Introduction
Stroke is the fifth leading cause of death in the
United States, affecting >795,000 individuals each year
(1). Elderly patients with
underlying chronic and neurodegenerative conditions such as
Alzheimer's and Parkinson's disease exhibit a higher risk of
ischemic strokes that often lead to long term disability (2). Partial or complete blockage of the
cerebral arteries during cerebral ischemia restricts blood flow and
affects the metabolic needs of the brain (3). As a result, neuronal function is
impaired and cell death occurs due to excessive glutamate,
inadequate supply of oxygen and glucose, collapse in energy
dependent processes, increase in reactive oxygen species (ROS) and
disruption of the blood-brain barrier (BBB) (3,4).
Glutamate is an amino acid excitatory
neurotransmitter involved in neuronal development, learning, memory
and aging (5,6). However, high concentrations of
glutamate released under pathologic stimuli overwhelms neurons and
contributes to increased oxidative stress and neuronal damage
(7). Glutamate neurotoxicity
occurs when a high concentration of extracellular glutamate
inhibits the glutamate/cystine antiporter, lowering the level of
cystine in the cell. Cystine is the precursor of glutathione (GSH),
and upon depletion, elevates intracellular ROS resulting in
oxidative stress (8). Accumulation
of ROS leads to an increase in the level of Ca2+ in the
mitochondrial matrix (9). High
levels of Ca2+ sensitizes the formation of the
mitochondrial permeability transition pore (MPTP), leading to the
collapse of the mitochondrial membrane potential (MMP) and cell
death (9-11).
Furthermore, binding of glutamate to the corresponding receptors
activates downstream signalling pathways, such as the MAPK
signalling pathway, which serves a central role in neuronal
plasticity, survival, death and memory formation (12). Moreover, an increase in Erk1/2
mediated by ROS further exacerbates neurotoxicity (13). In contrast, a previous study
reported that Erk1/2 serves a key role in the survival of neurons
during post ischemic recovery (13,14).
Perturbation of cellular Ca2+ homeostasis and increased
oxidative stress activates the JNK and p38 signalling pathways, and
promotes neuronal cell death (15).
B355252, a phenoxythiophene sulfonamide compound,
exerts a number of neuroprotective effects. It was initially
identified from a proprietary library of chemical compounds as a
potentiator of nerve growth factor (NGF)-primed neurite outgrowth
and elongation in a neuronal cell model (16). In the presence of sub-physiological
concentrations of NGF, B355252 promotes differentiation and
production of axon-like processes in pheochromocytoma cell line
PC12, and its derivative neuroscreen-1(16). Previous studies have reported the
anti-apoptotic and antioxidant properties of B355252 in chemical
models of ischemia and Parkinson's disease in vitro
(17-19).
In the present study, B355227 (molecular weight, 494.3 DA), a
substituted analogue of B355252, synthesized by our medicinal
chemists during the SAR studies of B355252, was shown to readily
cross the BBB in an in vitro assay. B355227 exerted
neuroprotective effects in a glutamate-induced oxidative stress
model, and protected HT22 cells from oxidative stress via the
modulation of a number of key effectors, such as GSH, ROS and
Ca2+ overload. In addition, B355227 downregulated the
activation of Erk1/2, JNK and P38 protein families that were
reported to serve key roles in ischemia (18).
Materials and methods
Cell culture
Mouse hippocampal HT22 and endothelial bEND.3 cell
lines were purchased from the American Type Culture Collection.
Cells were maintained at 37˚C in growth media (GM) consisting of
DMEM (Cytiva) supplemented with 10% FBS (Cytiva), 1%
penicillin/streptomycin (Cytiva) and 1% L-glutamine (Lonza Group,
Ltd.) in a humidified incubator with 5% CO2. For all
experiments, cells were incubated at 37˚C in a humidified incubator
with 5% CO2, unless indicated otherwise.
Nuclear Magnetic Resonance
(1H and 13C) of B355227
B355227 was synthesized and characterized by the
Medicinal Chemistry Group, Department of Pharmaceutical Science,
North Carolina Central University (Durham, USA). All solvents and
reagents were obtained from commercial sources (16). Proton nuclear magnetic resonance
(1H NMR) spectra and carbon nuclear magnetic resonance (13C NMR)
spectra were recorded on a Varian VNMRS-500 (500 MHz)
spectrometer.
Assessment of cell viability
Cell viability was assessed using the redox
indicator dye resazurin sodium salt (Sigma-Aldrich; Merck KGaA).
HT22 cells were seeded in 96-well plates at a density of
2x104 cells/well and incubated overnight at 37˚C. Next
day, the cells were treated with drugs (B355227 with and without
glutamate) in GM at 37˚C for 24 h. Resazurin was added to the wells
to a final concentration of 1%, and cells were incubated at 37˚C
for 3 h. Fluorescence was measured using a PHERAstar microplate
reader (BMG Labtech), at an excitation wavelength of 540 nm, and an
emission wavelength of 590 nm.
Measurement of trans-endothelial
electrical resistance (TEER)
bEND.3 cells were seeded at a density of
1.3x106 cells/well in Transwell inserts and cultured for
12 days in GM at 37˚C in 5% CO2. The TEER was measured
daily using End-Ohm chopstick electrodes connected to an EVOM
resistance meter (World Precision Instruments, Inc.). The TEER
value of each well was calculated by subtracting the resistance of
blank inserts without cells from the sample inserts with cells.
Values were expressed as Ω cm2.
Evaluation of in vitro barrier
function
Following the incubation of bEND.3 cells in
Transwell inserts for 12 days as previously described, cells were
washed and incubated in serum-free media (SFM) containing DMEM, 1%
L-glutamine and 1% penicillin/streptomycin for 1 h at 37˚C. SFM was
replaced with 1 ml of 1X Hanks' Buffered Saline Solution (HBSS;
Lonza) at the apical and luminal sides of the Transwell insert,
followed by exposure to vehicle or 100 µg/ml Fluorescein
isothiocyanate-dextran (FITC-dextran ; 40 kDa; Sigma-Aldrich; Merck
KGaA) at 37˚C for 1 h. After incubation, media from the luminal
side of the Transwell insert was collected and the fluorescence was
measured using spectrophotometry at an excitation wavelength of 485
nm, and an emission wavelength of 520 nm. Subsequently, 20 µM
Caffeine (molecular weight, 194 kDa), a low molecular weight
compounds known to be permeable, and 20 µM Imatinib (molecular
weight, 493 kDa) and 20 µM Axitinib (molecular weight, 389 kDa),
compounds known to be impermeable to the BBB were used to further
validate the assay. Briefly, after 14 days in culture of
homogeneous bEND.3 monolayer culture and formation of BBBs, as
assessed by TEER value using End-Ohm chopstick electrodes connected
to an EVOM resistance meter, were pre-incubated in SFM at 37˚C for
1 h, which was subsequently replaced with 1 ml of 1X HBSS in both
chambers of the Trans-well plates. Vehicle or 20 µM of caffeine,
imatinib and axitinib were added to the cells and incubated at
37˚C. Samples were collected from the luminal side at 1, 2 and 3 h,
vacuum dried and dissolved in 300 µl of high-performance liquid
chromatography (HPLC) grade ultra-pure distilled water for further
analysis.
HPLC analysis
A total of 40 µl of luminal side HBSS sample
collected after exposure of bEND.3 cells to B355227 were analyzed
at room temp (22˚C) using Agilent 1220 Infinity II LC reverse-phase
system detector (Agilent Technologies, Inc.) with diode array
variable wavelength (190-600 nm). Separation of analytes was
achieved in mobile phase consisting of 10 to 95%
acetonitrile/H2O with 0.1% formic acid, at a flow rate
of 0.5 ml/min for 7 min on Waters Sunfire OBD using C18 column
(3x100 mm; 5 µM; Waters Corporation).
