Introduction
Thalassemia is an inherited disease associated with reduced synthesis of one or more globin chains and resulting in a defective hemoglobin (Hb) production and earlier damage to red blood cells (1,2). Hemoglobin E (HbE)/β-thalassemia is the most common severe thalassemia in South and Southeast Asian countries (3). Clinical burden of HbE/β-thalassemia varies markedly, and ranges from mild or asymptomatic anemia (β-thalassemia trait) to severe anemia (HbE/β-thalassemia major), which requires blood transfusions (4). Microcytic hypochromic anemia and iron overload represent the most frequent complications and major challenges in managing thalassemia (5). Iron is essential to numerous biological processes, such as cellular respiration and oxygen transport (6). Iron homeostasis must be maintained to avoid iron overload and iron deficiency, which can have severe consequences (7). Cardiomyopathy, which is a common cause of death, can be caused by cardiac iron overload in patients with transfusion-dependent thalassemia (8,9). Patients with non-transfusion-dependent thalassemia are also at high risk for iron overload and its consequences, such as liver cirrhosis (10). Severe thalassemia patients require blood transfusion to improve anemia via suppression of erythropoiesis (11,12). Because HbE/β-thalassemia is associated with increased serum iron, regular evaluation of iron level in these patients is therefore required (13,14).
Hepcidin (HEPC) is a hormone that is produced by the liver and released into the circulation (15,16). HEPC interacts with ferroportin1 (FPN1), a HEPC-receptor and an iron exporter protein, which is situated on the plasma membrane of iron exporting cells, such as duodenal enterocytes, hepatocytes, spleen cells (17) and erythropoietic cells (18). FPN1 exports iron from cells into the blood (19,20). FPN1/HEPC complex inhibits the ability of these cells to export iron (21). Therefore, higher HEPC expression will reduce iron export from storage cells into the blood via suppression of FPN1(18). HEPC production is regulated by erythropoiesis (22). Increased erythropoiesis reduces HEPC production and increases iron level due to the high release of iron from storage cells and enteric absorption for Hb synthesis (23).
The present study is a comparative cross-sectional study, in which a total of 260 participants, including 65 HbE/β thalassemia patients (pretransfusion), 65 HbE trait and β-thalassemia trait (patients' parents) and 130 healthy controls were involved for the evaluations of iron profile, HEPC gene expression and serum HEPC level. The aim of the present study was to investigate the association between HEPC expression and serum levels and iron overload among HbE/β-thalassemia patients.
Materials and methods
Samples recruitment and ethics
The present study is a comparative cross-sectional study that included a total of 260 blood samples (n=260) collected from 65 HbE/β-thalassemia patients and 65 parents at the Hematology Department of Hospital Sultanah Nur Zahirah, Terengganu. A total of 130 blood samples were collected from healthy postgraduate students at the Universiti Sultan Zainal Abidin (UniSZA), Gong Badak Campus, in Terengganu, Malaysia, who were included as the controls. All blood samples were collected between August 2019 and July 2020. The study was approved by UniSZA Human Research Ethics Committee [(approval no. UniSZA.C/2/UHREC/628-2 J1d.2) (73)] and the Medical Research and Ethics Committee [(approval no. KKM/NIHSEC/P19-1143) (11)]. The procedures were in accordance with the ethical standards and each participant to this study was given a participant information sheet (PIS) and an informed consent form (ICF) prior to blood samples collection. Both PIS and ICF were written in Bahasa Melayu and English languages to allow better understanding from all participants. Briefly, 5 ml of blood were collected from each participant by a trained nurse staff. The inclusion criteria were as follows: i) Transfusion dependent HbE/β-thalassemia patients (pretransfusion) and their parents (HbE trait and β-thalassemia trait); ii) healthy participants with normal hematological blood profile and hemoglobin analysis; and iii) transfusion dependent HbE/β-thalassemia patients who required lifelong regular blood transfusion. The exclusion criteria were as follows: i) Patients or parents with iron deficiency anemia (IDA) or alpha thalassemia trait (DNA analysis for alpha globin gene was performed); and ii) healthy participants with low hemoglobin and red cell indices, abnormal hemoglobin variants.
Laboratory investigations
Capillary electrophoresis and the VARIANTTM II β-Thalassemia Short Program Reorder Pack (Bio-Rad Laboratories, Inc.), which is a fully automated high-performance liquid chromatography (HPLC) system, were used to confirm the patient's diagnosis and ensure that healthy controls were thalassemia-free. Complete blood counts [red blood cells (RBCs), Hb, packed cell volume (PCV), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC) and red cell distribution width (RDW)] were analyzed using SYSMEX XN-1000 automated Hematology Analyzer (Sysmex America, Inc.). Iron profile [serum iron, total iron binding capacity (TIBC) and unsaturated iron binding capacity (UIBC)] was determined using UniCel® DxI 600 Access® Immunoassay (Beckman Coulter Inc.) and serum ferritin level was estimated using UniCel® DxI 800 (B) Access® Immunoassay (Beckman Coulter Inc.). Serum HEPC level was determined using Human HEPC ELISA Kit (cat. no. E-EL-H0077; Elabscience Biotechnology Co., Ltd.) according to the manufacturer's protocol.
RNA extraction and cDNA synthesis
Total RNA was extracted from the peripheral blood samples of all participants using QIAamp RNA Blood Mini Kit (Qiagen GmbH) according to the manufacturer's protocol. RNA purity and concentration were evaluated via a NanoPhotometer® NP80 (Implen GmbH).
