiviruses, all rats were treated as described previously to
establish the asthmatic rat model. After the asthmatic rat model
had been established for 24 h, inspiratory and expiratory
resistances were detected.
Statistical analysis
Each assay was performed at least three times.
Statistical analysis was performed using GraphPad Prism 7.0
(GraphPad Software, Inc.). Data are presented as the mean ±
standard deviation. The differences between two groups or among
multiple groups were assessed by Student's t-test or one-way ANOVA
followed by Tukey's post hoc test, respectively. P<0.05 was
considered to indicate a statistically significant difference.
Results
TUG1 expression is increased in the
airway tissues of the rat asthma model
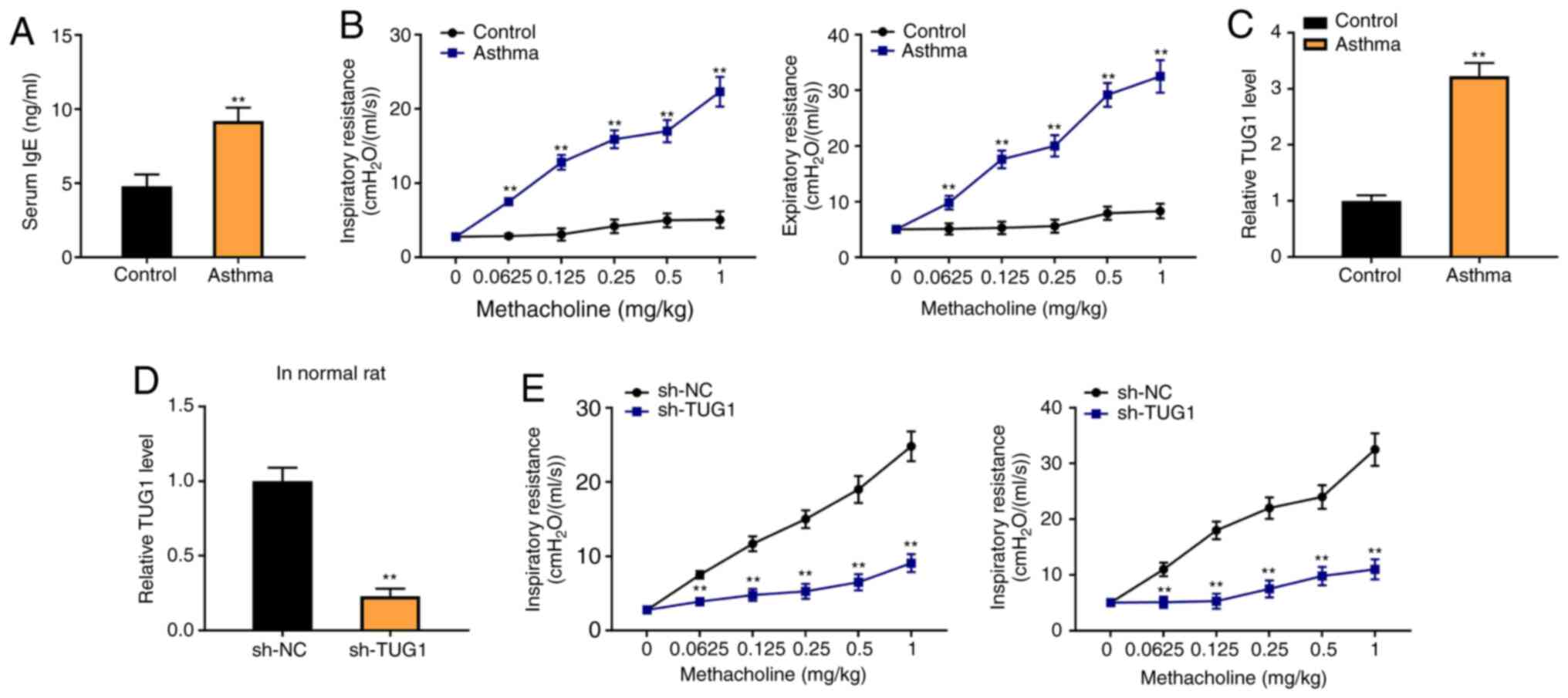
To determine the association between TUG1 expression
and asthma, a rat asthma model was constructed. The results from
ELISA indicated that the level of serum IgE was significantly
increased in rats in the asthma group compared with the control
group (P<0.01; Fig. 1A). The
inspiratory resistance and expiratory resistance were both higher
in the asthma group than in the control group (P<0.01; Fig. 1B). Moreover, RT-qPCR revealed that
the TUG1 expression was increased in the Asthma group, compared
with the control group (P<0.01; Fig.
1C). The results indicated that sh-TUG1 markedly decrease the
inspiratory resistance and expiratory resistance of the rat asthma
model, compared with the sh-NC group (P<0.01, Fig. 1D).