Compound permeability assay
An in vitro BBB permeability assay was
performed using bEND.3 cells that had been incubated in Transwell
inserts for 12 days as previously described, when maximum TEER was
observed in culture. Cells were treated with vehicle or 20 µM
B355227 (determined using a dose response toxicity assay) in 1 ml
GM at the apical side of the chamber. A total of 1 ml GM was added
to the luminal side of the chamber and incubated for 1 h at 37˚C in
5% CO2. Following incubation, media from the luminal
side was retrieved and centrifuged at room temp for 10 min at 150 x
g to remove cell debris. The supernatant was referenced as
conditioned media (CM) and was used in further neuroprotection
assays.
Immunofluorescence
Following the BBB permeation assay in bEND.3 cells,
expression of zonula occludens-1 (ZO-1) protein was analyzed using
immunofluorescence, as previously described (20). Cells employed in the permeability
assay were washed with PBS and fixed in 4% formaldehyde at room
temp for 15 min. The cells were blocked in 10% donkey serum
(Rockland Immunochemicals, Inc.) for 1 h at room temp and incubated
overnight at 4˚C with anti-rabbit anti-ZO-1 antibody (1:1,000; cat.
no. 40-2200; Thermo Fisher Scientific, Inc.) diluted 200-fold.
Following primary incubation, the cells were washed with PBS and
incubated with Alexa Fluor 488-conjugated donkey anti-rabbit IgG
secondary antibody (1:1,000; cat. no. A21206; Thermo Fisher
Scientific, Inc.) room temperature for 1 h. Cells were washed three
times with PBS and mounted on slides with VECTASHIELD (Vector
Laboratories, Inc.). Images of the stained cells were captured
using a FV3000 confocal laser scanning microscope (magnification,
x40; Olympus Corporation).
Neuroprotection assay with CM
HT22 cells (2x104 cells/well) grown
overnight at 37˚C in 96-well plates were exposed to 100 µl CM in
the presence or absence of 5 mM glutamate (Sigma-Aldrich; Merck
KGaA). Following 24 h incubation at 37˚C in 5% CO2, cell
viability was assessed using the resazurin assay, as previously
described.
Analysis of GSH
Determination of reduced GSH was performed as
described by Gliyazova et al (18) using MCB glutathione detection kit
(cat. no. 30019; Biotium, Inc.). Briefly, a total of
6.5x105 HT22 cells grown overnight at 37˚C in 64-mm
plates were treated with B355227 with and without glutamate for 24
h. Cells were subsequently counted, and 1x106 cells were
lysed and centrifuged at 700 x g for 5 min at 22˚C. A total of 5 µl
cell lysate was mixed with 5 µl of 10 mM MCB and 2 µl of
glutathione-S-transferase (GST) reagent according to the protocol
provided with the MCB glutathione detection kit, followed by 30 min
incubation at 37˚C. Fluorescence was measured using a PHERAStar
multipurpose fluorescence reader (BMG LabTech GmbH) at an
excitation wavelength of 394 nm, and an emission wavelength of 490
nm.
Determination of ROS
Intracellular ROS was determined using
2',7'-dichlorofluorescein diacetate (H2DCFDA; cat. no.
D399; Thermo Fisher Scientific, Inc.). A total of 2x104
HT22 cells were cultured overnight at 37˚C in 96-well plates and
treated with B355227 in the presence or absence of glutamate for 8
h. The cells were subsequently washed with phosphate buffered
saline (PBS) and incubated with 10 µM H2DCFDA in phenol
red-free DMEM (cat. no. 21063029; Thermo Fisher Scientific, Inc.)
for 30 min at 37˚C. Cells were washed again in PBS and the
fluorescence was measured using a PHERAStar microplate reader (BMG
LabTech) at an excitation wavelength of 485 nm, and an emission
wavelength of 520 nm. After the fluorometric measurements, the
cells were lifted from each well using trypsin-EDTA (Thermo Fisher
Scientific, Inc.) and counted in the Vi-CELL XR cell viability
analyser (Beckman Coulter, Inc.) to determine the total number of
cells. The data was normalized by dividing the total fluorescence
intensity at every time-point by the total number of cells.
Measurement of Ca2+
Changes in intracellular Ca2+ levels were
measured fluorometrically using Fura-2 AM dye (cat. no. 47989;
Sigma-Aldrich; Merck KGaA). HT22 cells were seeded at
6.5x105 in 64-mm plates and cultured overnight at 37˚C
were treated with B355227 with and without glutamate for 24 h at
37˚C. Following incubation, the cells were detached with
trypsin-EDTA, counted in the Vi-CELL and 1x106 cells
from each treatment group (untreated control, glutamate, B355227
and B355227 + glutamate groups) were incubated with 2.5 M Fura-2 AM
in phenol red-free DMEM (cat. no. 21063029; Thermo Fisher
Scientific, Inc.) for 30 min at 37˚C. Cells were washed with PBS,
resuspended in phenol-free DMEM containing 10% FBS and incubated at
37˚C. After 30 min incubation, cells were sequentially excited at
340 and 380 nm using a Fluoromax-4 spectrofluorometer (HORIBA
Scientific) and the peak emission signal was measured at 510 nm.
Changes in Ca2+ concentration were measured using the
ratio of the emission signals obtained, and the results were
interpreted as relative values.
Measurement of the MMP
HT22 (6x106 cells) cultured overnight at
37˚C on cover slips and exposed to 5 µM B355227 (Medicinal
Chemistry Group, North Carolina Central University) with and
without 5 mM glutamate (Sigma-Aldrich; Merck KGaA) were treated
with 200 nM MitoTracker Red CMXRos dye (Thermo Fisher Scientific,
Inc.) in SFM for 30 min at 37˚C. The cells were washed with PBS and
re-incubated in GM for 15 min at 37˚C. Following incubation, the
cells were fixed with 4% formaldehyde in PBS for 15 min at room
temp, and cell nuclei were stained with 10 µg/ml DAPI in PBS for 5
min at room temp. Cover slips were mounted on the slides and images
were captured using a Zeiss LSM-800 confocal scanning microscope
(Zeiss GmbH) at an excitation wavelength of 579 nm, and an emission
wavelength of 599 nm (magnification, x60). The images were analysed
with ImageJ software, version 1.52 (National Institutes of Health),
and the fluorescence intensity normalized as follows: Corrected
total fluorescence (CTCF)=integrated density - (area of collected
cell x mean fluorescence of background reading).
Immunoblot analysis
Immunoblots were performed according to a previous
study by Pokharel et al (21). Briefly, HT22 cells
(1x107) treated for 24 h at 37˚C with 5 mM glutamate
supplemented with or without 5 µM B355227 were lysed in RIPA buffer
containing protease inhibitor cocktail (Thermo Fisher Scientific,
Inc.). The protein content was quantified using a Pierce BCA
protein assay kit (Thermo Fisher Scientific, Inc). A total of 30 µg
protein were electrophoresed on 4-12% SDS-PAGE and transferred to
PVDF membranes. The membranes were subsequently blocked with
Intercept (PBS) blocking buffer (cat. no. 927-70001; LI-COR
Biosciences) for 1 h at room temp and incubated overnight at 4˚C
with the following primary antibodies obtained from Cell Signaling
Technology, Inc.: Anti-Erk1/2 (1:1,000; cat. no. 4695),
anti-phospho-Erk1/2 (1:2,000; cat. no. 9106), anti-p38 (1:1,000;
cat. no. 8690), anti-phospho-p38 (1:2,000; cat. no. 9216), anti-JNK
(1:1,000; cat. no. 9252), anti-phospho-JNK (1:2,000; cat. no.
