Introduction
Atherosclerosis is a chronic inflammatory disease of
the large and medium-sized arteries, and is one of the most
prominent causes of mortality worldwide (1-3).
Atherosclerosis has been identified as the most common pathological
process underlying diseases of the coronary, carotid and peripheral
arteries (4). Several types of
cells play crucial roles during the development of atherosclerosis,
including endothelial cells, intimal smooth muscle cells and
leukocytes (5,6). In normal vascular tissue, the normal
morphology and functional integrity of endothelial cells permits
them to engulf bacteria, as well as senescent and necrotic tissue.
However, the lack of morphological integrity and dysfunction of
vascular endothelial cells promotes inflammation and arterial
thrombosis (7). In addition,
damaged endothelial cells may undergo phenotypic changes in a
process known as endothelial-to-mesenchymal transition (EMT), which
allows endothelial cells to transform into mesenchymal- or
myofibroblast-like cells (8). EMT
is a complicated process characterized by the loss of specific
endothelial markers, such as platelet endothelial cell adhesion
molecule-1 and vascular epithelial calcitonin, and the acquisition
of mesenchymal markers (9).
Accumulating evidence has demonstrated that EMT is closely
associated with the pathological processes of atherosclerosis and
increases plaque calcification and plaque instability, leading to
vascular stenosis (narrowing of the lumen) and ischemia in
atherosclerotic tissues (10,11).
Semaphorins are a large class of secreted or
transmembrane proteins, which were originally identified as
regulators of neuronal growth (12). Previous studies have revealed that
semaphorins serve important roles in several physiological
processes, including angiogenesis, vascular development and immune
responses (13,14). Semaphorin 7A (Sema7A) is a class 7
semaphorin that has a glycophosphatidylinositol-anchored domain
(15). Numerous studies have shown
that Sema7A and its membrane receptor, β1 integrin, exerted
regulatory effects on neuronal growth, numerous cancer types and
the inflammatory response (16-18).
Furthermore, Sema7A and β1 integrin have been reported to be
associated with atherosclerosis progression in previous studies
(19,20). However, to the best of our
knowledge, research on the biological role of Sema7A in endothelial
cell injury and EMT-associated atherosclerosis is currently
limited.
The present study was undertaken to investigate the
expression of Sema7A and β1 integrin and their biological functions
in atherosclerosis, and examine the effects of Sema7A/β1 integrin
on the biological functions of oxidized low-density lipoprotein
(ox-LDL)-treated HUVECs. The aim was to determine whether Sema7A
can regulate cellular injury, apoptosis and EMT in ox-LDL-treated
HUVECs by binding to β1 integrin, and whether the Sema7A/β1
integrin axis may serve as a novel target for the treatment of
endothelial cell injury and dysfunction in atherosclerosis.
Materials and methods
Cell culture and treatment
HUVECs were obtained from the American Type Culture
Collection. Cells were cultured in DMEM supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
streptomycin/penicillin, and maintained with 5% CO2 at
37˚C. To induce inflammatory injury and EMT, HUVECs were pretreated
with 50 µg/ml ox-LDL (Peking Union-Biology Co. Ltd) for 24 h at
37˚.
Plasmid construction and cell
transfection
To knock down Sema7A expression, specific short
hairpin (sh)RNA targeting Sema7A (shRNA-Sema7A-1/2) and
corresponding negative control (NC) shRNA (shRNA-NC) were
synthesized by Shanghai Integrated Biotech Solutions Co., Ltd. To
overexpress β1 integrin, a pcDNA3.1 expression vector containing
the entire length of β1 integrin (Ov-β1 integrin) and empty vector
(Ov-NC) were constructed by Shanghai GenePharma Co., Ltd. These
recombinants were transfected into HUVECs at 37˚, using
Lipofectamine® 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After
transfection for 48 h, cells were harvested for subsequent
experiments. In addition, transfected HUVECs were treated with 50
µg/ml ox-LDL for 24 h at 37˚, and harvested as aforementioned.
Cell Counting Kit-8 (CCK-8) assay
The effects of Sema7A and β1 integrin on HUVEC
viability were detected using a CCK-8 assay. Briefly, HUVECs were
seeded into 96-well plates (5x103 cells/well) and
transfected with shRNA-Sema7A with or without Ov-β1 integrin and
corresponding NCs for 48 h prior to treatment with 50 µg/ml ox-LDL
for 24 h. Following the incubation, 10 µl CCK-8 reagent
(MedChemExpress) was added to each well and incubated for a further
2 h at 37˚C. The absorbance was measured at a wavelength of 450 nm
using a microplate reader (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted from HUVECs from different
treatment groups using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Total RNA was reverse transcribed into cDNA using a
HiScriptQ RT SuperMix for qPCR (Vazyme Biotech Co., Ltd.) according
to the manufacturer's protocol. qPCR was subsequently performed
using a SYBR Premix ExTaq kit (Takara Bio, Inc.) on an ABI Prism
7900 Real-Time PCR detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The qPCR thermocycling conditions were as
follows: 95˚C for 3 min, followed by 35 cycles of denaturation at
95˚C for 30 sec, annealing at 60˚C for 30 sec and extension at 72˚C
for 1 min. A final extension step at 72˚C for 7 min was performed
for each assay. Each treatment group was ran five times. GAPDH was
used as the endogenous control. The relative expression of the
target genes was quantified using the 2-ΔΔCq method
(21). The primer sequences used
are as follows: Sema7A: Forward, 5'-TTCAGCCCGGACGAGAACT-3' and
reverse, 5'-GAACCGAGGGATCTTCCCAT-3'; β1 integrin: Forward,
5'-AGGAATGTTACACGGCTGCT-3' and reverse, 5'-ACCAAGTTTCCCATCTCCAG-3';
and GAPDH: Forward, 5'-GAAAGCCTGCCGGTGACTAA-3' and reverse,
5'-GCATCACCCGGAGGAGAAAT-3'.
ELISA
HUVECs were transfected with shRNA-Sema7A or
shRNA-NC and Ov-β1 integrin or Ov-NC, followed by treatment with
ox-LDL for 24 h at 37˚C. Then, the levels of inflammatory factors,
including IL-1β, IL-6 and C-C motif chemokine ligand 2 (CCL2), in
the culture supernatant were measured using ELISA kits (cat. no.
H002, H007-1-1 and H318-1, respectively; Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer's
protocols.
Western blotting
Total protein was extracted from ox-LDL-induced
HUVECs transfected with shRNA-Sema7A with or without Ov-β1 integrin
and corresponding NCs using RIPA lysis buffer (CoWin Biosciences).