TUG1 inhibition attenuates the
viability and migration of PDGF-BB-induced ASMCs
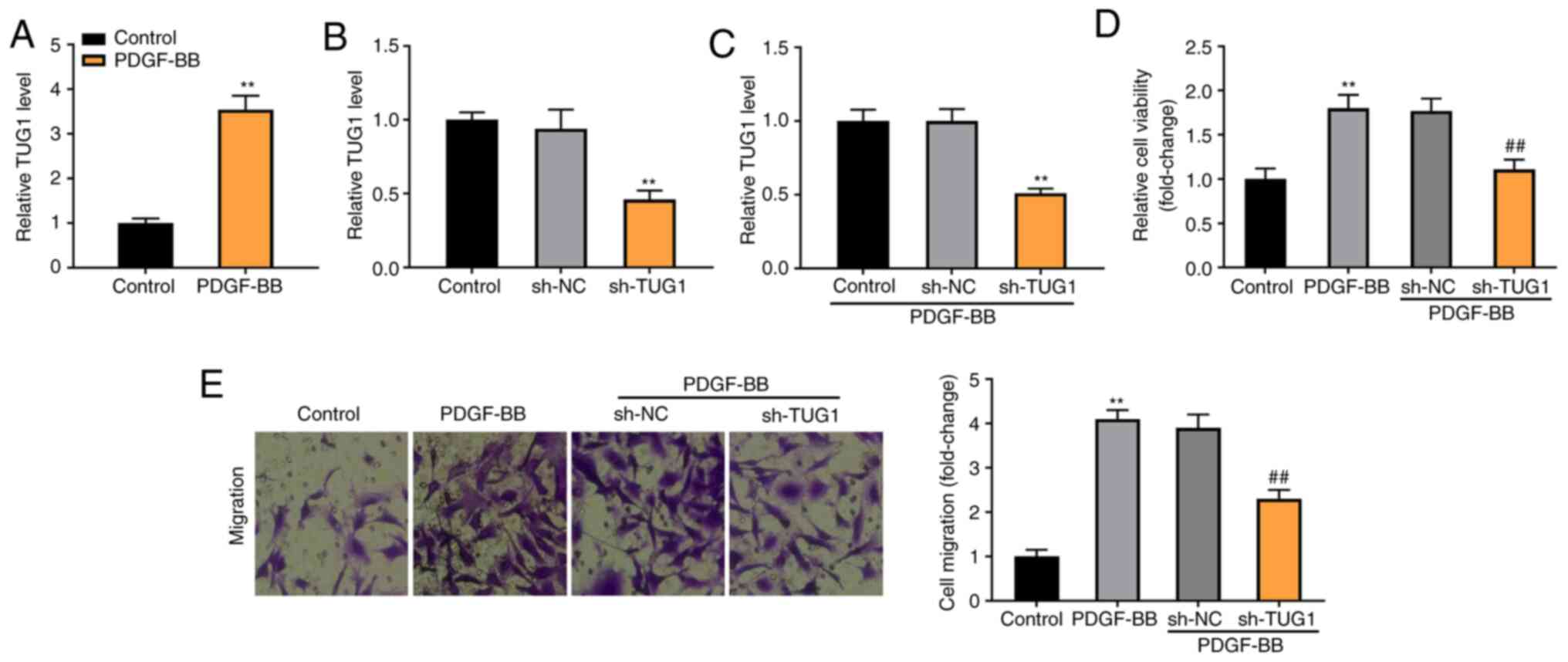
To establish the asthmatic model at the cellular
level, the ASMCs were extracted from the rats and induced by
PDGF-BB. As shown in Fig. 2A, TUG1
expression was significantly enhanced in the PDGF-BB-induced ASMCs,
compared with the control group (P<0.01). The expression of TUG1
was significantly reduced by the transfection of sh-TUG1 in ASMCs
(P<0.01, Fig. 2B). Furthermore,
sh-TUG1 successfully inhibited TUG1 expression in the
PDGF-BB-induced ASMCs (P<0.01; Fig.
2C). MTT and Transwell assays revealed that the viability and
migration of ASMCs were considerably increased in the PDGF-BB
group, in contrast with the Control group. Moreover, TUG1 knockdown
reduced the viability and migration of PDGF-BB-induced ASMCs,
compared with the sh-NC group (P<0.01; Fig. 2D and E).
miR-138-5p is a direct target of
TUG1
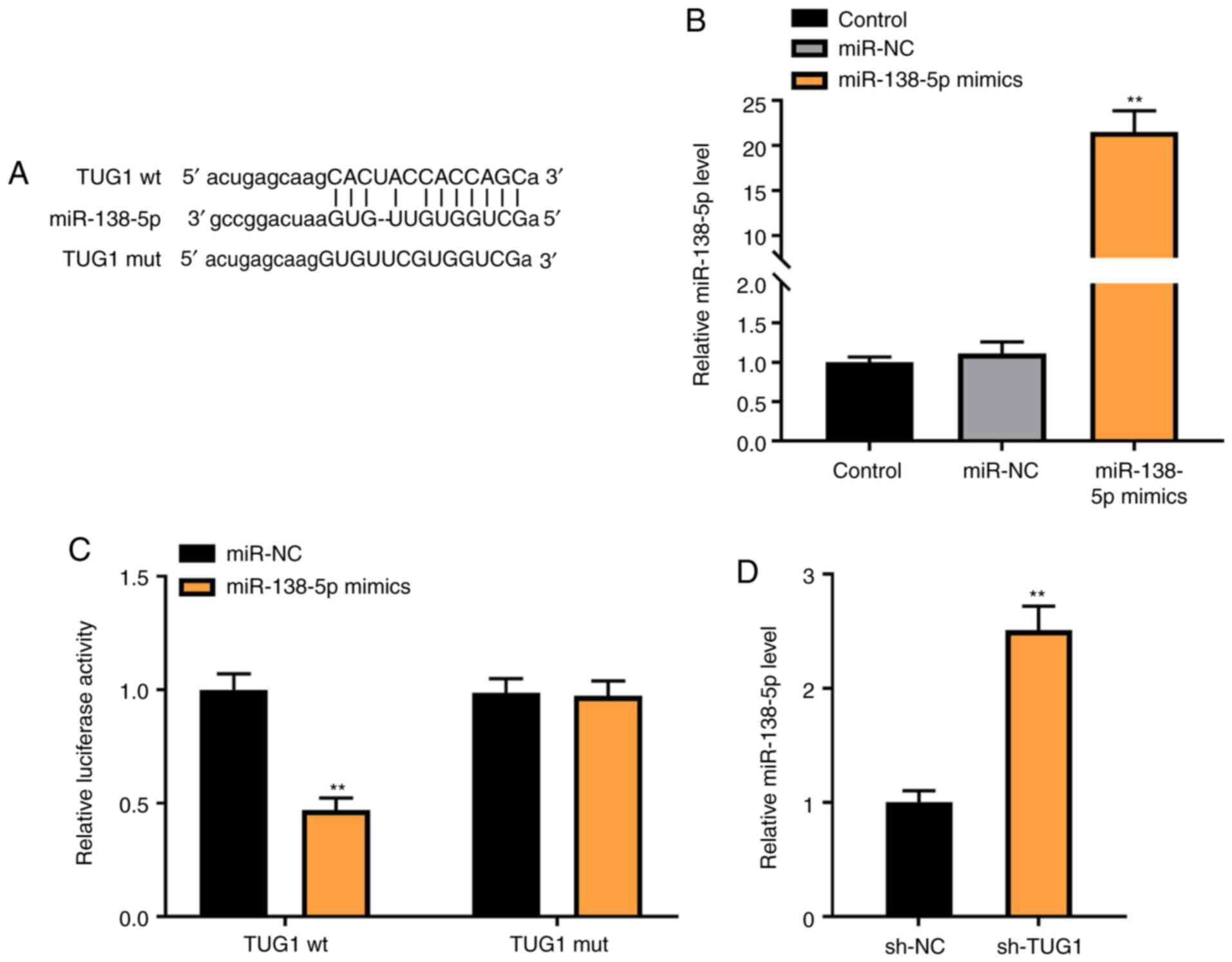
To investigate the mechanism of action underlying
the role of TUG1 in PDGF-BB-induced ASMCs, miR-138-5p was
identified as a potential target of TUG1 using Starbase (Fig. 3A). Transfection of miR-138-5p mimics