9255), anti-Bax (1:1,000; cat. no. 2772), anti-Bcl-2 (1:1,000; cat.
no. 3498) and anti-β-actin (1:1,000; cat. no. 3700) as reference
protein. The membranes were rinsed four times for 5 min at room
temperature in 1X TBST (0.2% Tween-20) and incubated at room
temperature with 15,000-fold dilution of IRDye 800CW-conjugated
donkey anti-rabbit IgG (cat. no. 926-32213; LI-COR Biosciences) or
20,000-fold dilution of IRDye 680LT-conjugated donkey anti-mouse
IgG (cat. no. 926-68022; LI-COR Biosciences) for 1 h. The membranes
were rinsed four times in 1X TBST for 5 min, and finally in 1X TBS
to remove residual Tween-20. Images were captured using an Odyssey
IR imaging system and signals were analysed using Odyssey 2.0
software (LI-COR Biosciences).
Statistical analysis
Statistical analyses were performed in GraphPad
Prism version 7 (GraphPad Software, Inc.). Data are presented as
the mean ± standard deviation of at least three biological
replicates in at least three independent experiments. Significant
differences between groups were determined using one-way ANOVA
followed by Tukey's multiple comparisons post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Structural characterization of
B355227
Synthesis of B355227, a substituted analogue of
B355252 was confirmed by NMR. Chemical shifts (δ) are reported in
parts per million (ppm) using tetramethylsilane as an internal
standard (spectral data not shown). Multiplicities were reported
using the following abbreviations: Br, broad; s, singlet; d,
doublet; t, triplet; q, quartet; m, multiplet. 1H NMR (CDCl3, 500
MHz) δ 2.97 (t, 4H, J=5.0 Hz), 3.12 (t, 4H, J=5.0 Hz), 3.76 (s,
3H), 4.18 (s, 2H), 6.52 (dd, 1H, J=2.0, 8.0 Hz), 6.64 (t, 1H, J=2.0
Hz), 6.73 (dd, 1H, J=2.0, 8.5 Hz), 6.76 (s, 1H), 6.78-6.83 (m, 2H),
7.22 (ABq, 2H, J=8.5 Hz), 7.28 (s, 1H); 13C NMR (CDCl3, 125 MHz) δ
45.7, 47.4, 49.5, 55.2, 105.2, 107.9, 111.2, 112.6, 113.4, 113.6,
120.1, 128.2, 129.8, 130.3, 130.7, 137.4, 153.3, 158.2, 158.7,
159.9. The values indicate estimated chemical shift numbers of
1H and 13C of B355227 in parts per million
(ppm).
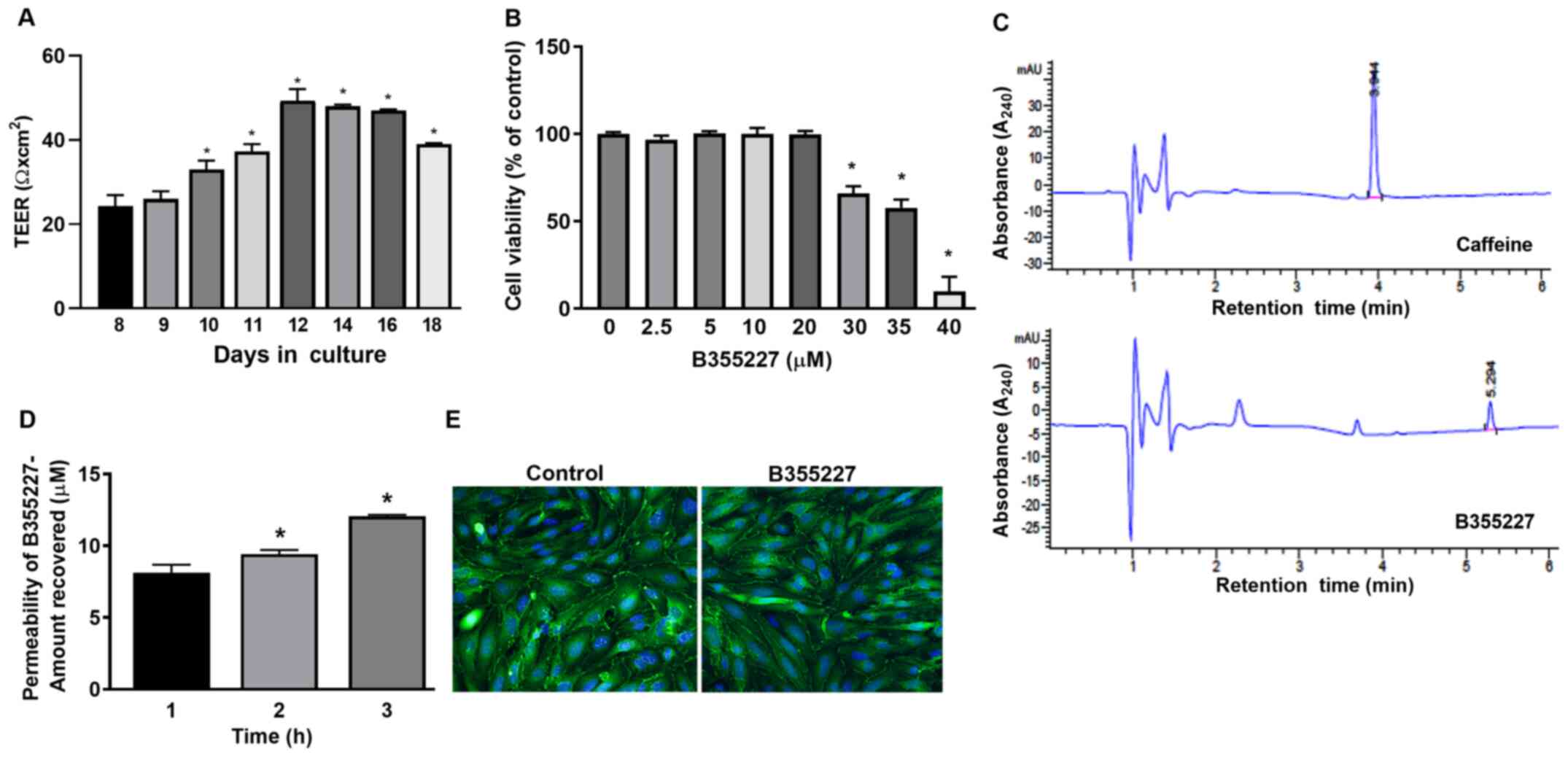
B355227 is permeable across bEND.3
cells in vitro
An in vitro permeability assay was performed
to determine the permeability of B355227 through the bEND.3 BBB
monolayer assay system. TEER was initially measured prior to the
permeability experiments to ensure model barrier integrity.
Expression levels of the ZO-1 protein were analyzed using
immunofluorescence to ensure cell confluency. A gradual and
significant rise in TEER values was observed after 10 days in
culture compared with day 8, attaining a maximum of 50 Ω
cm2 on day 12, which remained at a high level until day
16, and thereafter declined on day 18 (Fig. 1A). TEER is a measure of electrical
resistance in cells and reflects the formation of functional
barriers. A rise in TEER value is associated with an increase in
claudin-5 and ZO-1 expression at the contact points between cells
(22). Immunofluorescence detected
similar pattern of ZO-1 localization on the membrane contact points
of bEND.3 cells treated with B35227 compared with untreated
control, which confirmed the integrity of tight junctions in the
cells treated with 20 µM B355227 (Fig.
1E).
The maximum nontoxic dose of B355227 in bEND.3 cells
was established by evaluating the viability of the cells after
treatment with varying concentrations (2.5-40 µM) of the compound
for 24 h. B355227 exhibited no adverse effect on bEND.3 cells at
concentrations up to 20 µM, but exhibited dose-dependent toxicity
at concentrations ≥30 µM (Fig.
1B). Following the establishment of the highest non-toxic
concentration of B355227 as 20 µM, the permeability assay was
performed to determine the capacity of B355227 to cross the bEND.3
BBB. B355227 was detected in the luminal chamber using HPLC
(Fig. 1C), and a time-dependent
increase in compound concentration in the luminal component of the
Transwell plate was observed (Fig.