Reverse transcription quantitative (RT-q)PCR
GoTaq 2-Step RT-qPCR (Promega Corporation) was used to synthesize cDNA from RNA samples (100 ng) according to the manufacturer's protocol. All PCR amplifications were conducted using 2 µl of cDNA mixed with 20 µl of GoTaq PCR master mix, following a 2-step amplification protocol as follows: A starting denaturing step for 2 min at 95˚C, followed by 40 cycles of denaturation for 15 sec at 95˚C, and annealing and extension for 1 min at 60˚C by using 1-Step RT-qPCR Systems (Applied Biosystems). Data were analyzed by 1-Step One Software v2.3 (Applied Biosystems). Beta actin (β-actin) was used as the reference gene. The relative expression levels of HPEPC and FPN1 were normalized to endogenous control and were expressed as 2-ΔΔCq (24). The sequences of the primers used are listed in Table I. All experiments were performed in triplicates.
 |
Table I
Sequences of the primers used for reverse transcription quantitative analysis.
|
Table I
Sequences of the primers used for reverse transcription quantitative analysis.
| Gene name |
Forward primer |
Reverse primer |
Accession no. |
| Hepcidin |
5'-TTTCCCACAACAGACGGGAC-3' |
5'-AGCTGGCCCTGGCTCC-3' |
NM_032541 |
| Ferroportin1 |
5'-TTGCCGGAGTCATTGCTGCTA-3' |
5'-TGGAGTTCTGTACACCATT-3' |
NM_016917 |
| β-actin |
5'-GAGCGCGGCTACAGCTT-3' |
5'-TCCTTAATGTCACGCACGATTT-3' |
NM_007393 |
Statistical analysis
All statistical analyses were performed using SPSS software (version 20; IBM Corp.). Kruskal Wallis was applied for data comparison and P<0.05 was considered to indicate a statistically significant difference. Multiple Mann-Whitney Test with Bonferroni correction was applied for 2-group comparisons and the level of significance was set at 0.017.
Results
Demographic data
The study included 65 transfusion-dependent HbE/β-thalassemia patients, 65 parents and 130 healthy controls. All groups comprised 100 men and 160 women. The majority of HbE/β-thalassemia patients were aged between 11 and 32 years, while parents were aged between 39 and 56 years. Furthermore, the median age of healthy controls was between 18 and 32 years (Table II).
|
Table II
Demographic data of HbE/β-thalassemia patients, parents and healthy controls.
|
Table II
Demographic data of HbE/β-thalassemia patients, parents and healthy controls.
| Characteristics |
Healthy controls (n=130) |
Parents (n=65) |
HbE/β-thalassemia patients (n=65) |
| Age range, years |
18-32 |
39-56 |
11-32 |
| Sex |
|
|
|
| Male, n (%) |
31 (23.8%) |
26 (40.0%) |
43 (66.2%) |
| Female, n (%) |
99 (76.25) |
39 (60.0%) |
22 (33.8%) |
Increased HbE and HbF in HbE/β-thalassemia patients
To confirm the diagnosis of HbE/β-thalassemia in patients, their parents and thalassemia-free controls, HPLC analysis and capillary electrophoresis was performed for all blood samples from all participants prior to further laboratory analysis. The results demonstrated a significant decrease in HbA in HbE/β-thalassemia patients compared with parents (P=0.001) and healthy controls (P<0.001; Table III). However, there was no significant difference in HbA of parents compared to healthy controls (P=0.056). Furthermore, there was a significant increase in HbF (P=0.001) and HbE (P<0.001) in HbE/β-thalassemia patients compared with their parents and healthy controls. The results also showed a significant increased HbF in parents compared to healthy controls (P=0.006).
|
Table III
Comparison between medians of hemoglobin (Hb) variants in HbE/β-thalassemia patients, parents and healthy controls.
|
Table III
Comparison between medians of hemoglobin (Hb) variants in HbE/β-thalassemia patients, parents and healthy controls.
| Studied variables |
Healthy controls (n=130) Median (Iq) |
Parents (n=65), median (Iq) |
HbE/β-thalassemia patients (n=65), median (Iq) |
X2-statistica (df) |
P-valueb |
Reference range |
| HbA, % |
96.20 (4.03) |
81.80c (20.60) |
2.30 (2.35) |
203.9(2) |
0.001 |
>90.0 |
| HbA2, % |
2.80 (0.50) |
4.80 (0.50) |
4.80 (2.15) |
175.8(2) |
0.001 |
2.0-3.3 |
| HbF, % |
0.30 (0.11) |
0.80c (0.65) |
47.2 (11.60) |
181.8(2) |
0.001 |
<1.0 |
| HbE, % |
0.00 |
0.20c (1.10) |
44.6 (10.58) |
89.00(2) |
0.000 |
- |
Reduced Hb concentration and red cell indices in thalassemia patients
There was a significant decrease in RBCs count, Hb concentration and red cell indices (except RDW) in HbE/β-thalassemia patients and their parents (P=0.001) compared with healthy controls. The results also revealed a significant decrease in Hb concentration of parents compared to healthy controls (P=0.003) (Table IV).
|
Table IV
Comparison between median haematological parameters in HbE/β-thalassemia patients, parents and healthy controls.
|
Table IV
Comparison between median haematological parameters in HbE/β-thalassemia patients, parents and healthy controls.
