Introduction
Due to the tremendous efforts of researchers and
clinicians during the past few decades, in numerous parts of the
world early diagnosis of cancer is readily achievable. Survival
from cancer has considerably increased as a result of systematic
screening and the development of novel cancer treatments with
improved efficacy. Thus, given the growing number of cancer
survivors, the opportunity arose to study the genetic background of
these patients, environmental exposure to carcinogens, the
long-term side effects of cancer treatments and their risk of
developing subsequent primary cancers.
According to the studies carried out, to date,
multiple primary cancers occur with a frequency ranging from 2 to
17% (1). As anticipated, their
prevalence follows an ascendant trend (2). The development of multiple cancers is
dictated by a complicated interplay between a variety of factors,
both patient-dependent (genetic predisposition, immune
deficiencies, hormonal dysfunctions) and external (infections,
exposure to ultraviolet radiation, ionizing radiation, smoking,
alcohol consumption, dietary factors). Chemotherapy and/or
radiotherapy for previous cancers greatly increase the risk for the
development of subsequent neoplasia, either hematologic
malignancies or solid tumors (3,4).
Despite the notable progress in oncologic treatment,
pancreatic cancer (PC) still portends an extremely poor prognosis,
with a five-year survival rate after radical surgery of 10% for
node-positive disease, which is the case in approximately two
thirds of patients and 30% for node-negative disease (5,6). This
is one of the reasons the appearance of a second or multiple
cancers in PC patients was long considered highly improbable
(7). However, a search of the
literature reveals several reports of single or multiple
extra-pancreatic cancers, located in the digestive tract, lung,
breast, prostate, kidney, and skin arising in patients previously
or consequently diagnosed with PC (7).
Although a series of studies (8,9)
concluded that the risk of PC is increased in families with
atypical multiple mole melanoma syndrome (FAMMM), also named
dysplastic nevus syndrome, other studies have not confirmed this
hypothesis (7,10). The controversy and puzzle regarding
a potential association between PC and melanoma in some patients
have been recently untangled owing to in-depth genetic studies
(11-15).
The case of a young patient diagnosed and treated
for PC is presented, in whom digital dermoscopic follow-up of
melanocytic nevi proved to be lifesaving, as it revealed the rapid
change of two nevi that acquired highly atypical features. The
common genetic background of PC and hereditary melanoma and the
optimal approach for these patients is also discussed.
Case report
A 38-year-old female patient was referred to the
Department of Dermatology, ‘Elias’ Emergency University Hospital,
in November 2019, for the clinical and dermoscopic assessment of
multiple pigmented nevi. The patient had been recently diagnosed
with PC with duodenal invasion and had undergone
pancreaticoduodenectomy, followed by the initiation of systemic
chemotherapy with 5-fluorouracil, irinotecan, oxaliplatin and
folinic acid (the FOLFIRINOX regimen). The study was approved by
the Ethics Committee of Elias Emergency University Hospital,
Bucharest, Romania (approval no. 4902). Written informed consent
was provided by the patient.
The patient had a positive family history for
oncologic diseases on the paternal side. Three paternal aunts had
been diagnosed with solid neoplasms: One had succumbed shortly
after being diagnosed with a digestive cancer (the patient was not
aware of the exact location of the neoplasm), another had been
diagnosed with breast cancer at a young age and the third with
thyroid cancer.
The physical examination was within normal limits,
except for the presence of numerous atypical melanocytic nevi
located on the trunk and limbs.
Close digital dermoscopic monitoring of the atypical
nevi, at three-month intervals was initially performed. Contrary to
the advice of the dermatologist, the patient missed several
scheduled control visits and only appeared a year later, when
significant changes in size, shape and structure could be observed
on digital dermoscopic examination in two melanocytic nevi, located
in the umbilical region and on the inferior abdominal integument,
respectively.