1D). A total of ~8 µM of B355227 was recovered in the luminal
side of the Transwell after 1 h exposure of cells to 20 µM of
B355227 compared with samples taken after addition of the compound
without incubation (Fig. 1D). The
quantity recovered increased to 9 µM and 12 µM at 2 and 3 h,
respectively (Fig. 1D). Following
the permeability assay, the cells were stained with anti-ZO-1
antibody to confirm that detection of B355227 in the luminal side
was a result of diffusion or paracellular transport across the
membrane and not due to a compromised or leaky membrane. No
difference was observed in the levels of ZO-1 expression in the
control group compared with the drug treated cells, which indicated
the passage of B355227 through the BBB in the experimental model
(Fig. 1E).
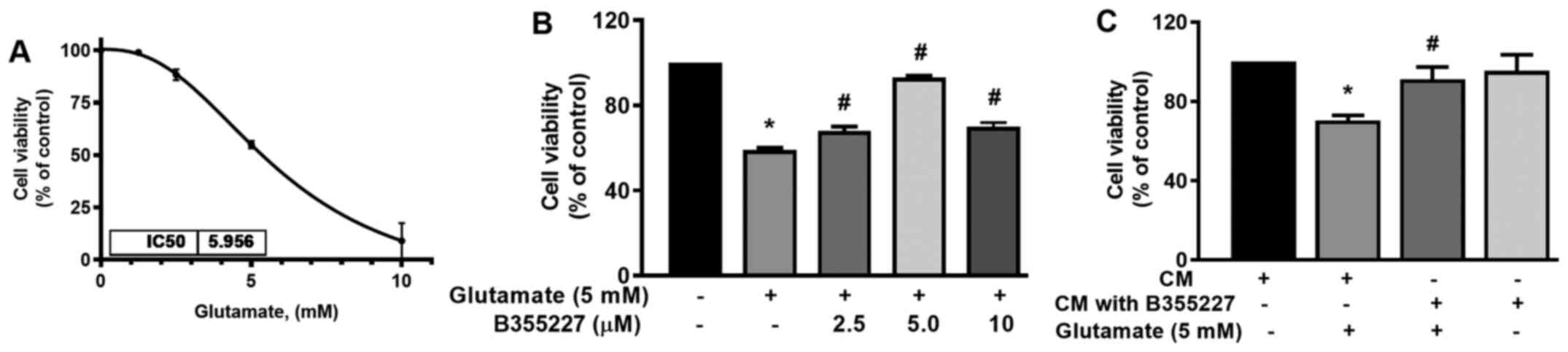
B355227 protects against
glutamate-induced toxicity
The neuroprotective effect of B355227 against
glutamate-induced toxicity was assessed in HT22 cells. The
IC50 of glutamate was established in HT22 cells to
determine the optimal concentration required for subsequent
experiments. The IC50 of glutamate in the cell line was
5.96 mM (Fig. 2A). Thus, cells
were treated with increasing concentrations of B355227 (2.5, 5 and
10 µM) in the presence of 5 mM glutamate, and assessed for
retention of viability. The results of the present study
demonstrated that 5 mM glutamate significantly reduced the
viability of HT22 cells by 40% compared with untreated control
(Fig. 2B). Following co-treatment
of 2.5, 5 or 10 µM B355227 with 5 mM glutamate, there was a
significant dose-dependent increase in cell viability by 10, 30 and
10%, respectively, compared with cells treated solely with
glutamate (Fig. 2B). Furthermore,
to validate the bioactivity of B355227, HT22 cells were treated
with CM recovered from the luminal side of the assay chamber
following the B355227 permeability assay, in the presence or
absence of 5 mM glutamate. Cells exposed to glutamate in CM
demonstrated an increased survival of ~20% compared with the cells
treated with glutamate alone (Fig.
2C). Furthermore, cells exposed to B355227 CM for 24 h revealed
no evidence of toxicity, and no significant difference was observed
in cell viability compared with cells exposed to the control
CM.
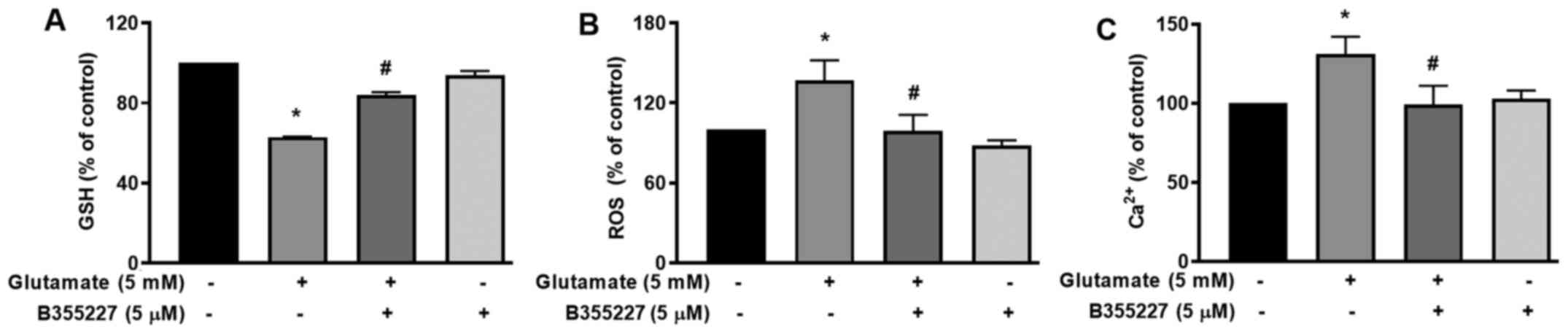
B355227 increases GSH, reduces ROS and
Ca2+, and blocks dissipation of the MMP in HT22
cells
The cellular mechanisms underlying the protective
actions of B355252 in HT22 cells undergoing glutamate-induced
oxidative stress were investigated. GSH, an antioxidant that plays
a role in scavenging ROS in response to oxidative stress (23) was quantified. The results of the
present study revealed a significant decrease of ~40% in cellular
glutathione content following exposure to glutamate for 24 h,
compared with the untreated control cells (Fig. 3A). In the presence of B355227, the
decline in GSH triggered by glutamate treatment was substantially
mitigated, while the compound alone exhibited no effect on GSH
compared with the control cells (Fig.
3A). As GSH serves a key role in the detoxification of H2O2 and
modulates ROS (24), ROS levels in
cells was investigated using the oxidative stress indicator,
H2DCFDA. The results of the present study indicated a
1.4-fold increase in ROS accumulation in cells exposed to
glutamate, compared with the untreated cells (Fig. 3B). Treatment with 5 µM B355227
considerably reduced the glutamate-induced increase of ROS by
~1.4-fold, which suggested that B355227 possessed antioxidant
activity.
The mutual interplay of ROS and Ca2+
signalling serves a crucial role in controlling functional changes
that accompany a number of pathophysiological events that include
cellular dysfunction leading to neurodegenerative disorders,
cardiovascular diseases and cancer (25,26).
Therefore, to determine whether B355227 exerted its neuroprotective
effects through modulation of Ca2+, HT22 cells were
exposed to glutamate alone or in conjunction with B355227.
Following exposure to glutamate, the intracellular Ca2+
levels increased by ~25%, compared with the control cells (Fig. 3C). However, when co-treated with
B355227, the glutamate-dependent increase in Ca2+ was
attenuated, and the intracellular Ca2+ levels returned
to a level comparable to those observed in untreated cells
(Fig. 3C).