| Studied variables |
Healthy controls (n=130), median (Iq) |
Parents (n=65), median (Iq) |
HbE/β-thalassemia patients (n=65), median (Iq) |
X2-statistica (df) |
P-valueb |
Reference range |
| RBCSs, x1012/l |
4.54 (0.51) |
5.40 (0.84) |
3.00 (0.710) |
163.5(2) |
0.001 |
4.50-5.50 |
| Hb, g/l |
132.0 (15.0) |
121.0 (19.00) |
75.10 (22.90) |
166.8(2) |
0.001 |
120-150 |
| PCV, l/l |
0.400 (0.05) |
0.390 (0.040) |
0.220 (0.04) |
147.5(2) |
0.001 |
0.350-0.450 |
| MCV, fl |
88.0 (4.93) |
64.90 (14.85) |
62.30 (6.85) |
189.3(2) |
0.001 |
83.0-101.0 |
| MCH, pg |
28.95 (1.80) |
20.50 (4.10) |
20.40 (4.10) |
143.9(2) |
0.001 |
27.0-32.0 |
| MCHC, g/l |
329.5 (12.25) |
319.0 (24.0) |
301.0 (36.50) |
125.04(2) |
0.001 |
315.0-345.0 |
| RDW, % CV |
13.20 (1.60) |
15.70 (4.30) |
24.60 (6.40) |
172.7(2) |
0.001 |
11.6-14.8 |
Increased serum ferritin in thalassemia patients
The results from iron profile revealed a significant increase in serum ferritin in HbE/β-thalassemia patients and their parents (P<0.001) compared with healthy controls. However, serum ferritin level was >20 times higher in HbE/β-thalassemia patients than in healthy volunteers (Table V). Serum iron and transferrin saturation were also significantly increased in HbE/β-thalassemia patients (P=0.001) compared with parents and healthy controls. Serum iron in parents were significantly increased compared to healthy controls (P=0.016). However, TIBC and UIBC were significantly decreased in HbE/β-thalassemia patients compared with parents and healthy controls (P=0.001; Table V and Fig. 1).
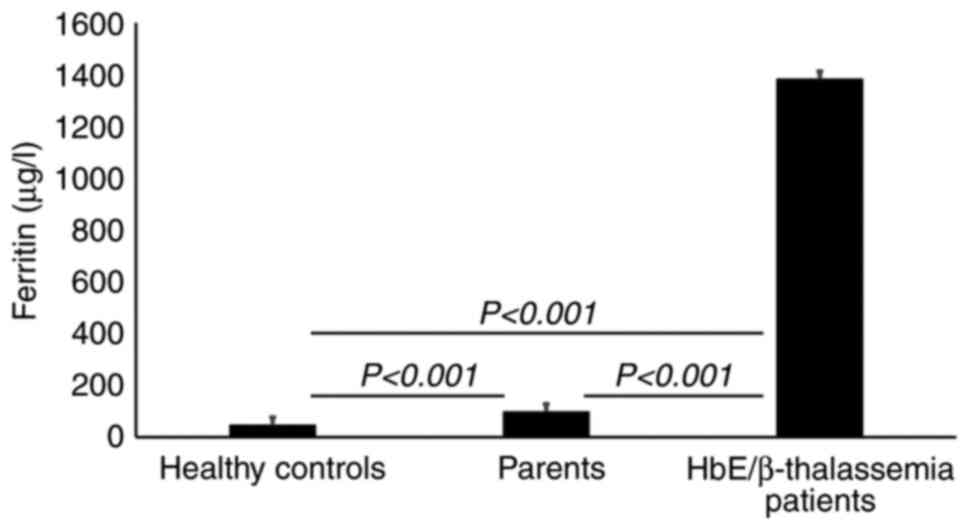 |
Figure 1
Serum ferritin concentrations. Results are presented as the median ferritin concentrations and show a significant increased ferritin level in HbE/β-thalassemia patients and their parents (P<0.001) compared with healthy controls. HbE/β-thalassemia, hemoglobin E/β-thalassemia.
|
|
Table V
Median iron profile and hepcidin levels in HbE/β-thalassemia patients, parents and healthy controls.
|
Table V
Median iron profile and hepcidin levels in HbE/β-thalassemia patients, parents and healthy controls.
| Studied variables |
Healthy controls (n=130), median (Iq) |
Parents (n=65), median (Iq) |
HbE/β-thalassemia median (Iq) |
X2-patients (n=65), statistica (df) |
P-valueb |
Reference range |
| Iron, µmol/l |
18.60 (8.65) |
24.40 (6.70) |
32.90 (10.15) |
601.5(2) |
0.001 |
10.7-32.2 |
| UIBC, µmol/l |
45.95 (14.90) |
43.10 (12.60) |
22.30 (6.80) |
89.29(2) |
0.001 |
27.8-63.6 |
| TIBC, µmol/l |
60.45 (8.72) |
57.90 (12.00) |
37.10 (8.40) |
97.29(2) |
0.001 |
42.96-80.55 |
| Transferrin saturated, % |
23.50 (12.25) |
25.00 (12.00) |
54.80 (15.20) |
77.90(2) |
0.001 |
15-50% |
| Ferritin, µg/l |
46.20 (44.23) |
97.50 (80.60) |
1383.0 (1098.8) |
1324.0(2) |
0.000 |
11.0-306.8 |
| Hepcidin, ng/ml |
4.823 (5.235) |
0.590 (0.056) |
0.0489 (0.043) |
101.2(2) |
0.001 |
0.16-10 |
| Hepcidin/ferritin |
0.3365 (0.467) |
0.033 (0.029) |
0.0115 (0.009) |
87.9(2) |
0.001 |
Up to 1 |
Reduced serum HEPC level in HbE/β-thalassemia patients and parents
Serum HEPC level was significantly reduced in HbE/β-thalassemia and their parents (P<0.001) compared with healthy controls (Table V and Fig. 2). However, there was no significant difference in the HEPC level between HbE/β-thalassemia and their parents (P=0.208).