Total protein was quantified using a Detergent Compatible Bradford
Protein assay kit (Beyotime Institute of Biotechnology) and the
proteins were separated via 10% SDS-PAGE. The separated proteins
were subsequently transferred onto PVDF membranes and blocked with
5% non-fat milk for 1 h at room temperature. The membranes were
then incubated with the following primary antibodies overnight at
4˚C: Anti-Sema7A (1:1,000; cat. no. ab255602), anti-β1 integrin
(1:1,000; cat. no. ab52971), anti-intracellular adhesion molecule
(ICAM)-1 (1:1,000; cat. no. ab282575), anti-vascular cell adhesion
molecule (VCAM)-1 (1:2,000; cat. no. ab134047), anti-Bax (1:2,000;
cat. no. ab182733), anti-cleaved caspase-3 (1:500; cat. no.
ab32042), anti-caspase-3 (1:5,000; cat. no. ab32351), anti-CD31
(1:1,000; cat. no. ab9498), anti-von Willebrand factor (vWF)
(1:1,000; cat. no. ab154193), anti-α-smooth muscle actin (α-SMA)
(1:1,000; cat. no. ab7817), anti-Snail family transcriptional
repressor 2 (Slug) (1:1,000; cat. no. ab51772), anti-zinc finger
E-box binding homeobox 1 (ZEB1) (1:500, ab203829) and anti-GAPDH
(1:1,000; cat. no. ab8245; all from Abcam). Following primary
antibody incubation, the membranes were washed four times with
0.05% PBS-Tween-20 and incubated with secondary antibodies [goat
anti-rabbit IgG, HRP, cat. no. 7074; or goat anti-mouse IgG, HRP,
cat. no. 7076; Cell Signaling Technology (1:2,000)] for 1 h at room
temperature. Protein bands were visualized using an ECL detection
system (Beyotime Institute of Biotechnology) and semi-quantified
using ImageJ software (version 1.49; National Institutes of
Health).
Flow cytometry
The effects of the knockdown of Sema7A and Ov-β1
integrin on cell apoptosis were evaluated using flow cytometry.
Briefly, HUVECs were transfected with shRNA-Sema7A with or without
Ov-β1 integrin for 48 h, then induced for 24 h at 37˚C with ox-LDL.
The cells were harvested, washed and resuspended in binding buffer,
then incubated with 5 µl Annexin V-FITC for 15 min and 10 µl PI (10
mg/ml) for 5 min in the dark. Apoptotic cells were detected by a
flow cytometry (Accuri C6; BD Biosciences) and analyzed using
FlowJo software (version 10.2; FlowJo LLC).
Immunofluorescence staining
Transfected or control HUVECs were plated into
six-well plates with coverslips at a density of 4x105
cells/well and allowed to reach 70-80% confluence. Subsequently,
the cells were fixed with 4% paraformaldehyde for 30 min and
incubated with 0.1% Triton X-100 for permeabilization for 7 min,
both at room temperature. The slides were then blocked with 5%
skimmed milk diluted in 0.01 M PBS for 1 h at room temperature, and
incubated with an anti-α-SMA primary antibody (1:100; cat. no.
ab184675; Abcam) overnight at 4˚C. Following incubation, the slides
were washed four times with PBS and incubated with a goat
anti-mouse IgG H&L secondary antibody (1:200; cat. no.
ab150113; Abcam) for 2 h at room temperature. Nuclei were
counterstained with DAPI (Vector Laboratories, Inc.) for 5 min at
room temperature. Finally, the slides were observed using a
fluorescence microscope (magnification, x200; Olympus
Corporation).
Statistical analysis
Statistical analysis was performed using SPSS
version 19.0 software (IBM Corp.) and GraphPad Prism version 5.0
(GraphPad Software, Inc.) was used to draw the graphs. Data are
presented as the mean ± SD from at least three independent
experiments. Statistical differences between multiple groups were
determined using one-way ANOVA followed by Tukey's multiple
comparisons test, while statistical differences between two groups
were determined using an unpaired, two-tailed Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Sema7A and β1 integrin expression is
upregulated in ox-LDL-induced HUVECs
ox-LDL plays a key role in endothelial cell injury
during the progression of atherosclerosis (22). To simulate lipid
accumulation-induced dysfunction of the endothelium in
atherosclerosis, HUVECs were treated with 50 µg/ml ox-LDL for 24 h.
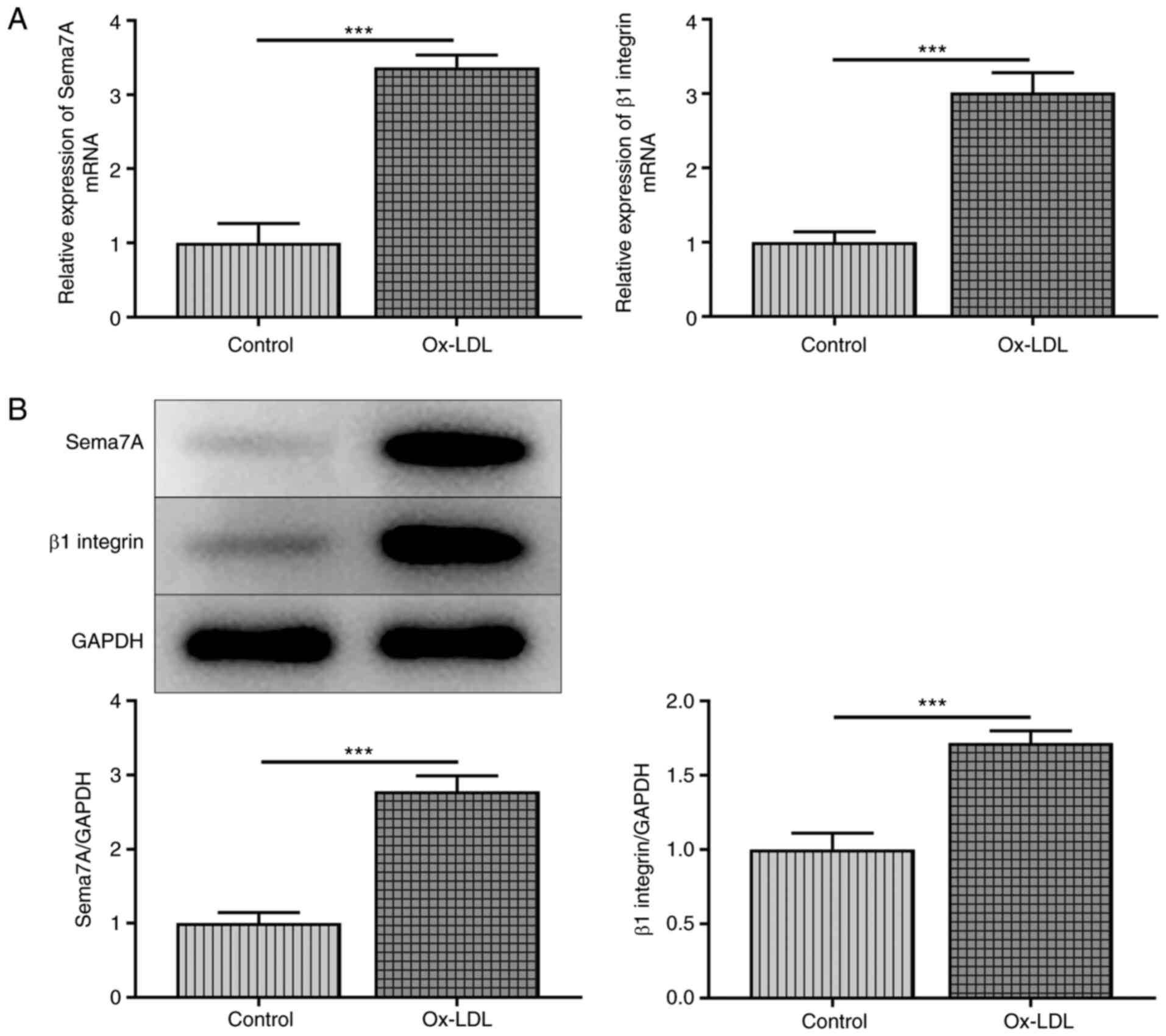
As shown in Fig. 1A, the mRNA
expression levels of Sema7A and β1 integrin were significantly in
HUVECs upregulated following ox-LDL treatment. Similarly, ox-LDL
upregulated the protein expression levels of Sema7A and β1 integrin
in HUVECs compared with the control group (Fig. 1B). These data suggested that the
aberrant expression of Sema7A and β1 integrin in HUVECs was
associated with ox-LDL stimulation.