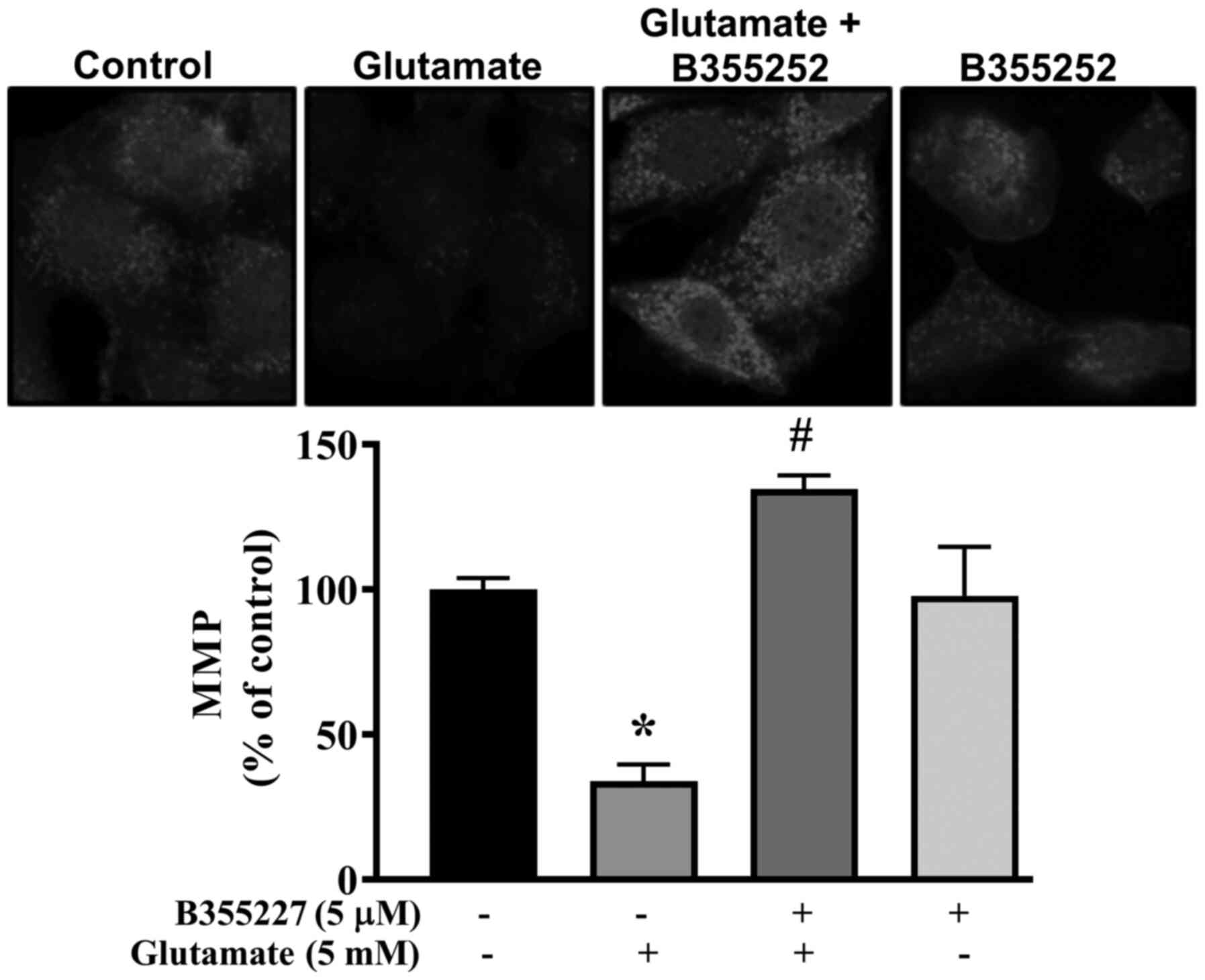
Collapse of the MMP is associated with cell death in
glutamate-induced oxidative injury (27). Thus, the MMP in HT22 cells exposed
to glutamate with or without B355227 was investigated using
MitoTracker Red CMXRos dye. Exposure of HT22 cells to glutamate
resulted in significant disruption of the MMP, indicated by a ~60%
decrease in fluorescence intensity compared with untreated control
cells (Fig. 4). In contrast, the
signal intensity of the mitochondrial dye was concentrated at a
significantly high level in cells co-treated with glutamate and
B355227 compared to cells treated with glutamate alone. Similar
values of fluorescence intensity were observed in cells treated
with B355227 alone and the untreated control cells.
Influence of B355227 on MAPK and
Bax/Bcl-2 signalling pathways
Proteins of the MAPK family serve a dynamic role in
glutamate-induced oxidative stress (28). To further explore the
neuroprotective effects of B355227 following glutamate treatment,
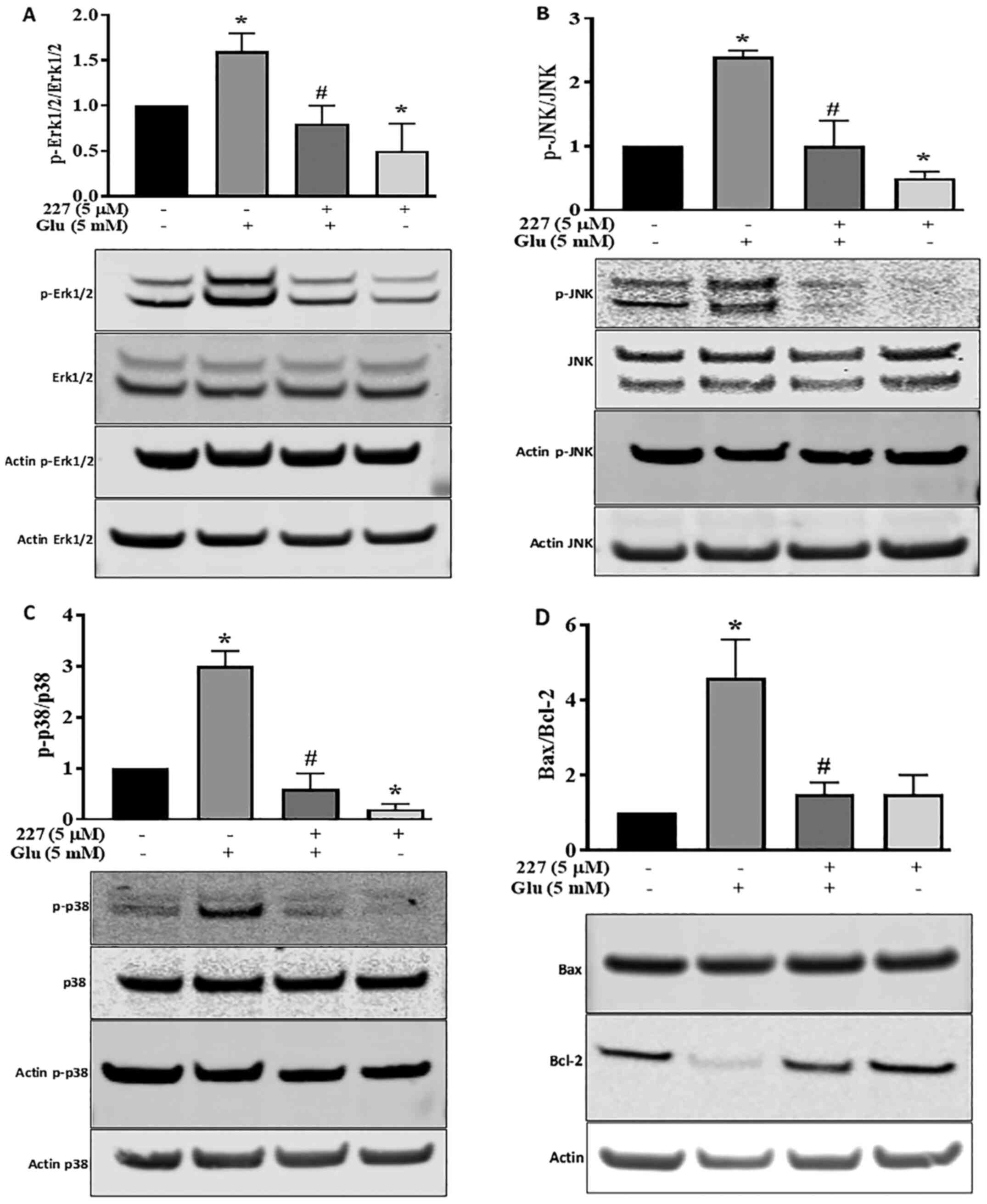
the MAPK pathway was investigated. The phosphorylation of Erk1/2,
JNK and p38 proteins, along with the expression levels of
pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins were
determined. Quantitative analysis of western blotting bands
demonstrated a significant increase in Erk1/2 phosphorylation
induced by glutamate, compared with the untreated control HT22
cells. However, the increase in Erk1/2 phosphorylation was reversed
following co-incubation with glutamate and B355227 (Fig. 5A). Incubation of HT22 cells with
B355227 alone exhibited no difference in the phosphorylation of
Erk1/2 compared with cells co-treated with glutamate and B355227,
but significantly decreased the phosphorylation of Erk1/2 compared
with untreated cells.