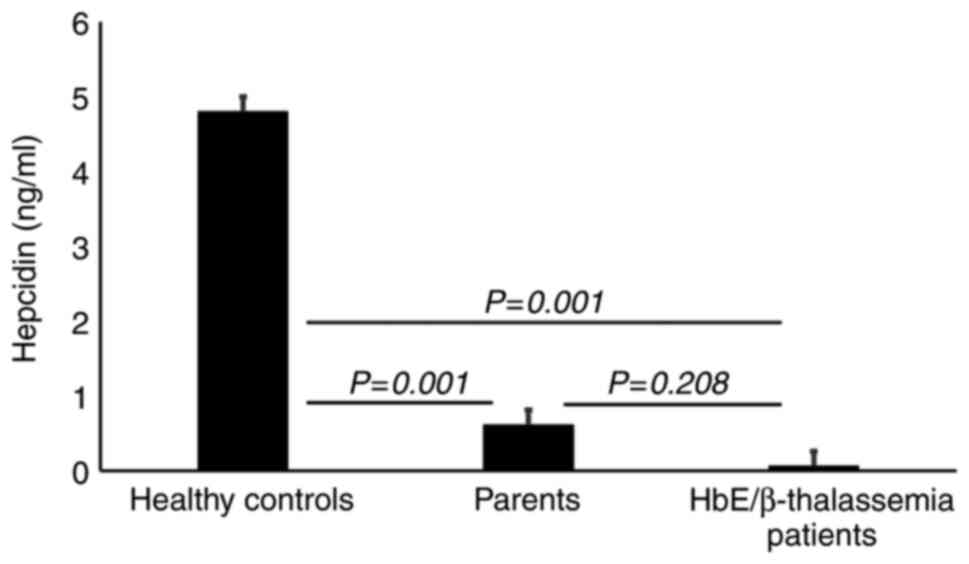 |
Figure 2
Serum hepcidin concentrations. Results show a significantly lower HEPC concentration in HbE/β-thalassemia patients (P=0.001) compared with their parents and healthy controls. Results also demonstrate a significant decrease in HEPC concentration in parents (P=0.001) compared with healthy controls. HbE/β-thalassemia, hemoglobin E/β-thalassemia.
|
HEPC downregulation in HbE/β-thalassemia patients and parents
Six blood samples of each group were randomly selected for gene expression analysis by RT-qPCR. The results demonstarted a significant lower HEPC gene expression in HbE/β-thalassemia patients and their parents (P=0.001) compared with healthy controls. However, there was no significant difference in HEPC gene expression between HbE/β-thalassemia patients and their parents (P=0.208; Fig. 3).
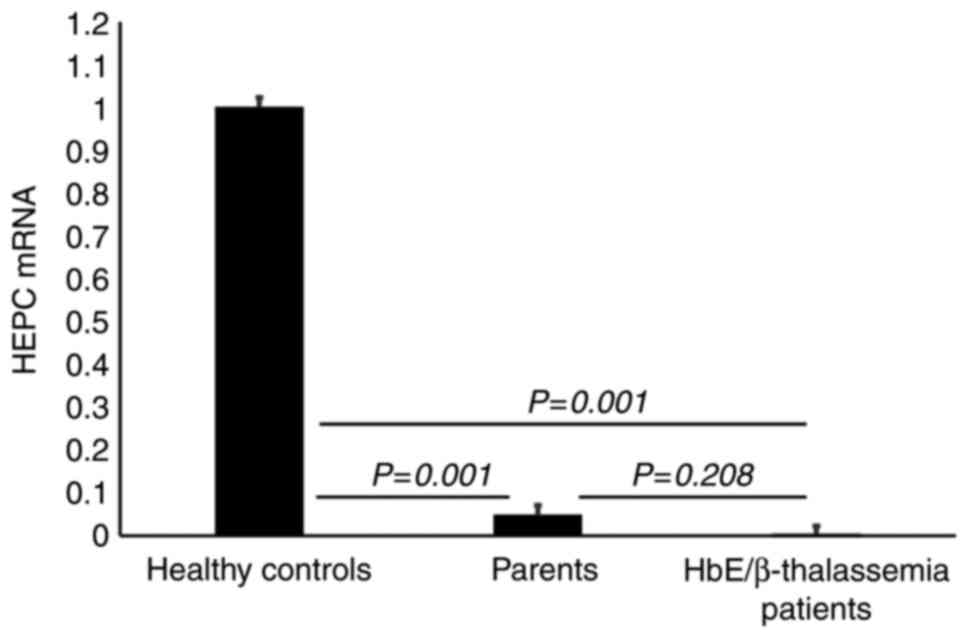 |
Figure 3
Gene expression analysis of HEPC. Results display a significant downregulation of HEPC gene expression in HbE/β-thalassemia patients and their parents (P=0.001) compared with healthy controls. However, there was no significant difference in HEPC gene expression in HbE/β-thalassemia patients (P=0.208) compared to their parents. HEPC, hepcidin; HbE/β-thalassemia, hemoglobin E/β-thalassemia.
|
Upregulated FPN1 in HbE/β-thalassemia patients and parents
The results from RT-qPCR demonstrated a significant upregulation (P=0.001) of FPN1 gene in HbE/β-thalassemia patients and their parents (7-folds and 3-folds higher, respectively) compared with healthy volunteers (Fig. 4).