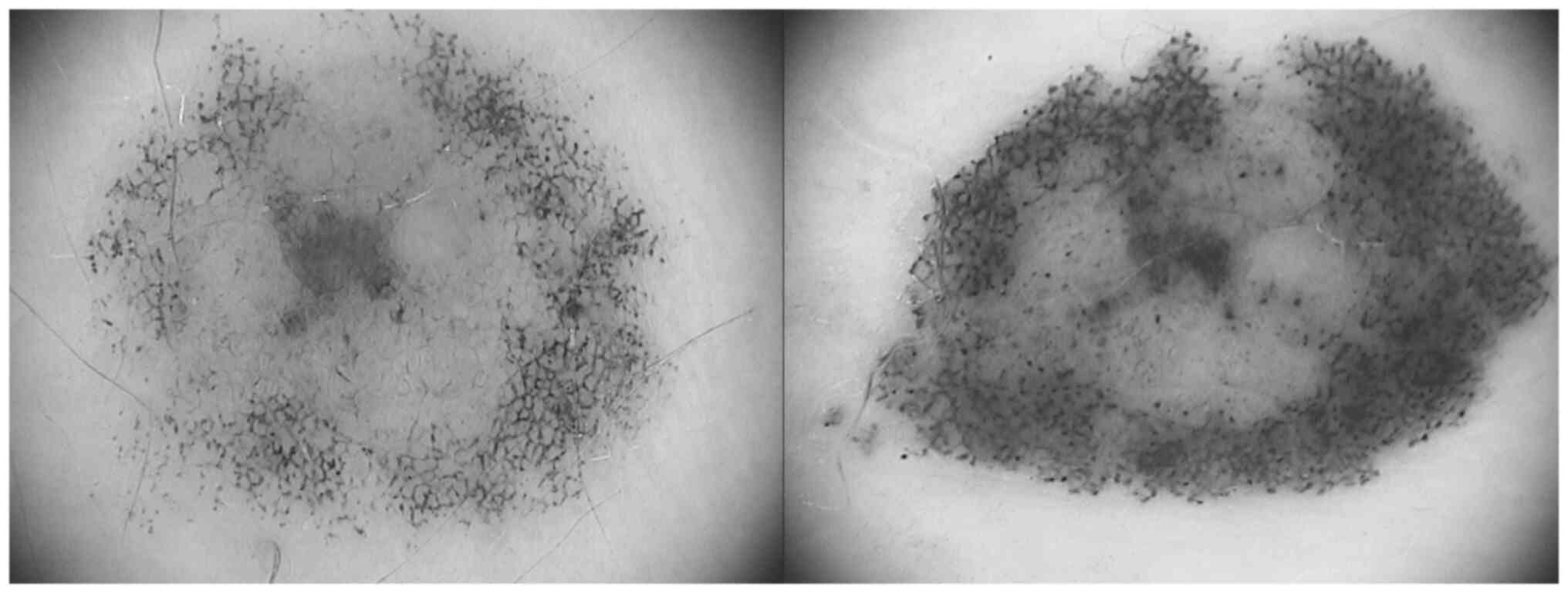
The melanocytic lesion located in the periumbilical
region was a reticular-homogenous nevus with a diameter of ~7.5 mm.
It presented marked asymmetry, irregular borders, atypical pigment
network, uneven pigmentation with multiple areas of
hyperpigmentation and structureless, hypopigmented areas,
irregularly distributed brown and black globules and dots, as well
as pseudopods (Fig. 1). Compared
with the previous examination, the nevus had increased in size, had
changed its shape and exhibited intensification of pigmentation. It
had also gained the striking atypical features aforementioned.