The involvement of JNK and p38 pathways in ischemia
have been previously reported (29). Therefore, the expression levels and
phosphorylation of JNK and p38 in HT22 cells were determined
following treatment with either glutamate or B355227 alone, or in
combination. Glutamate exposure increased the phosphorylation of
JNK by 2.3-fold, and exposure to B355227 diminished the
phosphorylation of JNK in the presence of glutamate (Fig. 5B). Similarly, glutamate treatment
elevated P38 the phosphorylation, which was reduced below the basal
level when treated in combination with B355227 (Fig. 5C). Interestingly, when HT22 was
treated with B355227 alone the phosphorylation of JNK was reduced
by ~2-fold, and the phosphorylation of p38 was reduced by a factor
>5-fold, compared with the untreated control cells respectively
(Fig. 5B and C).
The results of the present study indicated that an
improvement in neuronal viability following treatment with B355227
was associated with a reduction in the glutamate-induced increase
of intracellular Ca2+, protection of mitochondrial
potential and decreased MAPK pathway activation. Thus, the effects
of B355227 on anti-apoptotic Bcl-2 and pro-apoptotic Bax proteins
were investigated. The results demonstrated that treatment with
glutamate alone reduced the expression levels of Bcl-2, but exerted
no effects on the expression levels of Bax, resulting in a 4.2-fold
increase in the pro-apoptotic Bax/Bcl-2 ratio compared with
untreated cells (Fig. 5D). In
contrast, the glutamate-induced reduction in Bcl-2 was abolished
following co-treatment with glutamate and B355227, leading to
~4-fold decrease in the Bax/Bcl-2 ratio, similar to the observed
Bax/Bcl-2 ratio in the control group. Treatment with B355227 alone
increased the expression levels of Bcl-2 and Bax compared with the
untreated controls. However, the ratio of pro-apoptotic Bax/Bcl-2
remained at a similar level to the ratio observed in the control
group (Fig. 5D).
Discussion
Restricted passage of drugs through the BBB limits
the choice of drug for the treatment of neurodegenerative diseases,
and presents a major challenge for the development of new drugs
targeting these disorders (30).
The endothelial lining of cerebral micro-vessels controls the
paracellular permeability of solute across the membrane and
selectively allows the passage of compounds via transmembrane
diffusion, absorption endocytosis or saturable transporters
(31). An in vitro BBB
model is being utilized for the screening of drugs that have the
potential to permeate the BBB in vivo (22). In the present study, a bEND.3
monolayer in an in vitro BBB model was established to screen
a series of analogues of B355252(16), a phenoxythiophene compound.
Confirmation of chemical structure is an important step in compound
synthesis workflow, because it verifies information about the
structure of organic molecules. One compound, B355227 identified
from the series was permeable through the BBB in vitro and
possessed neuroprotective properties. The structure was confirmed
by proton and carbon NMR spectral analyses, which substantiated the
positions of its proton and carbon peaks, and thus established the
structural identity of the compound. Using the experimental model
of the BBB in the present study, a TEER value of ~50 Ω
cm2 was established. A wide range of variation in TEER
values have been reported in previous studies, ranging from 30-140
Ω cm2 in bEND.3 cells (32,33).
However, the present study demonstrated that the attained TEER
value of ~50 Ω cm2 was sufficient to exclude the passage
of low molecular weight imatinib and axitinib. Additionally, the
results of Transwell assays demonstrated that visualization of
continuous ZO-1 expression in the cytoplasmic border of bEND.3
cells highlighted the formation of proper tight junctions between
cells in the Transwell culture system. ZO-1, claudin, occludin and
junctional adhesion protein form a multiprotein complex with gap
junction and adherence junction proteins, and play a central role
in maintaining membrane integrity (34). The importance of ZO-1 in the
formation of notochord, neuronal tube and allantois has been
identified in a knockdown study using mice (35).
Hypoxia and glucose deprivation in ischemia
initiates a complex cascade of molecular events including depletion
of GSH, elevated ROS and an increase in Ca2+ influx
(36). Oxidative stress-mediated
elevation of ROS has been highlighted as a major contributor of
glutamate-induced oxidative injury (37). The results of the present study
demonstrated that B355227 protected HT22 cells from
glutamate-induced cell injury, with an optimal concentration ~5 µM.
Although there was significant protection at 10 µM B355227 compared
with glutamate-only treated cells, cell viability was reduced when
compared with cells treated with 5 µM B355227. The specific
mechanisms underlying the decrease in cell viability are yet to be
elucidated, as cells treated with B355227 alone demonstrated no
toxicity at a concentration of 20 µM. We hypothesized that
increasing the amount of B355227 to 10 µM in the presence of a high
concentration of glutamate increased the stress burden on the
metabolic activity of cells, and interfered with the antioxidant
capacity of B355227, thus leading to a loss of effectiveness in
cell protection.
Results of the present study demonstrated that the
levels of ROS decreased in glutamate-treated cells as early as 8 h
following treatment with B355227. In addition, B355227 prevented
the glutamate-induced reduction of GSH, demonstrating the
antioxidant and neuroprotective properties of the compound. A
decrease in the cellular levels of GSH leads to the accumulation of
mitochondrial ROS and Ca2+ overload, thus contributing
to cell death, which is prevented by increasing the intracellular
GSH (38). Similarly, inhibition
of the Ca2+ channel is often used in the treatment of
stroke, as it protects cells from glutamate-induced oxidative
stress (39,40). The results of the present study
also demonstrated markedly reduced levels of Ca2+ in
cells exposed to glutamate in the presence of B355227, which
indicates a role for B355227 in mitigating the elevation of
Ca2+ during glutamate toxicity. Furthermore, an increase
in mitochondrial Ca2+ during ischemia has been
associated with the collapse of the MMP, involving poly
(ADP-ribose) polymerase in the early phase, and formation of the
MPTP in the late phase (41).
Increase in the MPTP causes rupture in the outer mitochondrial
membrane, releasing apoptosis inducing factor (AIF) from the inner
mitochondrial space (42).
Moreover, inhibition of glutamate-dependent MMP depolarization
using B355227 involves AIF-1, as observed in our previous study
using B355252(18). Although both
caspase-dependent and -independent pathways have been proposed in
glutamate-mediated toxicity, contrasting findings have been
reported in a number of studies, which were dependent on the HT22
cell line used (43-45).
Glutamate induced necrosis at relatively early time points (before
12 h) and apoptosis at late time points (12-24 h) in HT22 cells
(46). Mitochondrial oxidative
stress and dysfunction were identified as essential events required
for the induction of apoptosis. However, apoptosis occurs in HT22
cells through an ATP-independent process, involving the release of
mitochondrial AIF, which catalyzes DNA fragmentation (46). Data from our previous study
demonstrated that the parent compound, B355252, inhibits the
glutamate-induced increase in AIF (18), which suggests that B355227 may
protect HT22 from cell death by partially modulating AIF
activity.