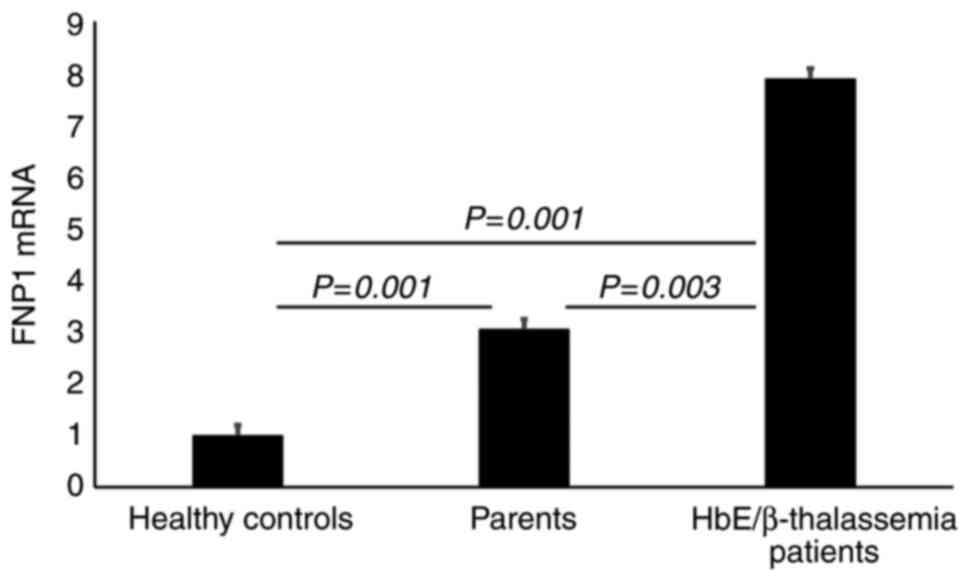 |
Figure 4
Gene expression analysis of FPN1. Results show a significant upregulation of FPN1 gene expression (P=0.001) in HbE/β thalassemia patients (>7-fold change) and their parents (>3-fold change) compared with healthy controls (P=0.001). Results also display a significant upregulation of FPN1 gene expression in HbE/β-thalassemia patients (P=0.003) compared with their parents. HbE/β-thalassemia, hemoglobin E/β-thalassemia.
|
Discussion
HbE/β-thalassemia remains one of the most common inherited blood disorders, which is characterized by an abnormal β-globin chain production. HbE/β-thalassemia is prevalent worldwide and represents a major health problem in numerous countries, such as Southeast Asian countries like Malaysia (3,25). The complication arising from this blood disorder is not only due to ineffective erythropoiesis but also to iron overload due to increased gastrointestinal iron absorption and blood transfusions (26). Iron overload in β-thalassemia patients is a major cause of mortality and morbidity (27).
Iron deposition leads to a marked cellular damage and organ dysfunction (28,29). Excess iron deposition is associated with cardiac hypertrophy and dilatation (30,31). Long term iron deposition also damages thyroid, parathyroid and adrenal glands (32). HEPC is a key regulator of iron homeostasis produced by hepatocytes and regulating intestinal iron absorption (16,33). FPN1 serves a key role in regulating dietary iron absorption in the duodenum, whereas serum HEPC levels control the concentration of FPN1 on the basolateral cell surface of enterocytes (34). The interplay between the iron exporter FPN1 and HEPC is critical for iron homeostasis, inflammation and anemia (35).
In the present study, the diagnosis of HbE/β-thalassemia patients, their parents and thalassemia-free healthy controls was confirmed by capillary electrophoresis and HPLC analysis. As a novelty of the present study, gene expression of HEPC and FPN1 together with serum HEPC level were studied in HbE//β-thalassemia patients and their parents and compared with those in healthy controls. The results revealed a significant downregulation of HEPC in HbE/β-thalassemia patients and their parents (P=0.001) compared with healthy controls. The downregulated HEPC was associated with a significant increase in FPN1 expression level in HbE/β-thalassemia patients and their parents (P=0.001) compared with healthy controls. These findings were similar to previous studies reporting that HEPC expression in β-thalassemia is downregulated in β-thalassemia mice (36,37). However, FPN1 is upregulated in β-thalassemia mice (38). The expression of fibroblast growth factor 23 (FGF23) was also reported as a primary regulator of hepcidin expression (39-41). Subsequently, the FGF23 expression level and its association with HEPC expression level in HbE/β-thalassemia patients will be further investigated.
Serum HEPC level, complete blood count and iron profile were also investigated in all participants from the present study. The results revealed that serum HEPC levels were significantly lower in HbE/β-thalassemia patients (pre-transfusion) and their parents (P=0.001) compared with healthy controls. These findings were supported by a previous study reporting that serum HEPC levels were decreased in β-thalassemia trait patients compared with healthy controls (42). However, the present findings were opposite to those from a previous study, in which serum HEPC levels were similar in β-thalassemia patients and healthy controls, which was attributed to the blood transfusion (11). Furthermore, HEPC was shown to be upregulated by increasing the body's iron but it downregulated due to ineffective erythropoiesis (33).
The serum HEPC levels were shown to be decreased in patients with β-thalassemia major compared with healthy controls (43). Furthermore, serum iron is increased in patients with β-thalassemia major compared with β-thalassemia trait patients and healthy controls (11,12). The increase in serum ferritin in β-thalassemia patients is mainly due to the suppression of HEPC caused by ineffective erythropoiesis which then increases iron absorption (44). Consistent with these results, the findings from the present study revealed a significantly lower HEPC serum level combined with a higher ferritin serum level in HbE/β-thalassemia patients and their parents compared with healthy volunteers. In addition, estimation of serum ferritin is a critical tool to evaluate the total body iron content (45). HEPC/ferritin ratio is therefore a marker of iron overload (42,46). Furthermore, the present study demonstrated that the HEPC/ferritin ratio was significantly decreased in HbE/β-thalassemia patients and their parents (P=0.001) compared with healthy controls. These findings were similar to a previous study in which HEPC/ferritin ratio is significantly decreased in β-thalassemia patients compared with controls (47).