markedly increased the expression of miR-138-5p in ASMCs
(P<0.01; Fig. 3B). The relative
luciferase activity was decreased in ASMCs co-transfected with
miR-138-5p mimics and TUG1 Wt, compared with cells co-transfected
with miR-NC and TUG1 Wt (P<0.01; Fig. 3C). As shown in Fig. 3D, downregulation of TUG1 markedly
increased miR-138-5p expression (P<0.01).
miR-138-5p decreases the viability and
migration of PDGF-BB-induced ASMCs
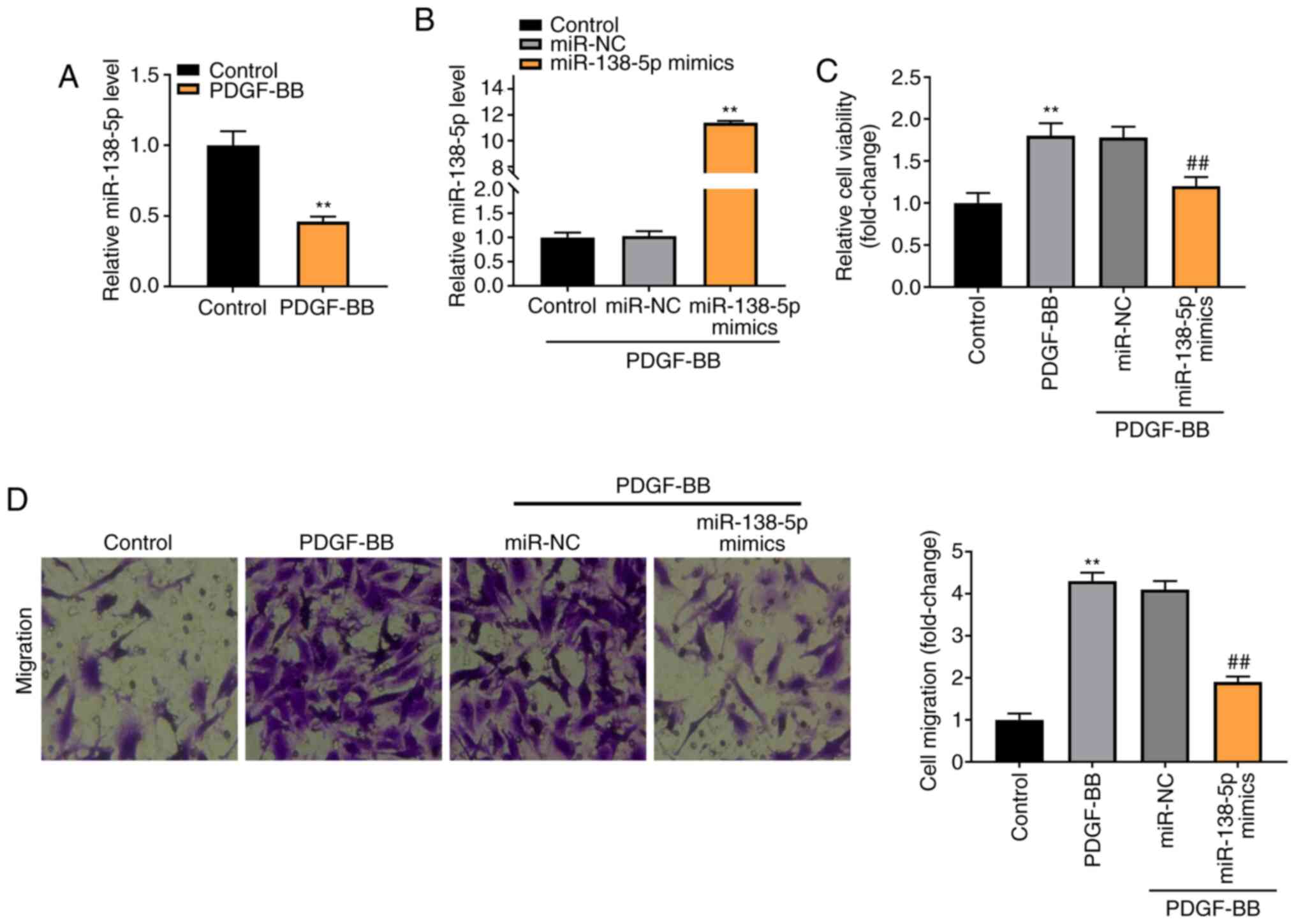
As shown in Fig. 4A,
expression of miR-138-5p was notably downregulated in the PDGF-BB
group, compared with the Control group (P<0.01). To verify the
regulatory effect of miR-138-5p on PDGF-BB-induced ASMCs,
miR-138-5p expression was increased by the transfection of
miR-138-5p mimics into ASMCs (P<0.01, Fig. 4B). As shown in Fig. 4C and D, the viability and migration of ASMCs
were markedly enhanced in the PDGF-BB group compared with the
Control group (P<0.01). The transfection of miR-138-5p mimics
notably decreased the viability and migration of PDGF-BB-induced
ASMCs compared with the miR-NC group (P<0.01).