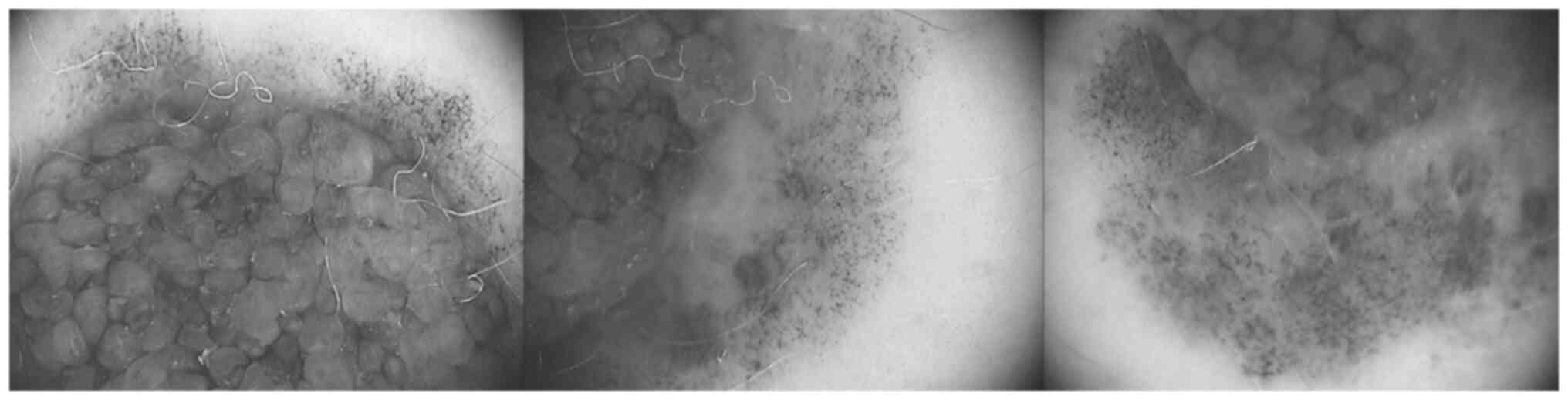
The second changing nevus was a compound nevus,
approximately 1 cm in diameter, located in the inferior abdominal
area. It exhibited asymmetry, was ill-defined, had irregular
borders and pigment variegation. The junctional component displayed
an atypical pigment network, with focal hyperpigmentation, and
irregularly distributed brown and black globules and dots (Fig. 2). Similar to the previously
described nevus, it had increased in size and had slightly changed
its shape.
The described nevi were surgically excised with a
0.5 cm safety margin.
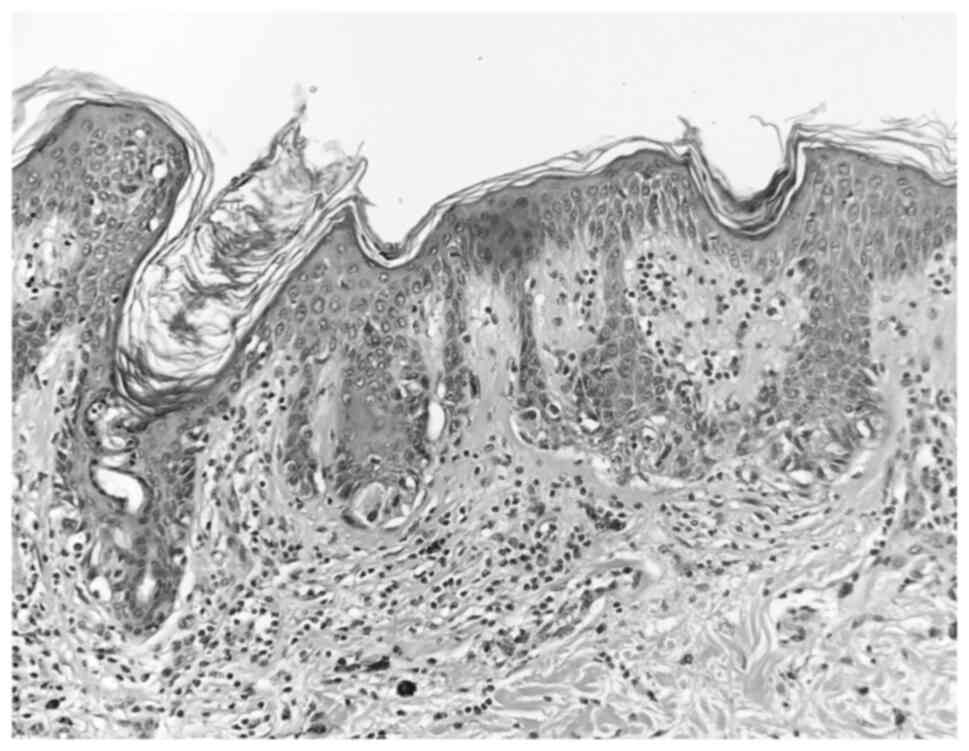
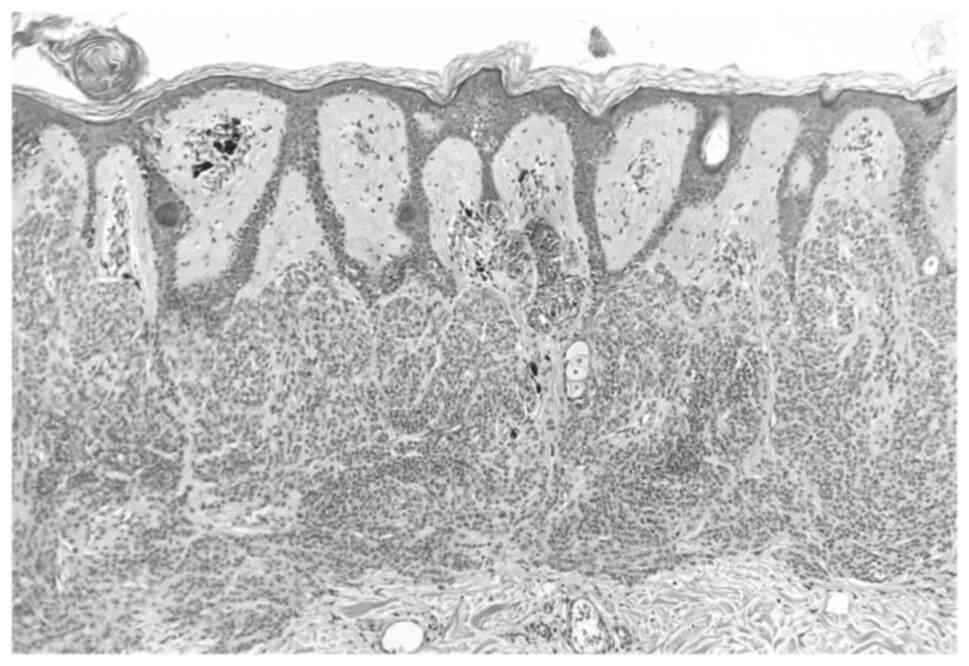
The histopathologic examination was performed with
the following parameters: The nevi were fixed with 10% neutral
buffered formalin at 21˚C for 24 h on histopathological sections of
a 4-µm thickness. Subsequently, staining was performed with
hematoxylin at 21˚C for 2 min and eosin at 21˚C for 30 sec. A light
microscope (Olympus BX43; Olympus Corporation) was used for
observation. The histopathological examination revealed two
completely excised dysplastic compound nevi (Figs. 3 and 4).
The melanocytic nevi of the patient were carefully
monitored thereafter, but no further changes have been noted.
Discussion
According to the numerous studies carried out thus
far, an increased risk of melanoma was observed in patients
diagnosed with non-melanoma skin cancers, hematologic malignancies,
nervous system neoplasms, testicular and breast cancer (11). Common genetic abnormalities,
immunological dysfunctions or exposure to environmental risk
factors may all play a role.