Sema7A knockdown and Ov-β1 integrin
ameliorate ox-LDL-induced impaired HUVEC viability
To determine the role of Sema7A and β1 integrin in
atherosclerosis, the effects of Sema7A and β1 integrin on the
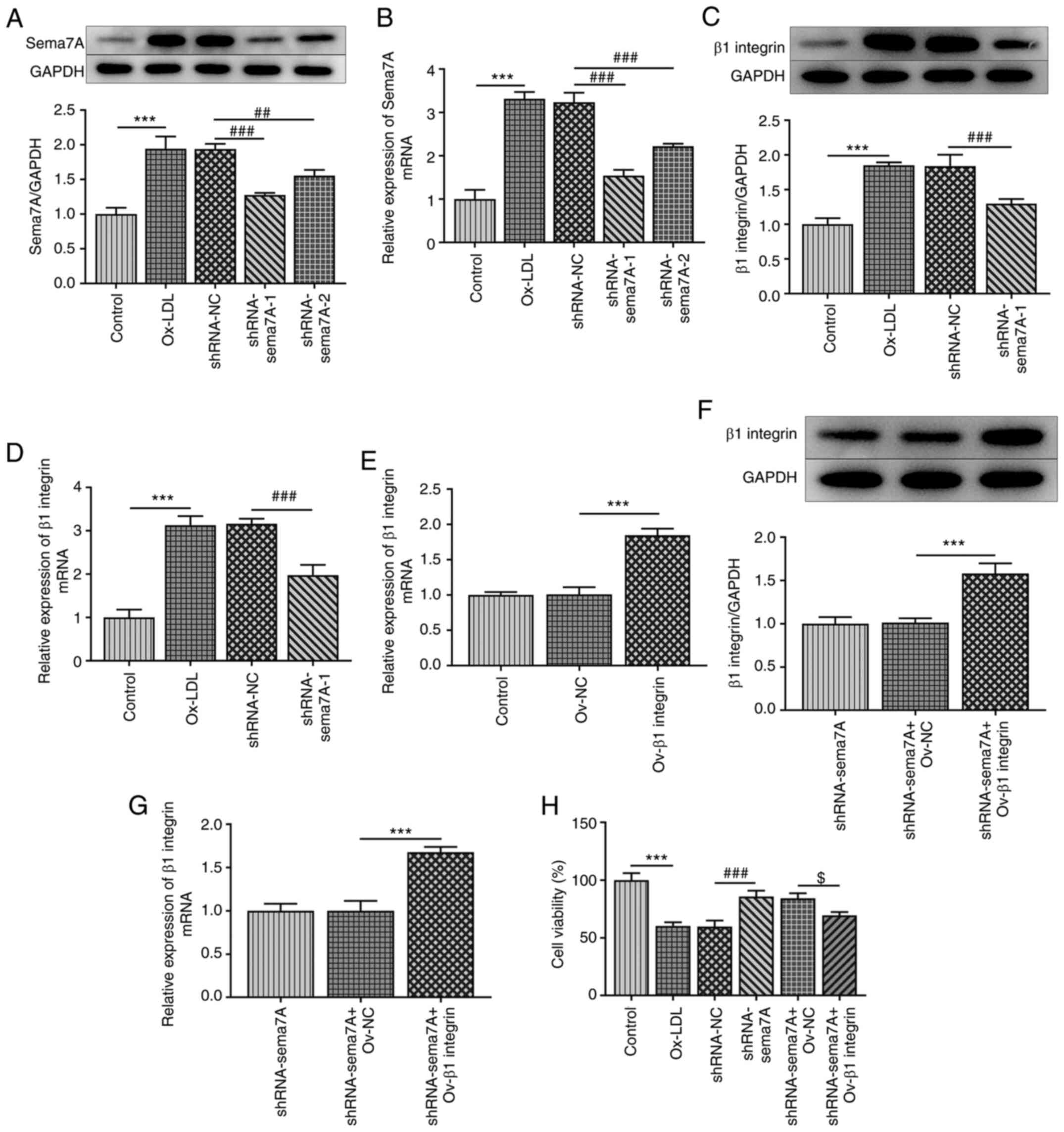
activities of HUVECs were investigated. As presented in Fig. 2A and B, Sema7A-knockdown vectors were
transfected into HUVECs and the transfection efficiency was
evaluated using RT-qPCR analysis and western blotting. According to
the results, shRNA-Sema7A-1 exhibited the best interference
efficiency, and was thus selected for the following experiments.
Subsequently, the effect of Sema7A knockdown on β1 integrin
expression was determined. The results revealed that ox-LDL
treatment notably upregulated the mRNA and protein expression level
of β1 integrin compared with the control group, while transfection
with shRNA-Sema7A-1 inhibited the ox-LDL-induced upregulated
expression of β1 integrin (Fig. 2C
and D). Furthermore, β1 integrin
overexpression vectors were transfected into Sema7A-knockdown cells
and the transfection efficiency was investigated (Fig. 2E-G). The results of the CCK-8 assay
revealed that cell viability was markedly repressed following
ox-LDL induction, while the knockdown of Sema7A alleviated the
suppressive effects of ox-LDL on cell viability. However, the
overexpression of β1 integrin reversed the Sema7A knockdown-induced
increase in HUVEC viability (Fig.