ROS plays a central role in glutamate-mediated
oxidative stress and displays a synergistic association with
Erk1/2(47). However, in ischemia,
Erk1/2 plays key roles in both the survival and apoptosis of cells,
depending on the time and concentration of glutamate exposure
(48,49). In the present study, an increase in
the levels of Erk1/2 phosphorylation was revealed following 24 h
glutamate exposure. Moreover, Erk1/2 phosphorylation was markedly
reduced following B355227 co-exposure, suggesting an apoptotic role
of Erk1/2. Consistent with the results of previous studies, the
present study reported that the levels of p-Erk1/2 were increased
following glutamate treatment in HT22 cells, leading to cell death
(18,49,50).
Following treatment with B355227, the phosphorylation of Erk1/2
decreased, leading to an increase in the levels of cell survival.
Furthermore, an increase in the levels of ROS is associated with
the levels of JNK and p38 phosphorylation, leading to apoptosis and
cell death (15). Increased JNK
phosphorylation in ischemia is associated with neurodegenerative
diseases, and the corresponding inhibition was associated with
neuroprotection (51). In the
present study, the glutamate-induced increase in the
phosphorylation of JNK in HT22 cells was reversed following
co-exposure to B355227, thus protecting the cells from glutamate
neurotoxicity. In addition, a decrease in levels of phosphorylated
JNK and p38 was observed in cells treated with B355227 alone. The
results of our previous study demonstrated that B355252 stimulated
cell proliferation in cells treated with B355252 alone (18). Although the specific mechanisms
underlying the observed increase in cell proliferation are yet to
be elucidated, regulation of JNK and P38 may play a key role in the
process. Thus, further research is required to understanding the
underlying mechanisms.
The results of the present study demonstrated that
the ratio of Bax/Bcl-2 was increased in cells following glutamate
treatment, as a result of decreased levels of Bcl-2. In contrast,
the ratio of Bax/Bcl-2 in HT22 cells exposed to B355227 alone, or
co-treated with B355227 and glutamate was similar to that observed
in the control group. Thus, the neuroprotective effects of B355227
may also be partially mediated by an increase in the anti-apoptotic
Bcl-2 protein, or an interference with the mechanisms underlying
the reduction of Bcl-2 following exposure to glutamate. Changes in
anti-apoptotic protein expression levels have been observed in
neuronal cell models of glutamate-induced neurotoxicity, and in
animal models of ischemic stroke exposed to small molecules and
extracts of medicinal plants with antioxidant and neuroprotective
properties (52,53). The suppression of Ca2+
and subsequent decreases in the levels of JNK and p38
phosphorylation, coupled with the differential regulation of
Bax/Bcl-2 expression highlight the neuroprotective attributes of
B355227.
In conclusion, the findings of the present study
demonstrate that B355227 is permeable in vitro, and exerts
protection against glutamate-induced neurotoxicity through
antioxidant and anti-apoptotic activities. These activities include
a decrease in the levels of ROS, an increase in the levels of GSH,
maintenance of intracellular Ca2+ homeostasis,
prevention of the collapse of the MMP, reduction of Bcl-2
expression levels and modulation of the phosphorylation of MAPK-
associated proteins in the cell. Collectively, the results of the
present study revealed the neuroprotective nature of B355227, and
highlighted it as a potential therapeutic compound to target
oxidant-dependent mechanisms underlying disorders of the central
nervous system.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the National
Institutes of General Medical Sciences of the National Institutes
of Health (grant no. SC3GM116667).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GCI and SP conceived and designed the experiments.
SP and NSG performed the experiments. SP and GCI analyzed the data
and wrote the manuscript. SP and GCI confirmed the authenticity of
all the raw data. ALW and SRD synthesized the small molecule
B355227 used in the study. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Barthels D and Das H: Current advances in
ischemic stroke research and therapies. Biochim Biophys Acta Mol
Basis Dis. 1866(165260)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lee CH and Lee SH: General facts of
stroke. In: Stroke Revisited: Pathophysiology of Stroke: From Bench
to Bedside. Lee SH (ed). Springer, Singapore, pp3-10, 2020.
|
|
3
|
Deb P, Sharma S and Hassan KM:
Pathophysiologic mechanisms of acute ischemic stroke: An overview
with emphasis on therapeutic significance beyond thrombolysis.
Pathophysiology. 17:197–218. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mergenthaler P, Dirnagl U and Meisel A:
Pathophysiology of stroke: Lessons from animal models. Metab Brain
Dis. 19:151–167. 2004.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Jansson LC and Åkerman KE: The role of
glutamate and its receptors in the proliferation, migration,
differentiation and survival of neural progenitor cells. J Neural
Transm (Vienna). 121:819–836. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
McEntee WJ and Crook TH: Glutamate: Its
role in learning, memory, and the aging brain. Psychopharmacology
(Berl). 111:391–401. 1993.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Foran E and Trotti D: Glutamate
transporters and the excitotoxic path to motor neuron degeneration
in amyotrophic lateral sclerosis. Antioxid Redox Signal.
11:1587–1602. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bridges RJ, Natale NR and Patel SA: System
xc- cystine/glutamate antiporter: An update on molecular
pharmacology and roles within the CNS. Br J Pharmacol. 165:20–34.
2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Shih AY, Erb H, Sun X, Toda S, Kalivas PW
and Murphy TH: Cystine/glutamate exchange modulates glutathione
supply for neuroprotection from oxidative stress and cell
proliferation. J Neurosci. 26:10514–10523. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Angelova PR, Vinogradova D, Neganova ME,
Serkova TP, Sokolov VV, Bachurin SO, Shevtsova EF and Abramov AY:
Pharmacological sequestration of mitochondrial calcium uptake
protects neurons against glutamate excitotoxicity. Mol Neurobiol.
56:2244–2255. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Meredith GE, Totterdell S, Beales M and
Meshul CK: Impaired glutamate homeostasis and programmed cell death
in a chronic MPTP mouse model of Parkinson's disease. Exp Neurol.
219:334–340. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nozaki K, Nishimura M and Hashimoto N:
Mitogen-activated protein kinases and cerebral ischemia. Mol
Neurobiol. 23:1–19. 2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Satoh T, Nakatsuka D, Watanabe Y, Nagata
I, Kikuchi H and Namura S: Neuroprotection by MAPK/ERK kinase
inhibition with U0126 against oxidative stress in a mouse neuronal
cell line and rat primary cultured cortical neurons. Neurosci Lett.
288:163–166. 2000.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shioda N, Han F and Fukunaga K: Role of
Akt and ERK signaling in the neurogenesis following brain ischemia.
Int Rev Neurobiol. 85:375–387. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen RW, Qin ZH, Ren M, Kanai H,
Chalecka-Franaszek E, Leeds P and Chuang DM: Regulation of c-Jun
N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured
brain neurons: Roles in glutamate excitotoxicity and lithium
neuroprotection. J Neurochem. 84:566–575. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Williams AL, Dandepally SR, Gilyazova N,
Witherspoon SM and Ibeanu G: Microwave-assisted synthesis of
4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl)phenoxy)thiophene-2-sulfonamide(B-355252):
A new potentiator of Nerve Growth Factor (NGF)-induced neurite
outgrowth. Tetrahedron. 66:9577–9581. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chimeh U, Zimmerman MA, Gilyazova N and Li
PA: B355252, a novel small molecule, confers neuroprotection
against cobalt chloride toxicity in mouse hippocampal cells through
altering mitochondrial dynamics and limiting autophagy induction.
Int J Med Sci. 15:1384–1396. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gliyazova NS, Huh EY and Ibeanu GC: A
novel phenoxy thiophene sulphonamide molecule protects against
glutamate evoked oxidative injury in a neuronal cell model. BMC
Neurosci. 14(93)2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gliyazova NS and Ibeanu GC: The chemical
molecule B355252 is neuroprotective in an in vitro model of
Parkinson's disease. Cell Mol Neurobiol. 36:1109–1122.