Conversely, in melanoma patients, a series of second
primary cancers appear to develop with a higher frequency than
anticipated. Among these are non-melanoma skin cancers, nervous
system cancer, chronic lymphocytic leukemia, breast, renal,
thyroid, oropharyngeal, testicular, digestive tract, connective
tissue, and lung cancers (11).
Additional primary cancers have also been detected
in the setting of FAMMM, also known as dysplastic nevus syndrome
(8,12,16),
an autosomal dominant disease with incomplete penetrance and high
phenotypic heterogeneity (17).
Kindreds with FAMMM are not only predisposed to develop melanoma,
but also certain extracutaneous cancers, particularly pancreatic,
breast, lung, and lymphoreticular system cancer (8,16,17).
Genetic susceptibility is decisive for the
appearance of hereditary and sporadic melanomas. The most important
melanoma susceptibility gene is cyclin-dependent kinase inhibitor
2A (CDKN2A)/p16, harbored on chromosome 9p21. Mutations of this
gene have been detected in 20-60% of families predisposed to
hereditary melanoma (13-15,18).
Mutations in CDKN2A/ARF and other genes, encoding for
cyclin-dependent kinase 4 (CDK4; located on chromosome 12q14.1),
telomerase reverse transcriptase (TERT; located on chromosome
5p15.33), melanocyte inducing transcription factor (MITF; located
on chromosome 3p13), ubiquitin carboxyl-terminal hydrolase (BAP1;
located on chromosome 3p21.1), protection of telomeres 1 (POT1;
located on chromosome 7q31.33) are less frequently encountered
(18).
CDKN2A was also revealed to be implicated in
pancreatic tumorigenesis (10,19,20),
thus explaining the propensity for PC in FAMMM families.