2H).
Sema7A knockdown and Ov-β1 integrin
inhibit the ox-LDL-induced release of inflammatory cytokines and
adhesion factors in HUVECs
Inflammation and adhesion factors are closely
associated with the loss of morphological and functional integrity
of vascular endothelial cells in atherosclerosis (10). As shown in Fig. 3A, the production of IL-1β, IL-6 and
CCL2 was significantly increased in HUVECs induced with ox-LDL
compared with control cells. The knockdown of Sema7A abrogated the
secretion of IL-1β, IL-6 and CCL2, while the overexpression of β1
integrin enhanced the Sema7A knockdown-mediated decrease in the
levels of IL-1β, IL-6 and CCL2. Moreover, the results from RT-qPCR
and western blot analyses demonstrated that the stimulation with
ox-LDL markedly upregulated the expression levels of ICAM-1 and
VCAM-1 in HUVECs. The knockdown of Sema7A repressed the production
of these adhesion factors, while these effects were blocked by
Ov-β1 integrin (Fig. 3B and
C).
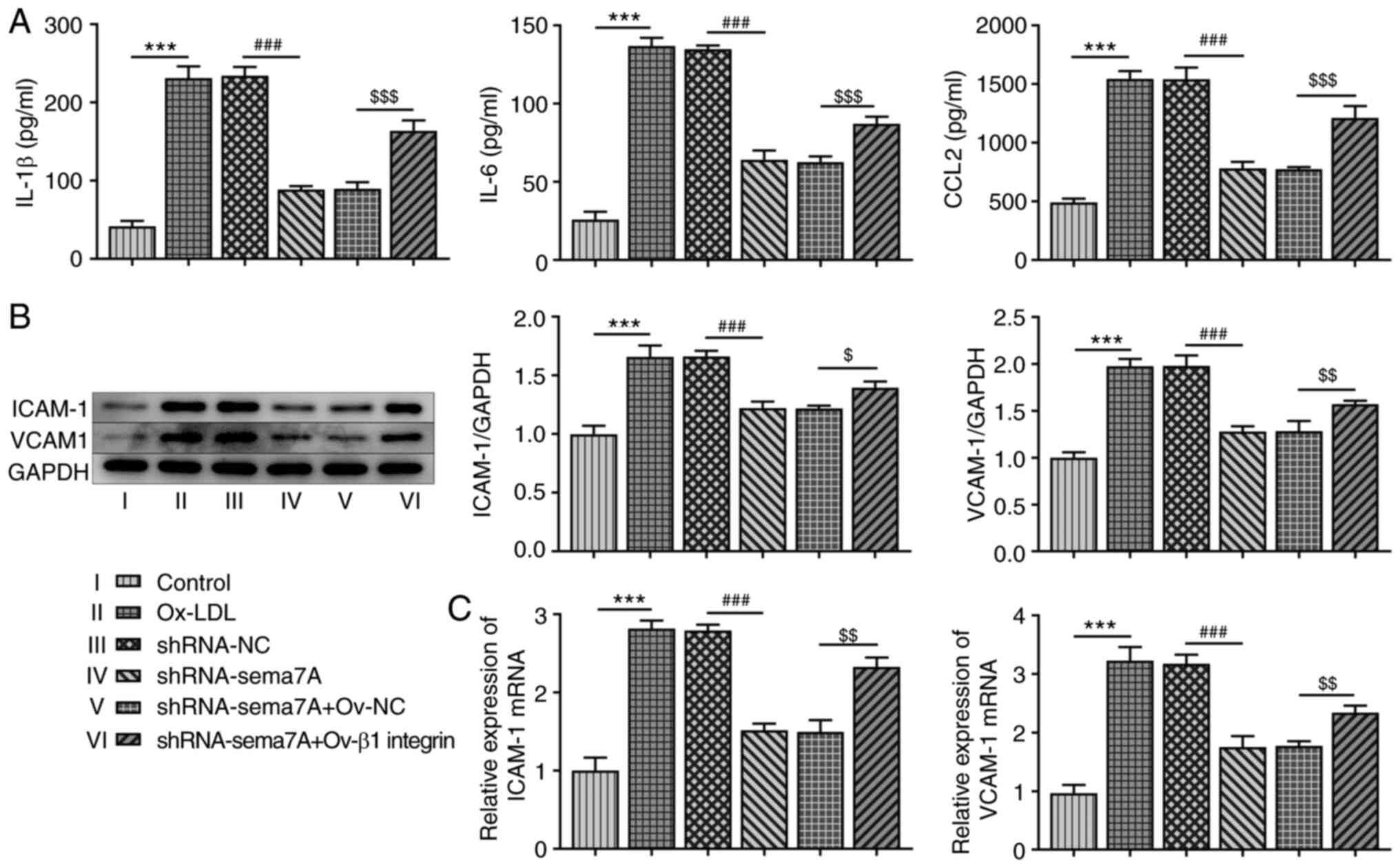 | Figure 3Effects of Sema7A and β1 integrin on
the levels of inflammatory cytokines and adhesion factors in
HUVECs. (A) IL-1β, IL-6 and CCL2 levels in ox-LDL induced HUVECs
were examined after transfection with shRNA-Sema7A in the presence
and absence of Ov-β1 integrin. Western blot assay and reverse
transcription-quantitative PCR were carried out to detect the (B)
protein levels and (C) mRNA expressions of ICAM-1 and VCAM-1 in
ox-LDL induced HUVECs transfected with shRNA-Sema7A in the presence
and absence of Ov-β1 integrin. Data are expressed as mean ± SD.