The results from the present study revealed a significant increase in serum iron (P=0.001) in HbE/β-thalassemia patients compared with their parents and healthy controls. However, there was a significant decrease in TIBC and UIBC (P=0.001) in HbE/β-thalassemia patients compared with the two other groups. These findings were in agreement with previous studies reporting that TIBC and UIBC are significantly decreased in β-thalassemia patients compared with healthy controls (48,49).
The present study reported an elevated serum level of ferritin, which is the most significant indicator for iron overload, in HbE/β-thalassemia patients and their parents compared with healthy controls. The elevation of ferritin was attributed to the enhanced iron absorption in gastrointestinal tract due to the significant reduction of HEPC level in these patients.
Hemoglobin A (HbA) is the most dominant type of hemoglobin found in healthy adults and is made up of two α- and two β-globin chains (50,51). The normal synthesis of β-globin chains was found to be reduced in HbE/β-thalassemia patients (2,52). Similarly, the results from the present study showed lower HbA levels in HbE/β-thalassemia patients compared to the reference values. A decreased synthesis of HbA causes anemia in HbE/β-thalassemia patients due to the lack of oxygen transportation to various parts of the body (53,54). The low level of HbA would eventually lead to needed blood transfusion in severe cases of HbE/β-thalassemia patients in order to meet the body's oxygen demand (4). The main characteristics of HbE/β-thalassemia patients are higher blood concentrations in HbA2, HbF and HbE (38). Due to the nature of HbE/β-thalassemia, the production of δ-globin chain increases with a reduction in β-globin chains, leading to a raised levels of HbA2 (25). In a normal healthy adult, the β-globin gene is expressed at low level during the early fetal life, followed by HbF switch to HbA within three to six months, as γ-globin chain is replaced by β-globin chain (55). However, the conversion of HbF to HbA is delayed in HbE/β-thalassemia patients due to prolonged expression of the γ-globin chain since the β-globin gene is impaired (25). Similarly, the findings from the present study demonstrated a significant increase in HbF level (P=0.001) in HbE/β-thalassemia patients as one of the dominant hemoglobin variants instead of HbA. Furthermore, the present study also reported a significant elevation of HbE level in HbE/β-thalassemia patients, which was not detected in healthy controls. HbE is the abnormal hemoglobin variant that is caused by point mutation in the β chain, where glutamic acid is replaced by lysine (52). The dominant hemoglobins in HbE/β-thalassemia patients were HbE and HbF compared with the dominant HbA found in healthy volunteers (56).
A previous study reported an impairment in normal β chain production in HbE/β-thalassemia patients (52). Similarly, the results from the present study showed a severe decrease in HbA level in HbE/β-thalassemia patients compared with a mild reduction of HbA level in the parents or normal HbA level in healthy controls. The results also demonstrated that the decrease in HbA level was associated with a significant decreased in RBCs, Hb, PCV, MCV, MCH and MCHC levels (P<0.001) in HbE/β-thalassemia patients compared to parents and healthy controls. Similarly, the hemoglobin level ranged from 3 to 11 g/dl depending on the phenotypes and severity of HbE/β-thalassemia patients (52). A lower hemoglobin level indicates a hypochromic state in HbE/β-thalassemia patients, where erythrocytes appear paler than normal (57). The anemic nature of HbE/β-thalassemia patients are characterized by low PCV, MCV and MCH levels (52).
Taken together, the findings from the present study reported a significant upregulation of FPN1 gene (P=0.001) associated with a significant downregulated of HEPC gene in patients with HbE/β-thalassemia and their parents (P=0.001) compared with healthy volunteers. The downregulation of HEPC was associated with a significantly lower serum HEPC level and a significant increase in serum ferritin level in HbE/β-thalassemia patients and their parents. These findings suggested that the suppression of HEPC loses its negative regulatory function on FPN1, which resulted in increased iron and ferritin levels in HbE/β-thalassemia patients and their parents due to increased iron absorption.
In conclusion, this study demonstrated a significant upregulation of FPN1 associated with a significant downregulation of HEPC in HbE/β-thalassemia patients, which may contribute to an elevated iron burden in HbE/β-thalassemia patients. The findings from the present study suggested that evaluation of HEPC and FPN1 expression levels may be considered as useful indicators of iron toxicity risks in patients with HbE/β-thalassemia and β-thalassemia, and both genes may subsequently be used as future therapeutic targets in these patients.
Acknowledgements
The authors would like to thank Dr Alawiyah Awang Abd Rahman and Dr Azly Sumanty Binti Ab Ghani [Hospital Sultanah Nur Zahirah (HSNZ)] for their cooperation and help to perform all medical laboratory analyses at HSNZ. The authors would also like to thank Ms. Kasmawati Binti Abu Bakar, Mrs and Norashikin Mustafa (Pediatric Ward, HSNZ) for their help in collecting blood samples from patients and data.
Funding
This study was supported by Universiti Sultan Zainal Abidin [grant no. UniSZA/2017/DPU/41(R0018-R345)].