CDKN2A codes for p16 INK4 and p14 alternative
reading frame (ARF). p16 INK4 inhibits the activity of cyclin
D1-CDK4 complex, which phosphorylates the retinoblastoma protein in
order to allow progression to the G1 phase of the cell
cycle (21). Therefore, CDKN2A/p16
impedes cell growth and acts as a tumor suppressor gene. In
Northern Europe, one of the most prevalent mutations of CDKN2A/p16
associated with the development of melanoma is the p16-Leiden
mutation, represented by the deletion of 19 bp in exon 2 that leads
to loss of the tumor-suppressive function of p16 INK4(22). Approximately 70% of p16-Leiden
mutation carriers are estimated to develop melanoma (23) and 15-20% of them are expected to
develop PC (24-26).
The great clinical heterogeneity that characterizes
FAMMM syndrome and the variations in associated cancers results not
only from the different types of CDKN2A mutations, but also from
the influences of other genetic and environmental factors (27). Given the extremely aggressive
behavior and poor prognosis of both melanoma and PC, the
identification of individuals at risk for one or both of these
cancers and their close surveillance is of utmost importance.
Hence, further research has led to the detection of multiple
genetic factors that modify the risk of melanoma and PC in
p16-Leiden mutation carriers, such as melanocortin 1 receptor gene
(MC1R) variants that influence the risk of melanoma (28,29),
rs36115365-C, a single-nucleotide polymorphism which controls TERT
expression and is associated with increased risk of PC and
decreased risk of melanoma (30,31),
mutations in glutathione S-transferase genes GSTM1 and
GSTT1(32), as well as in the
vitamin D receptor gene that appear to have a slight protective
effect against melanoma (33).
The benefits of regular skin checkup of FAMMM
kindreds are indisputable. On the other hand, mortality from PC
even exceeds mortality attributable to melanoma (34,35).
Screening for PC is considerably more complicated. There is no
universally accepted screening protocol for PC, but annual
laboratory tests, such as the determination of the serum level of
alkaline phosphatase, pancreatic enzymes and tumor markers
(carcinoembryonic antigen and CA19.9) and imagistic investigations
(abdominal ultrasound, computed tomography, or magnetic resonance
imaging) are recommended in high-risk individuals (26,36,37).
In conclusion, clinicians should be aware of the
strong association between FAMMM and PC. The reported case
underlines the importance of regular skin checkups and screening
for PC in patients with dysplastic nevus syndrome.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LGP, CG, CN and MMM conceptualized and designed the
study and the methodology. CG and MMM supervised the study. SN, TT
and MMM participated in the acquisition, analysis and
interpretation of data. CS, AMP, TT and CB researched the
literature and drafted the manuscript. LGP, CG, CN, SN and MMM
reviewed and edited the final manuscript. All authors read and
approved the final version of the manuscript and agree to be
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Elias Emergency University Hospital, Bucharest, Romania (approval
no. 4902). Written informed consent was provided by the
patient.
Patient consent for publication
The patient provided written informed consent for
the publication of the data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vogt A, Schmid S, Heinimann K, Frick H,
Herrmann C, Cerny T and Omlin A: Multiple primary tumours:
Challenges and approaches, a review. ESMO Open.
2(e000172)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Altekruse SF, Kosary CL, Krapcho M, Neyman
N, Aminou R, Waldron W, Ruhl J, Howlader N, Tatalovich Z, Cho H
(eds), et al: SEER Cancer Statistics Review, 1975-2007. National
Cancer Institute, Bethesda, MD, 2010. Available from: http://seer.cancer.gov/csr/1975_2007.
|
|
3
|
Leone G, Pagano L, Ben-Yehuda D and Voso
MT: Therapy-related leukemia and myelodysplasia: Susceptibility and
incidence. Haematologica. 92:1389–1398. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau
JYN, Wang JC and Liu LF: Roles of DNA topoisomerase II isozymes in
chemotherapy and secondary malignancies. Proc Natl Acad Sci USA.
104:11014–11019. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Allen PJ, Kuk D, Castillo CF, Basturk O,
Wolfgang CL, Cameron JL, Lillemoe KD, Ferrone CR, Morales-Oyarvide
V, He J, et al: Multi-institutional validation study of the
American joint commission on cancer (8th edition) changes for T and
N staging in patients with pancreatic adenocarcinoma. Ann Surg.