***P<0.001 vs. control; ###P<0.001 vs.
shRNA-NC; $P<0.05, $$P<0.01 and
$$$P<0.001 vs. shRNA-Sema7A + Ov-NC. Sema7A,
semaphorin 7A; ox-LDL, oxidized low-density lipoprotein; CCL2, C-C
motif chemokine ligand 2; shRNA, short hairpin RNA; Ov-,
overexpression vector; NC, negative control; ICAM-1, intracellular
adhesion molecule; VCAM, vascular cell adhesion molecule-1. |
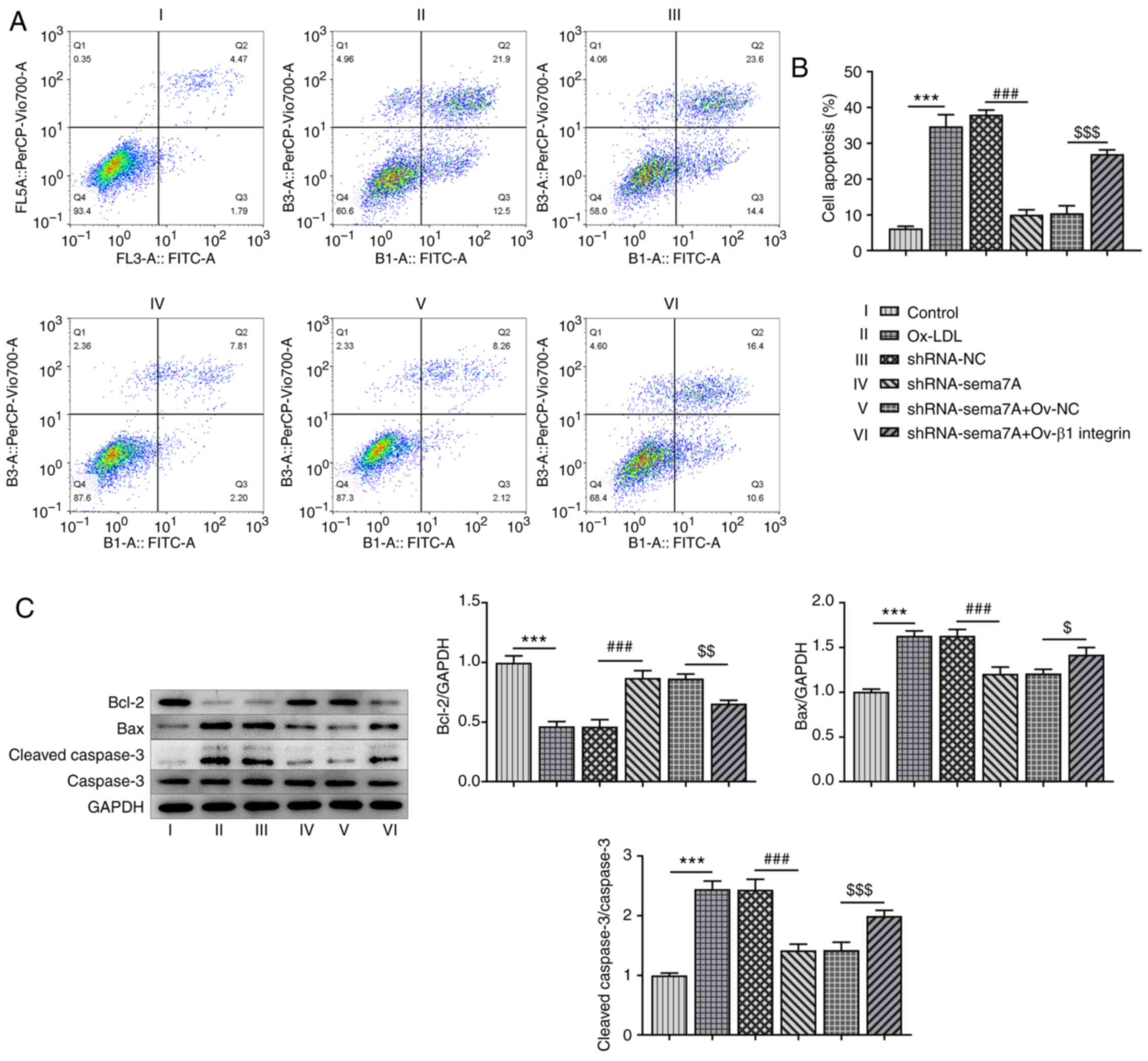
Sema7A knockdown and Ov-β1 integrin
prevent the apoptosis of HUVECs in response to ox-LDL
Next, the effects of Sema7A knockdown and Ov-β1
integrin transfection on the apoptosis of HUVECs were analyzed. As
illustrated in Fig. 4A and B, the apoptotic rate of HUVECs induced
with ox-LDL was markedly increased compared with the control cells.
The knockdown of Sema7A reduced ox-LDL-induced cell apoptosis,
while the overexpression of β1 integrin reversed the suppressive
effect of Sema7A knockdown on the apoptosis of HUVECs treated with
ox-LDL. Furthermore, the incubation of HUVECs with ox-LDL
downregulated Bcl-2 expression levels and upregulated the
expression levels of Bax and cleaved caspase-3. The knockdown of
Sema7A reversed the effects of ox-LDL, while transfection with
Ov-β1 integrin reversed the Sema7A knockdown-induced upregulation
of Bcl-2 protein expression levels and downregulation of Bax and
cleaved caspase-3 expression levels (Fig. 4C).
Sema7A knockdown and Ov-β1 integrin
attenuate ox-LDL-induced EMT in HUVECs
To further determine the functional role of Sema7A
and β1 integrin in the EMT of HUVECs treated with ox-LDL, the
protein and mRNA expression levels of EMT-related proteins were
analyzed using western blotting and RT-qPCR, respectively. As shown
in Fig. 5A and B, the expression levels of α-SMA, Slug and
ZEB1 were upregulated, while the expression levels of CD31 and vWF
were downregulated in HUVECs following induction with ox-LDL, and
these effects were reversed by the knockdown of Sema7A and
overexpression of β1 integrin. In addition, the results of the
immunofluorescence assay revealed that α-SMA expression levels were
significantly upregulated after HUVECs were induced with ox-LDL,
whereas they were downregulated by the knockdown of Sema7A
expression. However, the overexpression of β1 integrin inhibited
the effects of Sema7A knockdown on ox-LDL-induced EMT in HUVECs
(Fig. 5C).