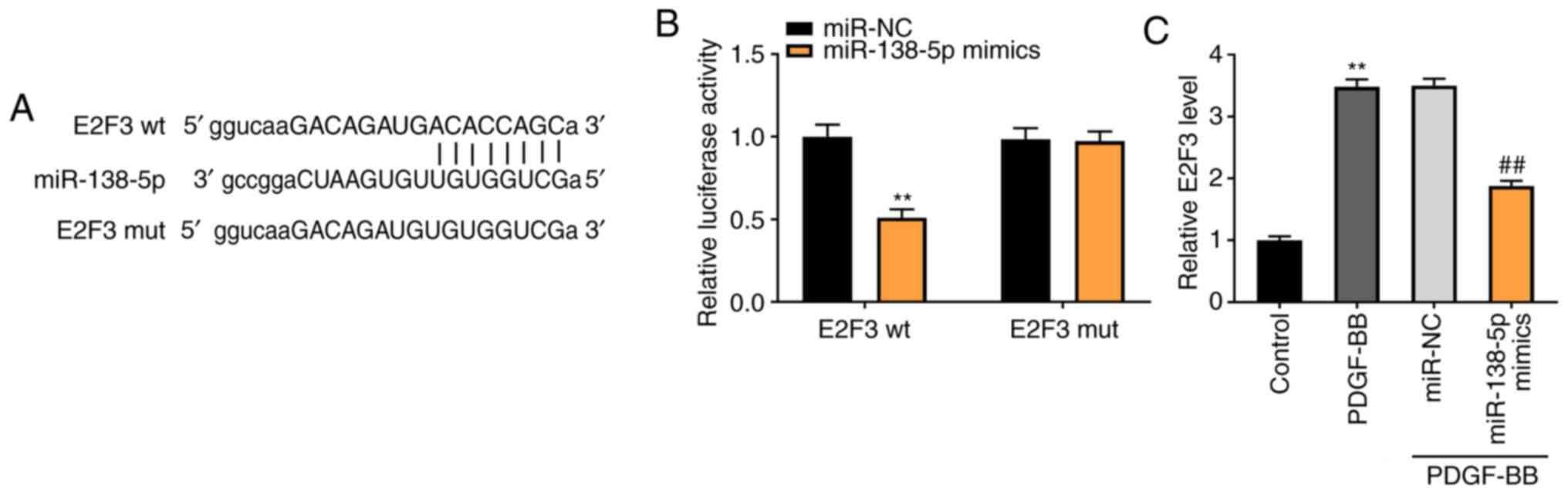
miR-138-5p modulates E2F3 expression
levels
To demonstrate the underlying mechanism by which
TUG1 mediated the growth of ASMCs, TargetScan was used to predict
the binding site for miR-138-5p on the 3' UTR of E2F3 (Fig. 5A). Using the luciferase reporter
assay, it was determined that miR-138-5p overexpression
considerably inhibited the luciferase activity of WT E2F3 3' UTR
reporter vector (P<0.01; Fig.
5B). As shown in Fig. 5C, the
E2F3 expression in the PDGF-BB group was higher than that in the
control group (P<0.01). Transfection of miR-138-5p mimics
notably decreased the E2F3 expression in PDGF-BB-induced ASMCs,
compared with the miR-NC group (P<0.01).
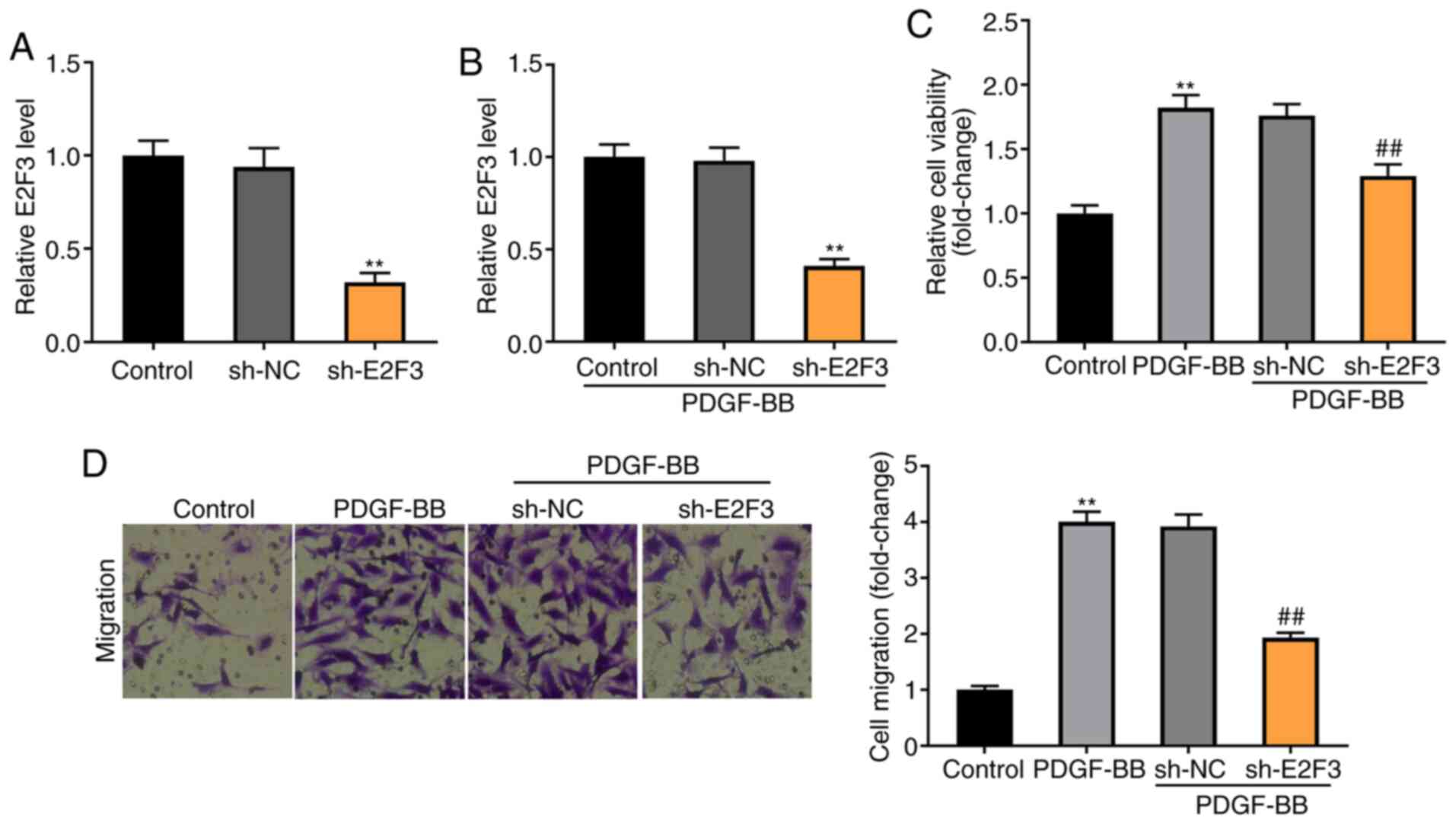
Knockdown of E2F3 decreases the
viability and migration of PDGF-BB-induced ASMCs
To explore the molecular mechanism by which E2F3
regulates the viability and migration of PDGF-BB-induced ASMCs,