265:185–191. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kang MJ, Jang JY, Chang YR, Kwon W, Jung W
and Kim SW: Revisiting the concept of lymph node metastases of
pancreatic head cancer: Number of metastatic lymph nodes and lymph
node ratio according to N stage. Ann Surg Oncol. 21:1545–1551.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ghiorzo P, Ciotti P, Mantelli M, Heouaine
A, Queirolo P, Rainero ML, Ferrari C, Santi PL, De Marchi R, Farris
A, et al: Characterization of ligurian melanoma families and risk
of occurrence of other neoplasia. Int J Cancer. 83:441–448.
1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bergman W, Watson P, de Jong J, Lynch HT
and Fusaro RM: Systemic cancer and the FAMMM syndrome. Br J Cancer.
61:932–936. 1990.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tucker MA, Fraser MC, Goldstein AM, Elder
DE, Guerry D IV and Organic SM: Risk of melanoma and other cancers
in melanoma-prone families. J Invest Dermatol. 100:350S–355S.
1993.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Goldstein AM, Fraser MC, Struewing JP,
Hussussian CJ, Ranade K, Zametkin DP, Fontaine LS, Organic SM,
Dracopoli NC, Clark WH Jr, et al: Increased risk of pancreatic
cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J
Med. 333:970–974. 1995.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Swerdlow AJ, Storm HH and Sasieni PD:
Risks of second primary malignancy in patients with cutaneous and
ocular melanoma in Denmark, 1943-1989. Int J Cancer. 61:773–779.
1995.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bergman W, Palan A and Went LN: Clinical
and genetic studies in six Dutch kindreds with the dysplastic
naevus syndrome. Ann Hum Genet. 50:249–258. 1986.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gabree M, Patel D and Rodgers L: Clinical
applications of melanoma genetics. Curr Treat Options Oncol.
15:336–350. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Goldstein AM, Chan M, Harland M,
Gillanders EM, Hayward NK, Avril MF, Azizi E, Bianchi-Scarra G,
Bishop DT, Bressac-de Paillerets B, et al: High-risk melanoma
susceptibility genes and pancreatic cancer, neural system tumors,
and uveal melanoma across GenoMEL. Cancer Res. 66:9818–9828.
2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Goldstein AM, Chan M, Harland M, Hayward
NK, Demenais F, Bishop DT, Azizi E, Bergman W, Bianchi-Scarra G,
Bruno W, et al: Features associated with germline CDKN2A mutations:
A GenoMEL study of melanoma-prone families from three continents. J
Med Genet. 44:99–106. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Lynch HT, Fusaro RM, Pester J, Oosterhuis
JA, Went LN, Rumke P, Neering H and Lynch JF: Tumour spectrum in
the FAMMM syndrome. Br J Cancer. 44:553–560. 1981.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fusaro RM, Lynch HT, Lynch JF and Madsen
NJ: Phenotypic variation and systemic cancer in the FAMMM syndrome.
Pigment Cell Res. 1:152–157. 1988.
|
|
18
|
Leachman SA, Lucero OM, Sampson JE,
Cassidy P, Bruno W, Queirolo P and Ghiorzo P: Identification,
genetic testing, and management of hereditary melanoma. Cancer
Metastasis Rev. 36:77–90. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Whelan AJ, Bartsch D and Goodfellow PJ:
Brief report: A familial syndrome of pancreatic cancer and melanoma
with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med.
333:975–977. 1995.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Caldas C, Hahn SA, da Costa LT, Redston
MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ and Kern
SE: Frequent somatic mutations and homozygous deletions of the p16
(MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 8:27–32.
1994.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Serrano M, Hannon GJ and Beach D: A new
regulatory motif in cell-cycle control causing specific inhibition
of cyclin D/CDK4. Nature. 366:704–707. 1993.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Gruis NA, van der Velden PA, Sandkuijl LA,
Prins DE, Weaver-Feldhaus J, Kamb A, Bergman W and Frants RR:
Homozygotes for CDKN2 (p16) germline mutation in Dutch familial
melanoma kindreds. Nat Genet. 10:351–353. 1995.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bishop DT, Demenais F, Goldstein AM,
Bergman W, Bishop JN, Bressac-de Paillerets B, Chompret A, Ghiorzo
P, Gruis N, Hansson J, et al: Geographical variation in the
penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst.