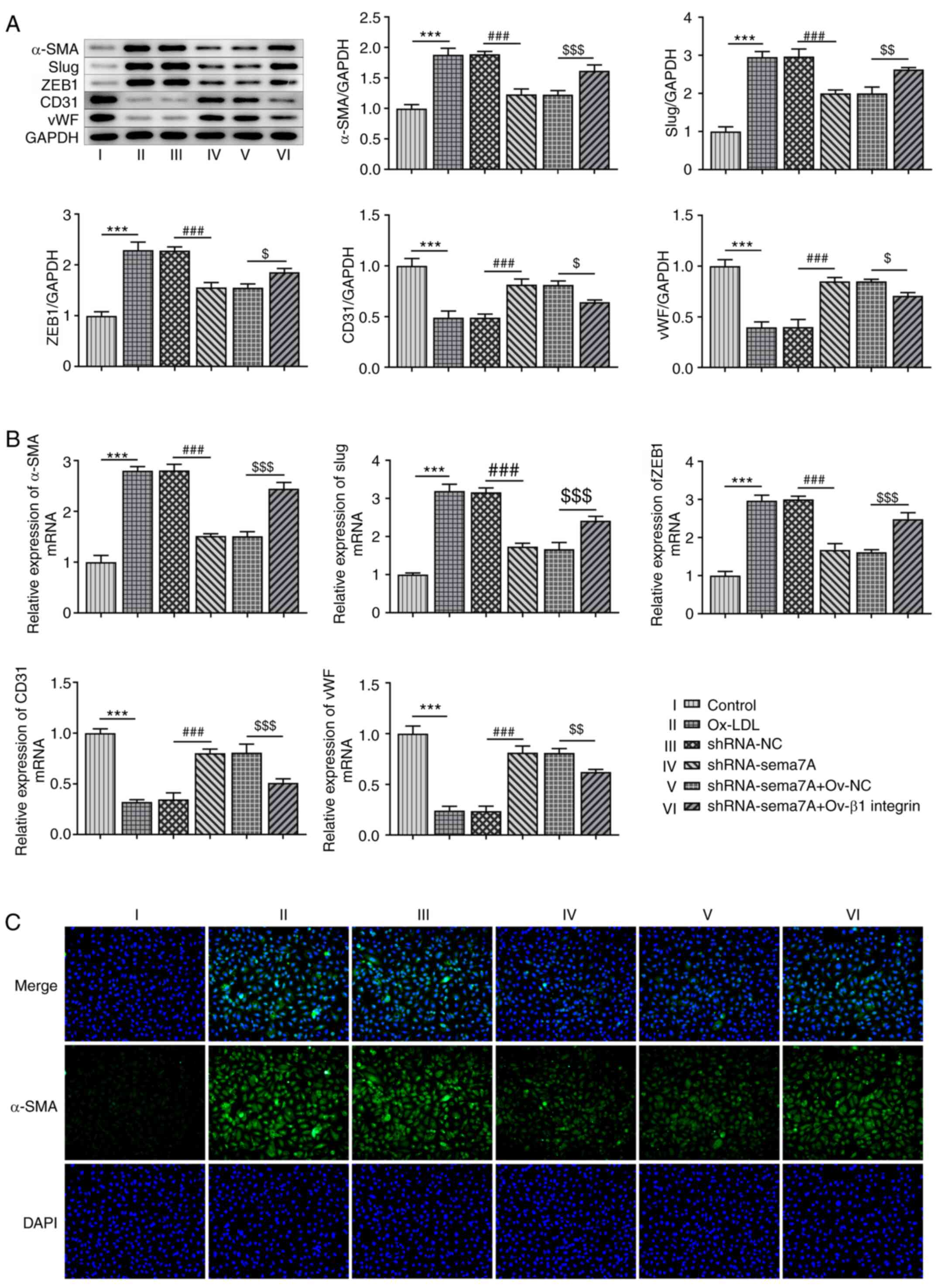 | Figure 5Effects of Sema7A and β1 integrin on
ox-LDL-induced epithelial-to-mesenchymal transition in HUVECs. (A)
Western blot assay was used to determine the protein levels of
α-SMA, Slug, ZEB1, CD31 and vWF in ox-LDL-induced HUVECs
transfected with shRNA-Sema7A in the presence and absence of Ov-β1
integrin. (B) Reverse transcription-quantitative PCR analysis was
used to measure mRNA expression of α-SMA, Slug, ZEB1, CD31 and vWF
were estimated by in ox-LDL-induced HUVECs transfected with
shRNA-Sema7A in the presence and absence of Ov-β1 integrin. (C)
Immunofluorescence assay was performed to assess α-SMA protein
levels in ox-LDL-induced HUVECs transfected with shRNA-Sema7A in
the presence or absence of Ov-β1 integrin. Data are expressed as
mean ± SD. ***P<0.001 vs. control;
###P<0.001 vs. shRNA-NC group; $P<0.05,
$$P<0.01, $$$P<0.001 vs. shRNA-Sema7A +
Ov-NC group. Sema7A, semaphorin 7A; ox-LDL, oxidized low-density
lipoprotein; α-SMA, α-smooth muscle actin; vWF, von Willebrand
factor; Slug, Snail family transcriptional repressor 2; ZEB1, zinc
finger E-box binding homeobox 1; shRNA, short hairpin RNA; Ov-,
overexpression vector; NC, negative control. |
Discussion
Atherosclerosis is the main cause of morbidity and
mortality in cardiovascular diseases, and is responsible for more
lives than all cancer types combined (23). The results of the present study
revealed that the expression of Sema7A and β1 integrin was
dysregulated ox-LDL-induced HUVECs. In addition, Sema7A and β1
integrin were serve key roles in ox-LDL-induced HUVECs by
regulating cell viability, inflammatory responses, apoptosis and
EMT. The knockdown of Sema7A expression increased cell viability,
inhibited the release of inflammatory factors and adhesion
molecules, suppressed cell apoptosis and inhibited the EMT process,
whereas the overexpression of β1 integrin reversed the effects of
Sema7A silencing in HUVECs induced with ox-LDL.
Endothelial cells exist in the walls of blood and
lymphatic vessels and have unique functions in vascular biology
(24). Vascular endothelial cell
injury is a common pathological basis of various cardiovascular
diseases and represents the early stage of atherosclerosis
(25). Hyperlipidemia, a
cardiovascular risk factor, promotes the accumulation of plasma LDL
in the arterial wall, which drives the formation of atherosclerotic
plaques, and induces endothelial cell injury and apoptosis during
the occurrence and development of atherosclerosis (26,27).
ox-LDL is a causal risk factor for atherosclerosis and binds to the
scavenger receptor in endothelial cells to promote the secretion of
inflammatory cytokines, inducing apoptosis and further promoting
EMT (28,29).
Vascular endothelial cell inflammation plays a
central role in all stages of atherosclerosis, from lesion
initiation to progression and destabilization (30). A previous study reported that ox-LDL
induced a local inflammatory response, which inflamed the
endothelium and upregulated the expression of adhesion molecules,
leading to the adhesion of circulating leukocytes to the
endothelium (31). Thus, in the
present study, ox-LDL was used to stimulate HUVECs to mimic
hyperlipidemia-induced endothelial cell injury. The results
demonstrated that ox-LDL treatment significantly reduced HUVEC
viability and induced the production of inflammatory factors,
including IL-1β, IL-6 and CCL2. In addition, the expression levels
of ICAM-1 and VCAM-1 were markedly upregulated after HUVECs were
exposed to 50 µg/ml ox-LDL for 24 h. The present study also
revealed that the knockdown of Sema7A abrogated the generation of
ox-LDL-induced inflammatory factors, IL-1β, IL-6 and CCL2, while
the overexpression of β1 integrin reversed these effects of
Sema7A-knockdown. As expected, the upregulated expression levels of
ox-LDL-induced ICAM-1 and VCAM-1 were downregulated by the
knockdown of Sema7A and recovered by Ov-β1 integrin transfection.