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
HANAJ and WRWT designed and conceptualized the study. Data collection and experiment implementation were performed by HKMS. Data were analysed by HANAJ, WRWT, II, MFJ, ASAW and HKMS. The manuscript was drafted by HANAJ and HKMS. HANAJ, WRWT, II, MFJ, ASAW and HKMS revised and reviewed the manuscript. Supervision was provided by HANAJ and WRWT. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by UniSZA Human Research Ethics Committee (UHREC) [approval no. UniSZA.C/2/UHREC/628-2 J1d.2(73)] and the Medical Research and Ethics Committee [approval no. KKM/NIHSEC/P19-1143(11)]. All participants provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Cao A and Galanello R: Beta-thalassemia. Genet Med. 12:61–76. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Aziz NA, Taib WW, Kharolazaman NK, Ismail I, Al-Jamal HAN, Jamil NWWA, Esa E and Ibrahim H: Evidence of new intragenic HBB haplotypes model for the prediction of beta-thalassemia in the Malaysian population. Sci Rep. 11(16772)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Haiyuni MY, Aziee S, Nasir A, Abdullah WZ and Johan MF: LARP2 DNA methylation in transfusion-dependent haemoglobin E-beta (HBE/β) and β-thalassaemia major patients. Mal J Med Health Sci. 15:46–53. 2019.
|
|
4
|
Fucharoen S and Weatherall DJ: The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2(a011734)2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Clark RJ, Tan CC, Preza GC, Nemeth E, Ganz T and Craik DJ: Understanding the structure/activity relationships of the iron regulatory peptide hepcidin. Chem Biol. 18:336–343. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mackenzie EL, Iwasaki K and Tsuji Y: Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid Redox Signal. 10:997–1030. 2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kaplan J, Ward DM and De Domenico I: The molecular basis of iron overload disorders and iron-linked anemias. Int J Hematol. 93:14–20. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Piperno A, Pelucchi S and Mariani R: Inherited iron overload disorders. Transl Gastroenterol Hepatol. 5(25)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mariani R, Trombini P, Pozzi M and Piperno A: Iron metabolism in thalassemia and sickle cell disease. Mediterr J Hematol Infect Dis. 1(e2009006)2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Taher AT and Saliba AN: Iron overload in thalassemia: Different organs at different rates. Hematology Am Soc Hematol Educ Program. 2017:265–271. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jones E, Pasricha SR, Allen A, Evans P, Fisher CA, Wray K, Premawardhena A, Bandara D, Perera A, Webster C, et al: Hepcidin is suppressed by erythropoiesis in hemoglobin E β-thalassemia and β-thalassemia trait. Blood. 125:873–880. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Locatelli F, Andrulli S, Pecchini F, Pedrini L, Agliata S, Lucchi L, Farina M, La Milia V, Grassi C, Borghi M, et al: Effect of high-flux dialysis on the anaemia of haemodialysis patients. Nephrol Dial Transplant. 15:1399–1409. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Olivieri NF, Muraca GM, O'Donnell A, Premawardhena A, Fisher C and Weatherall DJ: Studies in haemoglobin E beta-thalassaemia. Br J Haematol. 141:388–397. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hirsch RE, Sibmooh N, Fucharoen S and Friedman JM: HbE/β-thalassemia and oxidative stress: The key to pathophysiological mechanisms and novel therapeutics. Antioxid Redox Signal. 26:794–813. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Peslova G, Petrak J, Kuzelova K, Hrdy I, Halada P, Kuchel PW, Soe-Lin S, Ponka P, Sutak R, Becker E, et al: Hepcidin, the hormone of iron metabolism, is bound specifically to alpha-2-macroglobulin in blood. Blood. 113:6225–6236. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hassan Al-K, Azemin W-A, Dharmaraj S and Mohd K: Cytotoxic effect of hepcidin (th1-5) on human breast cancer cell line (mcf7). Jurnal Teknologi 77(3), 2015. doi:10.11113/jt.v77.6009. Accessed May 11, 2021.
|
|
17
|
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S and Andrews NC: The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1:191–200. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang DL, Senecal T, Ghosh MC, Ollivierre-Wilson H, Tu T and Rouault TA: Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts. Blood. 118:2868–2877. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, et al: Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 403:776–781. 2000.PubMed/NCBI View Article : Google Scholar
|
|
20
|
McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, et al: A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 5:299–309. 2000.PubMed/NCBI View Article : Google Scholar
|
|
21
|
De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G and Kaplan J: The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 18:2569–2578. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Pak M, Lopez MA, Gabayan V, Ganz T and Rivera S: Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 108:3730–3735. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ganz T: Hepcidin and iron regulation, 10 years later. Blood. 117:4425–4433. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Olivieri NF, Pakbaz Z and Vichinsky E: Hb E/beta-thalassaemia: A common & clinically diverse disorder. Indian J Med Res. 134(522)2011.PubMed/NCBI
|
|
26
|
Moradi G and Ghaderi E: Chronic disease program in Iran: Thalassemia control program. Chronic Diseases J. 1:98–106. 2013.