E2F3 was knocked down by the transfection of sh-E2F3 (P<0.01;
Fig. 6A). Knockdown of E2F3
significantly decreased the expression of E2F3 in PDGF-BB-induced
ASMCs (P<0.01; Fig. 6B). As
illustrated in Fig. 6C and D, the MTT and Transwell assays revealed
that the viability and migration of ASMCs were higher in the
PDGF-BB group compared with the control group (P<0.01), and
knockdown of E2F3 visibly decreased the viability and migration of
PDGF-BB-induced ASMCs compared with the sh-NC group
(P<0.01).
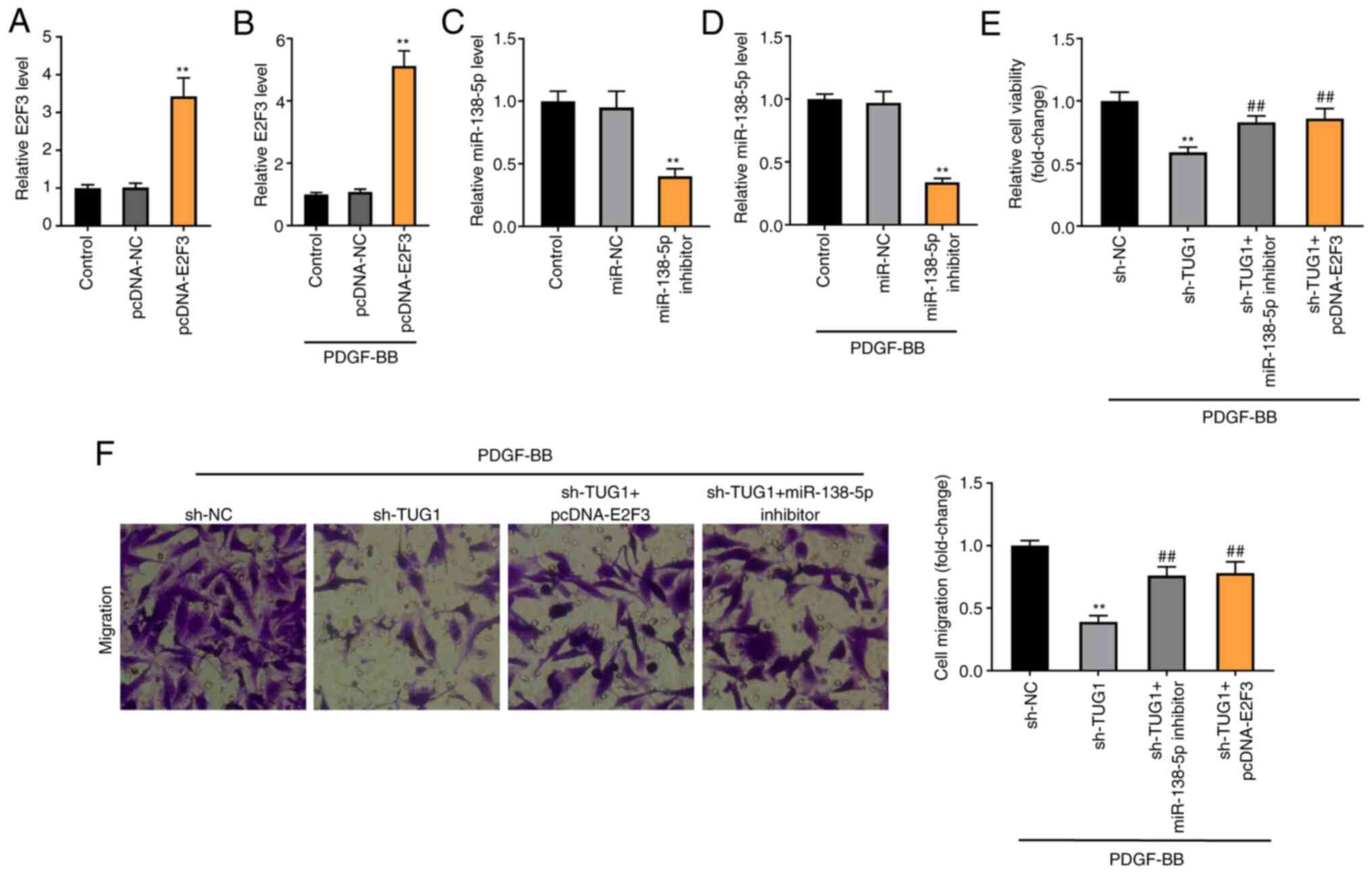
Knockdown of TUG1 decreases the
viability and migration of PDGF-BB-induced ASMCs via regulating
miR-138-5p/E2F3 axis
E2F3 expression was enhanced by the transfection of
pcDNA-E2F3 in both ASMCs and FDGF-BB-induced ASMCs (P<0.01;
Fig. 7A and B). miR-138-5p expression was inhibited by
the transfection of miR-138-5p inhibitor in both ASMCs and
FDGF-BB-induced ASMCs (P<0.01; Fig.