These data indicated that silencing of Sema7A may protect against
ox-LDL-induced inflammatory injury in endothelial cells by
regulating β1 integrin.
Inflammatory responses induced by injury stress
signals, such as ox-LDL, mediate further interactions between
monocytes and endothelial cells, resulting in increased apoptosis
of endothelial cells, which may initiate atherosclerosis (32-34).
Wu et al (35) found that
ox-LDL significantly induced HUVEC apoptosis, while the knockdown
of proprotein convertase subtilisin/kexin type 9 suppressed
apoptosis via the Bcl/Bax/caspase-9/caspase-3 signaling pathway. In
addition, Zhong et al (36)
demonstrated that myocardial infarction-associated transcript
promoted cell proliferation and inhibited apoptosis in
ox-LDL-induced atherosclerosis cell models by modulating the
microRNA-181b/STAT3 signaling axis. Thus, the suppression of
vascular endothelial cell apoptosis may promote pathological
remission of atherosclerosis (37).
In the present study, the apoptotic rate was increased in HUVECs
exposed to ox-LDL, which was accompanied with downregulated
expression levels of Bcl-2 and upregulated expression levels of Bax
and cleaved caspase-3. However, these changes were mitigated by the
knockdown of Sema7A, while β1 integrin-overexpression abolished the
inhibitory effects of Sema7A silencing, which is consistent with
previous reports.
EMT serves key roles in plaque instability and
atherosclerosis progression (38,39).
Previous studies have reported that the formation and instability
of atherosclerotic plaques are associated with endothelial cells
acquiring morphological and phenotypic properties of mesenchymal
cells, such as increased proliferation and migration, and the
secretion of leukocyte adhesion molecules, extracellular matrix and
matrix metalloproteinases (40). An
accumulating number of studies have found that Sema7A and its
receptor, β1 integrin, contributed to vascular endothelial cell
injury and the pathophysiology of atherosclerosis (19,20,41).
In the present study, the expression levels of α-SMA, Slug and ZEB1
were found to be upregulated, while the expression levels of CD31
and vWF were downregulated under the induction of ox-LDL. The
knockdown of Sema7A rescued the expression levels of EMT-related
proteins, while β1 integrin-overexpression counteracted the effects
of Sema7A silencing on EMT in HUVECs. These findings indicated that
the knockdown of Sema7A or overexpression of β1 integrin may
protect HUVECs against ox-LDL-induced EMT. In addition, the present
study focused on the regulatory role of Sema7A silencing in ox-LDL
HUVECs through modulating β1 integrin expression, but did not
explore whether Sema7A is involved in the regulation of β1 integrin
expression in ox-LDL HUVECs. Furthermore, the results of the
present study were not verified in animal experiments. Thus, the
relationship between Sema7A and β1 integrin and the underlying
mechanisms will be further investigated in vitro and in
vivo in future studies.
In conclusion, the findings of the present study
suggested that the knockdown of Sema7A/β1 integrin may protect
HUVECs from ox-LDL-induced injury by inhibiting the stimulation of
inflammatory responses, cell apoptosis and EMT. Therefore, Sema7A
and β1 integrin may represent novel targets for the treatment of
endothelial cell injury and dysfunction in atherosclerosis.
Collectively, the findings of the present study may provide
potential targets and therapeutic strategies against vascular
endothelial cell inflammation and EMT to promote the pathological
remission of atherosclerosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Medical Guidance (TCM)
Science and Technology Support Project of Science and Technology
Commission of Shanghai Municipality, China (grant no. 17401933700),
the Scientific Research Project of Shanghai Municipal Commission of
Health and Family Planning, China (grant no. 201640039) and the
Peak Plateau Subject of Shanghai University of Traditional Chinese
Medicine (Special Project for Clinical Talents), China (grant no.
171319).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XS and DL designed and supervised the experiments.
XS, JM, GY, HW and HL performed the experiments. HW analyzed the
data and HL searched the literature. XS, JM and DL wrote and
revised the manuscript. XS and DL have seen and can confirm the
authenticity of all the raw data. All the authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abdolmaleki F, Gheibi Hayat SM, Bianconi
V, Johnston TP and Sahebkar A: Atherosclerosis and immunity: A
perspective. Trends Cardiovasc Med. 29:363–371. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Taleb S: Inflammation in atherosclerosis.
Arch Cardiovasc Dis. 109:708–715. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Viola J and Soehnlein O: Atherosclerosis -
A matter of unresolved inflammation. Semin Immunol. 27:184–193.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Falk E: Pathogenesis of atherosclerosis. J
Am Coll Cardiol. 47 (Suppl 1):C7–C12. 2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Davies MJ, Woolf N, Rowles PM and Pepper
J: Morphology of the endothelium over atherosclerotic plaques in
human coronary arteries. Br Heart J. 60:459–464. 1988.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wolf D and Ley K: Immunity and
inflammation in atherosclerosis. Circ Res. 124:315–327.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han
X, Tang D and Chen R: research progress on the relationship between
atherosclerosis and inflammation. Biomolecules.
8(E80)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Correia AC, Moonen JR, Brinker MG and
Krenning G: FGF2 inhibits endothelial-mesenchymal transition
through microRNA-20a-mediated repression of canonical TGF-β
signaling. J Cell Sci. 129:569–579. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kovacic JC, Mercader N, Torres M, Boehm M
and Fuster V: Epithelial-to-mesenchymal and
endothelial-to-mesenchymal transition: From cardiovascular
development to disease. Circulation. 125:1795–1808. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chen PY, Qin L, Baeyens N, Li G, Afolabi
T, Budatha M, Tellides G, Schwartz MA and Simons M:
Endothelial-to-mesenchymal transition drives atherosclerosis
progression. J Clin Invest. 125:4514–4528. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Hansson GK and Hermansson A: The immune
system in atherosclerosis. Nat Immunol. 12:204–212. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Simpson KJ, Henderson NC, Bone-Larson CL,
Lukacs NW, Hogaboam CM and Kunkel SL: Chemokines in the
pathogenesis of liver disease: So many players with poorly defined
roles. Clin Sci (Lond). 104:47–63. 2003.PubMed/NCBI
|
|
13
|
Wannemacher KM, Wang L, Zhu L and Brass
LF: The role of semaphorins and their receptors in platelets:
Lessons learned from neuronal and immune synapses. Platelets.