2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Pokharel S, Kamli MR, Mir BA, Malik A, Lee
EJ and Choi I: Expression of transthyretin during bovine myogenic
satellite cell differentiation. In Vitro Cell Dev Biol Anim.
50:756–765. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pokharel S, Lee CH, Gilyazova N and Ibeanu
GC: Analysis of gene expression and neuronal phenotype in
neuroscreen-1 (NS-1) Cells. Int J Biomed Investig. 1:1–13.
2018.PubMed/NCBI
|
|
22
|
Brown RC, Morris AP and O'Neil RG: Tight
junction protein expression and barrier properties of immortalized
mouse brain microvessel endothelial cells. Brain Res. 1130:17–30.
2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Birben E, Sahiner UM, Sackesen C, Erzurum
S and Kalayci O: Oxidative stress and antioxidant defense. World
Allergy Organ J. 5:9–19. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Baquer NZ, Taha A, Kumar P, McLean P,
Cowsik SM, Kale RK, Singh R and Sharma D: A metabolic and
functional overview of brain aging linked to neurological
disorders. Biogerontology. 10:377–413. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Görlach A, Bertram K, Hudecova S and
Krizanova O: Calcium and ROS: A mutual interplay. Redox Biol.
6:260–271. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Feno S, Butera G, Vecellio Reane D,
Rizzuto R and Raffaello A: Crosstalk between Calcium and ROS in
Pathophysiological Conditions. Oxid Med Cell Longev.
2019(9324018)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kumari S, Mehta SL and Li PA: Glutamate
induces mitochondrial dynamic imbalance and autophagy activation:
Preventive effects of selenium. PLoS One. 7(e39382)2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Singer CA, Figueroa-Masot XA, Batchelor RH
and Dorsa DM: The mitogen-activated protein kinase pathway mediates
estrogen neuroprotection after glutamate toxicity in primary
cortical neurons. J Neurosci. 19:2455–2463. 1999.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yamazaki Y, Arita K, Harada S and Tokuyama
S: Activation of c-Jun N-terminal kinase and p38 after cerebral
ischemia upregulates cerebral sodium-glucose transporter type 1. J
Pharmacol Sci. 138:240–246. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pardridge WM: Drug transport across the
blood-brain barrier. J Cereb Blood Flow Metab. 32:1959–1972.
2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Banks WA: Characteristics of compounds
that cross the blood-brain barrier. BMC Neurol. 9 (Suppl
1)(S3)2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Koto T, Takubo K, Ishida S, Shinoda H,
Inoue M, Tsubota K, Okada Y and Ikeda E: Hypoxia disrupts the
barrier function of neural blood vessels through changes in the
expression of claudin-5 in endothelial cells. Am J Pathol.
170:1389–1397. 2007.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yang S, Mei S, Jin H, Zhu B, Tian Y, Huo
J, Cui X, Guo A and Zhao Z: Identification of two immortalized cell
lines, ECV304 and bEnd3, for in vitro permeability studies of
blood-brain barrier. PLoS One. 12(e0187017)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Stamatovic SM, Johnson AM, Keep RF and
Andjelkovic AV: Junctional proteins of the blood-brain barrier: New
insights into function and dysfunction. Tissue Barriers.
4(e1154641)2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Katsuno T, Umeda K, Matsui T, Hata M,
Tamura A, Itoh M, Takeuchi K, Fujimori T, Nabeshima Y, Noda T, et
al: Deficiency of zonula occludens-1 causes embryonic lethal
phenotype associated with defected yolk sac angiogenesis and
apoptosis of embryonic cells. Mol Biol Cell. 19:2465–2475.
2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Belov Kirdajova D, Kriska J, Tureckova J
and Anderova M: Ischemia-triggered glutamate excitotoxicity from
the perspective of glial cells. Front Cell Neurosci.
14(51)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Atlante A, Calissano P, Bobba A,
Giannattasio S, Marra E and Passarella S: Glutamate neurotoxicity,
oxidative stress and mitochondria. FEBS Lett. 497:1–5.
2001.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kho AR, Choi BY, Lee SH, Hong DK, Lee SH,
Jeong JH, Park KH, Song HK, Choi HC and Suh SW: Effects of
protocatechuic acid (PCA) on global cerebral ischemia-induced
hippocampal neuronal death. Int J Mol Sci. 19(19)2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Chen GJ and Yang MS: The effects of
calcium channel blockers in the prevention of stroke in adults with
hypertension: A meta-analysis of data from 273,543 participants in
31 randomized controlled trials. PLoS One. 8(e57854)2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ha JS and Park SS: Glutamate-induced
oxidative stress, but not cell death, is largely dependent upon
extracellular calcium in mouse neuronal HT22 cells. Neurosci Lett.
393:165–169. 2006.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Abramov AY and Duchen MR: Mechanisms
underlying the loss of mitochondrial membrane potential in
glutamate excitotoxicity. Biochim Biophys Acta. 1777:953–964.
2008.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Park JS, Park JH and Kim KY:
Neuroprotective effects of myristargenol A against
glutamate-induced apoptotic HT22 cell death. RSC Advances.
9:31247–31254. 2019.
|
|
44
|
Tan S, Wood M and Maher P: Oxidative
stress induces a form of programmed cell death with characteristics
of both apoptosis and necrosis in neuronal cells. J Neurochem.
71:95–105. 1998.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Weon JB, Yun BR, Lee J, Eom MR, Ko HJ, Lee
HY, Park DS, Chung HC, Chung JY and Ma CJ: Neuroprotective effect
of steamed and fermented Codonopsis lanceolata. Biomol Ther
(Seoul). 22:246–253. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fukui M, Song JH, Choi J, Choi HJ and Zhu
BT: Mechanism of glutamate-induced neurotoxicity in HT22 mouse
hippocampal cells. Eur J Pharmacol. 617:1–11. 2009.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Levinthal DJ and Defranco DB: Reversible
oxidation of ERK-directed protein phosphatases drives oxidative
toxicity in neurons. J Biol Chem. 280:5875–5883. 2005.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kim SH, Kim KY, Park SG, Yu SN, Kim YW,
Nam HW, An HH, Kim YW and Ahn SC: Mitochondrial ROS activates
ERK/autophagy pathway as a protected mechanism against
deoxypodophyllotoxin-induced apoptosis. Oncotarget.
8:111581–111596. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Sato K, Yamanaka Y, Asakura Y and Nedachi
T: Glutamate levels control HT22 murine hippocampal cell death by
regulating biphasic patterns of Erk1/2 activation: Role of
metabolic glutamate receptor 5. Biosci Biotechnol Biochem.
80:712–718. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Stanciu M, Wang Y, Kentor R, Burke N,
Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW,
et al: Persistent activation of ERK contributes to
glutamate-induced oxidative toxicity in a neuronal cell line and
primary cortical neuron cultures. J Biol Chem. 275:12200–12206.
2000.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Yang DD, Kuan CY, Whitmarsh AJ, Rincón M,
Zheng TS, Davis RJ, Rakic P and Flavell RA: Absence of
excitotoxicity-induced apoptosis in the hippocampus of mice lacking
the Jnk3 gene. Nature. 389:865–870. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
52
|
Lee HJ, Spandidos DA, Tsatsakis A, Margina
D, Izotov BN and Yang SH: Neuroprotective effects of
Scrophularia buergeriana extract against glutamate-induced
toxicity in SH-SY5Y cells. Int J Mol Med. 43:2144–2152.
2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhang G, Zhang T, Wu L, Zhou X, Gu J, Li
C, Liu W, Long C, Yang X, Shan L, et al: Neuroprotective effect and
mechanism of action of tetramethylpyrazine nitrone for ischemic
stroke therapy. Neuromolecular Med. 20:97–111. 2018.PubMed/NCBI View Article : Google Scholar
|