7C and D). To verify whether
TUG1 regulated the miR-138-5p/E2F3 axis in asthma, a feedback
experiment was performed using MTT and Transwell assay in
PDGF-BB-induced ASMCs. As illustrated in Fig. 7C and D, TUG1 knockdown visibly reduced the
viability and migration of PDGF-BB-induced ASMCs (P<0.01).
Moreover, miR-138-5p inhibition or E2F3 overexpression partially
reversed the inhibitory effects of sh-TUG1 on the viability and
migration of PDGF-BB-induced ASMCs (P<0.01).
Discussion
It has been documented that airway remodelling
accelerates structural changes to the airway in patients with
asthma, leading to irreversible or partially irreversible airflow
obstruction (24). In the present
study, the level of serum IgE, inspiratory resistance and
expiratory resistance were increased in a rat asthma model.
Previous studies have confirmed that the serum lgE level and airway
resistance are both elevated in asthma model rats (25,26).
These data indicated that the rat asthma model was constructed
successfully in the present study. Enhanced expression levels of
lncRNAs, including lncRNA TCF7(27), PVT1(28) and lncRNA LINC00882(29), have been discovered in cases of
asthma. In the present study, TUG1 expression was elevated in both
the rat asthma model and in PDGF-BB-induced ASMCs. Taking these
results into consideration, it can be hypothesized that TUG1 may be
a regulator of asthma.
Results from previous studies suggest that abnormal
growth of ASMCs leads to airway re-modelling in asthma via
thickening of the airway wall (30,31).
lncRNAs may serve as critical factors in the acceleration of the
asthma process. Zhang et al (26) suggested that lncRNA GAS5 binds to
miR-10a to upregulate BDNF expression, promoting ASMC proliferation
in asthma. Fan et al (27)
demonstrated that lncRNA TCF7 interacts with TIMMDC1 to facilitate
ASMC growth and migration in asthma by mediating AKT. Lin et
al (9) reported that TUG1
contributed to the proliferation and migration of ASMCs in asthma
through modulation of the miR-590-5p/FGF1 axis. In the present
study, TUG1 knockdown reduced the viability and migration of
PDGF-BB-induced ASMCs. The role of TUG1 was similar to that of the
aforementioned lncRNAs and suggests that TUG1 may be a potential
therapeutic target for asthma. To verify the anti-asthma effect of
TUG1 inhibition in vivo, an asthmatic rat model was
established. It was demonstrated that TUG1 knockdown inhibited
inspiratory and expiratory resistance in the model rats. These data
illustrate that TUG1 inhibition may alleviate the progression of
asthma in a rat model.
Several lncRNAs interact with miRNAs to regulate the
progression of asthma. Downregulation of lncRNA BCYRN1 interacts
with miR-150 to promote abnormal growth of ASMCs in asthmatic rats
(32). Silencing of lncRNA Malat1
suppresses proliferation and migration of PDGF-BB-induced ASMCs by
endogenously competing against miR-150(33). In the present study, miR-138-5p was
demonstrated to be a target of TUG1. An increasing number of
studies have shown that miR-138 is downregulated and serves a key
role in the regulation of asthma. miR-138 mediates a balance of
Th1/Th2 and attenuates CD4+ T cell proliferation by
targeting OX40L, which may inhibit the progression of asthma
(34). miR-138 expression is
decreased in human ASMCs, and miR-138 upregulation inhibits the
proliferation of ASMCs (14). In
the present study decreased miR-138-5p expression was observed in
PDGF-BB-induced ASMCs, and miR-138-5p inhibition partially reversed
the inhibitory effects of TUG1 knockdown on viability and migration
of PDGF-BB-induced ASMCs. These results demonstrated that TUG1 can
promote the abnormal growth of ASMCs via inhibition miR-138-5p in
asthma.