22:461–465. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Suzuki K, Kumanogoh A and Kikutani H:
Semaphorins and their receptors in immune cell interactions. Nat
Immunol. 9:17–23. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Gutiérrez-Franco A, Eixarch H, Costa C,
Gil V, Castillo M, Calvo-Barreiro L, Montalban X, Del Río JA and
Espejo C: Semaphorin 7A as a potential therapeutic target for
multiple sclerosis. Mol Neurobiol. 54:4820–4831. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pasterkamp RJ, Peschon JJ, Spriggs MK and
Kolodkin AL: Semaphorin 7A promotes axon outgrowth through
integrins and MAPKs. Nature. 424:398–405. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ma B, Herzog EL, Lee CG, Peng X, Lee CM,
Chen X, Rockwell S, Koo JS, Kluger H, Herbst RS, et al: Role of
chitinase 3-like-1 and semaphorin 7a in pulmonary melanoma
metastasis. Cancer Res. 75:487–496. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Suzuki K, Okuno T, Yamamoto M, Pasterkamp
RJ, Takegahara N, Takamatsu H, Kitao T, Takagi J, Rennert PD,
Kolodkin AL, et al: Semaphorin 7A initiates T-cell-mediated
inflammatory responses through alpha1beta1 integrin. Nature.
446:680–684. 2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hu S, Liu Y, You T and Zhu L: Semaphorin
7A promotes VEGFA/VEGFR2-mediated angiogenesis and intraplaque
neovascularization in ApoE-/- mice. Front Physiol.
9(1718)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
You T, Zhu Z, Zheng X, Zeng N, Hu S, Liu
Y, Ren L, Lu Q, Tang C, Ruan C, et al: Serum semaphorin 7A is
associated with the risk of acute atherothrombotic stroke. J Cell
Mol Med. 23:2901–2906. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang GL, Xia XL, Li XL, He FH and Li JL:
Identification and expression analysis of the MSP130-related-2 gene
from Hyriopsis cumingii. Genet Mol Res. 14:4903–4913.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Fang F, Yang Y, Yuan Z, Gao Y, Zhou J,
Chen Q and Xu Y: Myocardin-related transcription factor A mediates
OxLDL-induced endothelial injury. Circ Res. 108:797–807.
2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Stocker R and Keaney JF Jr: Role of
oxidative modifications in atherosclerosis. Physiol Rev.
84:1381–1478. 2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sun HJ, Hou B, Wang X, Zhu XX, Li KX and
Qiu LY: Endothelial dysfunction and cardiometabolic diseases: Role
of long non-coding RNAs. Life Sci. 167:6–11. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sun HJ, Zhu XX, Cai WW and Qiu LY:
Functional roles of exosomes in cardiovascular disorders: A
systematic review. Eur Rev Med Pharmacol Sci. 21:5197–5206.
2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Grover-Páez F and Zavalza-Gómez AB:
Endothelial dysfunction and cardiovascular risk factors. Diabetes
Res Clin Pract. 84:1–10. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Pober JS, Min W and Bradley JR: Mechanisms
of endothelial dysfunction, injury, and death. Annu Rev Pathol.
4:71–95. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang Y, Mu Q, Zhou Z, Song H, Zhang Y, Wu
F, Jiang M, Wang F, Zhang W, Li L, et al: Protective effect of
irisin on atherosclerosis via suppressing oxidized low density
lipoprotein induced vascular inflammation and endothelial
dysfunction. PLoS One. 11(e0158038)2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lee WJ, Ou HC, Hsu WC, Chou MM, Tseng JJ,
Hsu SL, Tsai KL and Sheu WH: Ellagic acid inhibits oxidized
LDL-mediated LOX-1 expression, ROS generation, and inflammation in
human endothelial cells. J Vasc Surg. 52:1290–1300. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Onat D, Brillon D, Colombo PC and Schmidt
AM: Human vascular endothelial cells: A model system for studying
vascular inflammation in diabetes and atherosclerosis. Curr Diab
Rep. 11:193–202. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Maugeri N, Rovere-Querini P, Baldini M,
Sabbadini MG and Manfredi AA: Translational mini-review series on
immunology of vascular disease: Mechanisms of vascular inflammation
and remodelling in systemic vasculitis. Clin Exp Immunol.
156:395–404. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Marui N, Offermann MK, Swerlick R, Kunsch
C, Rosen CA, Ahmad M, Alexander RW and Medford RM: Vascular cell
adhesion molecule-1 (VCAM-1) gene transcription and expression are
regulated through an antioxidant-sensitive mechanism in human
vascular endothelial cells. J Clin Invest. 92:1866–1874.
1993.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tsouknos A, Nash GB and Rainger GE:
Monocytes initiate a cycle of leukocyte recruitment when cocultured
with endothelial cells. Atherosclerosis. 170:49–58. 2003.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lutgens E, de Muinck ED, Kitslaar PJ,
Tordoir JH, Wellens HJ and Daemen MJ: Biphasic pattern of cell
turnover characterizes the progression from fatty streaks to
ruptured human atherosclerotic plaques. Cardiovasc Res. 41:473–479.
1999.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS
and Liu LS: PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL
via Bcl/Bax-caspase9-caspase3 pathway. Mol Cell Biochem.
359:347–358. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhong X, Ma X, Zhang L, Li Y, Li Y and He
R: MIAT promotes proliferation and hinders apoptosis by modulating
miR-181b/STAT3 axis in ox-LDL-induced atherosclerosis cell models.
Biomed Pharmacother. 97:1078–1085. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Gong L, Lei Y, Liu Y, Tan F, Li S, Wang X,
Xu M, Cai W, Du B, Xu F, et al: Vaccarin prevents ox-LDL-induced
HUVEC EndMT, inflammation and apoptosis by suppressing ROS/p38 MAPK
signaling. Am J Transl Res. 11:2140–2154. 2019.PubMed/NCBI
|
|
38
|
Evrard SM, Lecce L, Michelis KC,
Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d'Escamard V, Li
JR, Hadri L, Fujitani K, et al: Endothelial to mesenchymal
transition is common in atherosclerotic lesions and is associated
with plaque instability. Nat Commun. 7(11853)2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Boström KI, Yao J, Guihard PJ,
Blazquez-Medela AM and Yao Y: Endothelial-mesenchymal transition in
atherosclerotic lesion calcification. Atherosclerosis. 253:124–127.
2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wesseling M, Sakkers TR, de Jager SCA,
Pasterkamp G and Goumans MJ: The morphological and molecular
mechanisms of epithelial/endothelial-to-mesenchymal transition and
its involvement in atherosclerosis. Vascul Pharmacol. 106:1–8.
2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hu S, Liu Y, You T, Heath J, Xu L, Zheng
X, Wang A, Wang Y, Li F, Yang F, et al: Vascular Semaphorin 7A
upregulation by disturbed flow promotes atherosclerosis through
endothelial β1 integrin. Arterioscler Thromb Vasc Biol. 38:335–343.
2018.PubMed/NCBI View Article : Google Scholar
|