|
|
27
|
De Sanctis V, Soliman A and Yassin M: Iron overload and glucose metabolism in subjects with β-thalassaemia major: An overview. Curr Diabetes Rev. 9:332–341. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Rund D and Rachmilewitz E: Beta-Thalassemia. N Engl J Med. 353:1135–1146. 2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Handa P, Morgan-Stevenson V, Maliken BD, Nelson JE, Washington S, Westerman M, Yeh MM and Kowdley KV: Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am J Physiol Gastrointest Liver Physiol. 310:G117–G127. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Giesbrandt KJ, Bolan CW, Shapiro BP, Edwards WD and Mergo PJ: Diffuse diseases of the myocardium: MRI-pathologic review of cardiomyopathies with dilatation. AJR Am J Roentgenol. 200:W274–W282. 2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Buja LM and Roberts WC: Iron in the heart: Etiology and clinical significance. Am J Med. 51:209–221. 1971.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Guzelbey T, Gurses B, Ozturk E, Ozveren O, Sarsilmaz A and Karasu E: Evaluation of iron deposition in the adrenal glands of β thalassemia major patients using 3-tesla MRI. Iran J Radiol. 13(e36375)2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Coyne DW: Hepcidin: Clinical utility as a diagnostic tool and therapeutic target. Kidney Int. 80:240–244. 2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T and Kaplan J: Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 306:2090–2093. 2004.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ward DM and Kaplan J: Ferroportin-mediated iron transport: Expression and regulation. Biochim Biophys Acta. 1823:1426–1433. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Weizer-Stern O, Adamsky K, Amariglio N, Rachmilewitz E, Breda L, Rivella S and Rechavi G: mRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse models. Am J Hematol. 81:479–483. 2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Adamsky K, Weizer O, Amariglio N, Breda L, Harmelin A, Rivella S, Rachmilewitz E and Rechavi G: Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol. 124:123–124. 2004.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, Liu Y, Amariglio N, Rechavi G, Rachmilewitz EA, et al: Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 109:5027–5035. 2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Finberg KE: Ironing out an approach to alleviate the hypoferremia of acute inflammation. Haematologica. 106:326–328. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Stefanopoulos D, Nasiri-Ansari N, Dontas I, Vryonidou A, Galanos A, Psaridi L, Fatouros IG, Mastorakos G, Papavassiliou AG, Kassi E and Tournis S: Fibroblast growth factor 23 (FGF23) and klotho protein in beta-Thalassemia. Horm Metab Res. 52:194–201. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Renassia C and Peyssonnaux C: New insights into the links between hypoxia and iron homeostasis. Curr Opin Hematol. 26:125–130. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pasricha SR, Frazer DM, Bowden DK and Anderson GJ: Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: A longitudinal study. Blood. 122:124–133. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hendy OM, Allam M, Allam A, Attia MH, El Taher S, Eldin MM and Ali A: Hepcidin levels and iron status in beta-thalassemia major patients with hepatitis C virus infection. Egypt J Immunol. 17:33–44. 2010.PubMed/NCBI
|
|
44
|
Ganz T and Nemeth E: Hepcidin and iron homeostasis. Biochim Biophys Acta. 1823:1434–1443. 2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Hossaini SKE and reza Haeri M: Association between serum levels of hepcidin and ferritin in patients with thalassemia major and intermedia, the role of iron chelator. J Hematopathol. 12:143–147. 2019.
|
|
46
|
Jagadishkumar K, Yerraguntla N and Vaddambal MG: Serum hepcidin levels in children with beta Thalassemia major. Indian Pediatr. 55:911–912. 2018.PubMed/NCBI
|
|
47
|
Chauhan R, Sharma S and Chandra J: What regulates hepcidin in poly-transfused β-Thalassemia Major: Erythroid drive or store drive? Indian J Pathol Microbiol. 57:39–42. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ucler R, Kara E, Atmaca M, Olmez S, Alay M, Dirik Y and Bora A: A rare presentation of transfusional hemochromatosis: Hypogonadotropic hypogonadism. Case Rep Endocrinol. 2015(493091)2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Mishra AK and Tiwari A: Iron overload in Beta thalassaemia major and intermedia patients. Maedica (Bucur). 8:328–332. 2013.PubMed/NCBI
|
|
50
|
Abbass SAR and Defer IH: Some biochemical parameters in Iraqi patients with thalassemia and related with DM1. Int J Chem. 1:46–56. 2011.
|
|
51
|
Abdullah UYH, Ibrahim HM, Mahmud NB, Salleh MZ, Teh LK, Noorizhab MNFB, Zilfalil BA, Jassim HM, Wilairat P and Fucharoen S: Genotype-phenotype correlation of β-thalassemia in Malaysian population: Toward effective genetic counseling. Hemoglobin. 44:184–189. 2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Al-Hakeim HK, Auda FM and Ali BM: Lack of correlation between non-labile iron parameters, total carbonyl and malondialdehyde in major thalassemia. J Clin Biochem Nutr. 55:203–206. 2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Khera R, Singh T, Khuana N, Gupta N and Dubey AP: HPLC in characterization of hemoglobin profile in thalassemia syndromes and hemoglobinopathies: A clinicohematological correlation. Indian J Hematol Blood Transfus. 31:110–115. 2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Abdullah UYH, Ibrahim HM, Jassim HM, Salleh MZ, Kek TL, Fakhruzzaman Bin Noorizhab MN, Zilfalil BA, Wilairat P and Fucharoen S: Relative proteome quantification of alpha, beta, gamma and delta globin chains in early eluting peaks of Bio-Rad variant II® CE-HPLC of hemoglobin from healthy and beta-thalassemia subjects in Malaysia. Biochem Biophys Rep. 18(100635)2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Tatu T: Laboratory Diagnosis of β-Thalassemia and HbE. In: Beta Thalassemia. Marwa Z and Tamer H (eds). IntechOpen, 2020. doi:10.5772/intechopen.90317.
|
|
56
|
Lim WF, Muniandi L, George E, Sathar J, Teh LK and Lai MI: HbF in HbE/β-thalassemia: A clinical and laboratory correlation. Hematology. 20:349–353. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ivaldi G, Barberio G, Harteveld CL and Giordano P: HbA2 measurements in β-thalassemia and in other conditions. Thalassemia Rep. 4:45–48. 2014.
|