It has been documented that the expression of genes
in the E2F family is increased and these genes regulate the
progression of respiratory diseases. Inhibition of E2F-1 gene
expression can attenuate ASMC proliferation in asthma (35). E2F3 is highly expressed, and
knockdown of E2F3 reduces cell growth arrest and senescence in lung
cancer (36). In the present study,
loss-of-function experiments revealed that silencing of E2F3
reduced viability and migration of PDGF-BB-treated ASMCs,
suggesting that E2F3 silencing may attenuate the abnormal growth of
ASMCs. Prior studies have reported that lncRNAs serve as competing
endogenous RNAs to inhibit miRNA-mediated target gene
downregulation in the pathogenesis of asthma (37,38).
lncRNA PVT1 interacts with miR-203a to promote ASMC proliferation
and migration by increasing E2F3(20). TUG1 binds to miR-590-5p to increase
FGF1 expression, facilitating abnormal ASMC growth in asthma
(9). In the current study, E2F3 was
targeted by miR-138-5p, and miR-138-5p overexpression decreased
E2F3 expression in PDGF-BB-stimulated ASMCs. Considering the
interaction between TUG1 and miR-138-5p, it can be hypothesized
that TUG1 knockdown may inhibit E2F3 expression by increasing
miR-138-5p expression in PDGF-BB-stimulated ASMCs. Moreover,
feedback experiments indicated that E2F3 overexpression reversed
the inhibitory effects of TUG1 knockdown on viability and migration
of PDGF-BB-induced ASMCs. Taken together, these data suggest that
TUG1 may promote the viability and migration of ASMCs in asthma by
regulating the miR-138-5p/E2F3 axis.
In conclusion, TUG1 expression was upregulated in
asthmatic rats, and knockdown of TUG1 attenuated the progression of
asthma. Furthermore, TUG1 may contribute to the abnormal growth of
ASMCs by regulating the miR-138-5p/E2F3 axis. Therefore, TUG1 may
represent a promising therapeutic target for asthma.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
National Science and Technology Support Plan for the 12th Five-Year
Plan Period (grant no. 2012BAI04B01), the Hunan Key Laboratory of
Pediatric Emergency Medicine (grant no. 2018TP1028) and the Hunan
Provincial Health Commission Project (grant no. 20200611).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ conceived and designed the study, performed
experiments and analyzed the data and drafted the manuscript. CL
performed experiments and drafted the manuscript. PL, YC, LL and ZX
performed experiments and critically revised the manuscript. HZ and
CL confirm the authenticity of all the raw data. All authors read
and approved the final manuscript for publication.
Ethics approval and consent to
participate
Ethical approval was obtained from the Animal Ethics
Committee of Hunan Children's Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lambrecht BN, Hammad H and Fahy JV: The
cytokines of asthma. Immunity. 50:975–991. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Salter B, Pray C, Radford K, Martin JG and
Nair P: Regulation of human airway smooth muscle cell migration and
relevance to asthma. Respir Res. 18(156)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Israel E and Reddel HK: Severe and
difficult-to-treat asthma in adults. N Engl J Med. 377:965–976.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Nunes C, Pereira AM and Morais-Almeida M:
Asthma costs and social impact. Asthma Res Pract.
3(1)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Turner M, Galloway A and Vigorito E:
Noncoding RNA and its associated proteins as regulatory elements of
the immune system. Nat Immunol. 15:484–491. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Niu Y, Ma F, Huang W, Fang S, Li M, Wei T
and Guo L: Long non-coding RNA TUG1 is involved in cell growth and
chemoresistance of small cell lung cancer by regulating LIMK2b via
EZH2. Mol Cancer. 16(5)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu H, Zhou G, Fu X, Cui H, Pu G, Xiao Y,
Sun W, Dong X, Zhang L, Cao S, et al: Long noncoding RNA TUG1 is a
diagnostic factor in lung adenocarcinoma and suppresses apoptosis
via epigenetic silencing of BAX. Oncotarget. 8:101899–101910.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yang L, Liang H, Shen L, Guan Z and Meng
X: LncRNA Tug1 involves in the pulmonary vascular remodeling in
mice with hypoxic pulmonary hypertension via the
microRNA-374c-mediated Foxc1. Life Sci. 237(116769)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lin J, Feng X, Zhang J and Tong Z: Long
noncoding RNA TUG1 promotes airway smooth muscle cells
proliferation and migration via sponging miR-590-5p/FGF1 in asthma.
Am J Transl Res. 11:3159–3166. 2019.PubMed/NCBI
|
|
10
|
Yoo JK, Lee JM, Kang SH, Jeon SH, Kim CM,
Oh SH, Kim CH, Kim NK and Kim JK: The novel microRNA hsa-miR-CHA1
regulates cell proliferation and apoptosis in human lung cancer by
targeting XIAP. Lung Cancer. 132:99–106. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang Y, Xue Y, Liu Y, Song G, Lv G, Wang
Y, Wang Y, Li X and Yang L: MicroRNA-146a expression inhibits the
proliferation and promotes the apoptosis of bronchial smooth muscle
cells in asthma by directly targeting the epidermal growth factor
receptor. Exp Ther Med. 12:854–858. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gao Y, Wang B, Luo H, Zhang Q and Xu M:
miR-217 represses TGF-β1-induced airway smooth muscle cell
proliferation and migration through targeting ZEB1. Biomed
Pharmacother. 108:27–35. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang J, Wang HS and Su ZB: MicroRNA-142
inhibits proliferation and promotes apoptosis in airway smooth
muscle cells during airway remodeling in asthmatic rats via the
inhibition of TGF-β-dependent EGFR signaling pathway. Cell Physiol
Biochem. 47:1682–1695. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu Y, Yang K, Sun X, Fang P, Shi H, Xu J,
Xie M and Li M: MiR-138 suppresses airway smooth muscle cell
proliferation through the PI3K/AKT signaling pathway by targeting
PDK1. Exp Lung Res. 41:363–369. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yan Z, Bi M, Zhang Q, Song Y and Hong S:
LncRNA TUG1 promotes the progression of colorectal cancer via the
miR-138-5p/ZEB2 axis. Biosci Rep. 40(BSR20201025)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhu J, Shi H, Liu H, Wang X and Li F: Long
non-coding RNA TUG1 promotes cervical cancer progression by
regulating the miR-138-5p-SIRT1 axis. Oncotarget. 8:65253–65264.