94:894–903. 2002.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vasen HF, Gruis NA, Frants RR, van Der
Velden PA, Hille ET and Bergman W: Risk of developing pancreatic
cancer in families with familial atypical multiple mole melanoma
associated with a specific 19 deletion of p16 (p16-Leiden). Int J
Cancer. 87:809–811. 2000.PubMed/NCBI
|
|
25
|
de Snoo FA, Bishop DT, Bergman W, van
Leeuwen I, van der Drift C, van Nieuwpoort FA, Out-Luiting CJ,
Vasen HF, ter Huurne JA, Frants RR, et al: Increased risk of cancer
other than melanoma in CDKN2A founder mutation
(p16-Leiden)-positive melanoma families. Clin Cancer Res.
14:7151–7157. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vasen H, Ibrahim I, Ponce CG, Slater EP,
Matthäi E, Carrato A, Earl J, Robbers K, van Mil AM, Potjer T, et
al: Benefit of surveillance for pancreatic cancer in high-risk
individuals: Outcome of long-term prospective follow-up studies
from three European expert centers. J Clin Oncol. 34:2010–2019.
2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bergman W and Gruis N: Familial melanoma
and pancreatic cancer. N Engl J Med. 334:471–472. 1996.PubMed/NCBI
|
|
28
|
van der Velden PA, Sandkuijl LA, Bergman
W, Pavel S, van Mourik L, Frants RR and Gruis NA: Melanocortin-1
receptor variant R151C modifies melanoma risk in Dutch families
with melanoma. Am J Hum Genet. 69:774–779. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Demenais F, Mohamdi H, Chaudru V,
Goldstein AM, Newton Bishop JA, Bishop DT, Kanetsky PA, Hayward NK,
Gillanders E, Elder DE, et al: Association of MC1R variants and
host phenotypes with melanoma risk in CDKN2A mutation carriers: A
GenoMEL study. J Natl Cancer Inst. 102:1568–1583. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Fang J, Jia J, Makowski M, Xu M, Wang Z,
Zhang T, Hoskins JW, Choi J, Han Y, Zhang M, et al: Functional
characterization of a multi-cancer risk locus on chr5p15.33 reveals
regulation of TERT by ZNF148. Nat Commun. 8(15034)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Rachakonda S, Kong H, Srinivas N,
Garcia-Casado Z, Requena C, Fallah M, Heidenreich B, Planelles D,
Traves V, Schadendorf D, et al: Telomere length, telomerase reverse
transcriptase promoter mutations, and melanoma risk. Genes
Chromosomes Cancer. 57:564–572. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Fortes C, Mastroeni S, Melchi F, Anzidei
P, Innocenzi L, Giovinazzo R, Antonelli G, Pasquini P and
Venanzetti F: P5 polymorphisms in GSTM1, GSTT1, coffee consumption
and cutaneous melanoma risk. Melanoma Res. 20:e45–e46. 2010.
|
|
33
|
Randerson-Moor JA, Taylor JC, Elliott F,
Chang YM, Beswick S, Kukalizch K, Affleck P, Leake S, Haynes S,
Karpavicius B, et al: Vitamin D receptor gene polymorphisms, serum
25-hydroxyvitamin D levels, and melanoma: UK case-control
comparisons and a meta-analysis of published VDR data. Eur J
Cancer. 45:3271–3281. 2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hille ET, van Duijn E, Gruis NA, Rosendaal
FR, Bergman W and Vandenbroucke JP: Excess cancer mortality in six
Dutch pedigrees with the familial atypical multiple mole-melanoma
syndrome from 1830 to 1994. J Invest Dermatol. 110:788–792.
1998.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lynch HT, Smyrk T, Kern SE, Hruban RH,
Lightdale CJ, Lemon SJ, Lynch JF, Fusaro LR, Fusaro RM and
Ghadirian P: Familial pancreatic cancer: A review. Semin Oncol.
23:251–275. 1996.PubMed/NCBI
|
|
37
|
Brentnall TA, Bronner MP, Byrd DR, Haggitt
RC and Kimmey MB: Early diagnosis and treatment of pancreatic
dysplasia in patients with a family history of pancreatic cancer.
Ann Intern Med. 131:247–255. 1999.PubMed/NCBI View Article : Google Scholar
|