2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Humbert PO, Verona R, Trimarchi JM, Rogers
C, Dandapani S and Lees JA: E2F3 is crucial for normal cellular
proliferation. Genes Dev. 14:690–703. 2000.PubMed/NCBI
|
|
18
|
Cooper CS, Nicholson AG, Foster C, Dodson
A, Edwards S, Fletcher A, Roe T, Clark J, Joshi A, Norman A, et al:
Nuclear overexpression of the E2F3 transcription factor in human
lung cancer. Lung Cancer. 54:155–162. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Al Ahmed HA and Nada O: E2F3 transcription
factor: A promising biomarker in lung cancer. Cancer Biomark.
19:21–26. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yu X, Zhe Z, Tang B, Li S, Tang L, Wu Y,
Chen X and Fang H: α-Asarone suppresses the proliferation and
migration of ASMCs through targeting the lncRNA-PVT1/miR-203a/E2F3
signal pathway in RSV-infected rats. Acta Biochim Biophys Sin
(Shanghai). 49:598–608. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen G and Khalil N: TGF-beta1 increases
proliferation of airway smooth muscle cells by phosphorylation of
map kinases. Respir Res. 7(2)2006.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhou H, Wu Q, Wei L and Peng S:
Paeoniflorin inhibits PDGF-BB-induced human airway smooth muscle
cell growth and migration. Mol Med Rep. 17:2660–2664.
2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Najafi A, Masoudi-Nejad A, Ghanei M,
Nourani MR and Moeini A: Pathway reconstruction of airway
remodeling in chronic lung diseases: A systems biology approach.
PLoS One. 9(e100094)2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang XY, Zhang LX, Tian CJ, Tang XY, Zhao
LM, Guo YL, Cheng DJ, Chen XL, Ma LJ and Chen ZC: LncRNAs BCYRN1
promoted the proliferation and migration of rat airway smooth
muscle cells in asthma via upregulating the expression of transient
receptor potential 1. Am J Transl Res. 8:3409–3418. 2016.PubMed/NCBI
|
|
26
|
Zhang XY, Tang XY, Li N, Zhao LM, Guo YL,
Li XS, Tian CJ, Cheng DJ, Chen ZC and Zhang LX: GAS5 promotes
airway smooth muscle cell proliferation in asthma via controlling
miR-10a/BDNF signaling pathway. Life Sci. 212:93–101.
2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Fan M, Xu J, Xiao Q, Chen F and Han X:
Long non-coding RNA TCF7 contributes to the growth and migration of
airway smooth muscle cells in asthma through targeting TIMMDC1/Akt
axis. Biochem Biophys Res Commun. 508:749–755. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Austin PJ, Tsitsiou E, Boardman C, Jones
SW, Lindsay MA, Adcock IM, Chung KF and Perry MM: Transcriptional
profiling identifies the long noncoding RNA plasmacytoma variant
translocation (PVT1) as a novel regulator of the asthmatic
phenotype in human airway smooth muscle. J Allergy Clin Immunol.
139:780–789. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liu Z, Mei L and He Z: Long non-coding
RNA00882 contributes to platelet-derived growth factor-induced
proliferation of human fetal airway smooth muscle cells by
enhancing Wnt/beta-catenin signaling via sponging miR-3619-5p.
Biochem Biophys Res Commun. 514:9–15. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Keglowich LF and Borger P: The three A's
in asthma-airway smooth muscle, airway remodeling &
angiogenesis. Open Respir Med J. 9:70–80. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Perry MM, Baker JE, Gibeon DS, Adcock IM
and Chung KF: Airway smooth muscle hyperproliferation is regulated
by microRNA-221 in severe asthma. Am J Respir Cell Mol Biol.
50:7–17. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhang XY, Tang XY, Ma LJ, Guo YL, Li XS,
Zhao LM, Tian CJ, Cheng DJ, Chen ZC and Zhang LX: Schisandrin B
down-regulated lncRNA BCYRN1 expression of airway smooth muscle
cells by improving miR-150 expression to inhibit the proliferation
and migration of ASMC in asthmatic rats. Cell Prolif.
50(e12382)2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lin L, Li Q, Hao W, Zhang Y, Zhao L and
Han W: Upregulation of LncRNA Malat1 induced proliferation and
migration of airway smooth muscle cells via miR-150-eIF4E/Akt
signaling. Front Physiol. 10(1337)2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Huang L, Wang M, Chen Z, Yan Y, Gu W,
Zhang X, Tan J, Sun H and Ji W: MiR-138 regulates dendritic cells
mediated Th2-type immune response by regulating the OX40L
expression in asthma. Int J Clin Exp Pathol. 10:10979–10988.
2017.PubMed/NCBI
|
|
35
|
Amrani Y, Tliba O, Choubey D, Huang CD,
Krymskaya VP, Eszterhas A, Lazaar AL and Panettieri RA Jr:
IFN-gamma inhibits human airway smooth muscle cell proliferation by
modulating the E2F-1/Rb pathway. Am J Physiol Lung Cell Mol
Physiol. 284:L1063–L1071. 2003.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ren XS, Yin MH, Zhang X, Wang Z, Feng SP,
Wang GX, Luo YJ, Liang PZ, Yang XQ, He JX and Zhang BL:
Tumor-suppressive microRNA-449a induces growth arrest and
senescence by targeting E2F3 in human lung cancer cells. Cancer
Lett. 344:195–203. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Qiu YY, Wu Y, Lin MJ, Bian T, Xiao YL and
Qin C: LncRNA-MEG3 functions as a competing endogenous RNA to
regulate Treg/Th17 balance in patients with asthma by targeting
microRNA-17/ RORγt. Biomed Pharmacother. 111:386–394.
2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liang Z and Tang F: The potency of lncRNA
MALAT1/miR-155/CTLA4 axis in altering Th1/Th2 balance of asthma.
Biosci Rep. 40(BSR20190397)2020.PubMed/NCBI View Article : Google Scholar
|