Introduction
A fraction of circulating human albumin molecules
are non-covalently attached to either of the two recently
discovered albumin-associated O-glycoproteins (AOP1;107 kDa and
AOP2; 98 kDa) that are heavily O-glycosylated (1). Most, if not all, plasma
anti-α-galactoside (anti-Gal) and anti-β-glucoside (ABG) antibodies
are bound to albumin-associated AOP1 or AOP2 to form
antibody-AOP1/AOP2-albumin triplets (1) due to recognition of the serine- and
threonine-rich peptide sequences (STPS) on the O-glycoproteins as
surrogate ligands by either antibody (2). As a result, plasma anti-Gal and ABG
samples prepared by affinity chromatography (APAG and APABG
respectively) mostly consisted of triplets aforementioned (1). Whilst purified AOP1 and AOP2 occupy
all available binding sites on either antibody, albumin-bound AOP1
or AOP2 can occupy only some of these sites during triplet
formation, possibly due to steric hindrance (1). Utilizing the free binding sites
available on their antibodies, triplets can bind to affinity
chromatography matrices containing antibody-specific ligands
immobilized on them (1) and to
activated human macrophages (unpublished findings). This is
possibly because the ligands available on these two systems are
more accessible compared with those on albumin-bound AOP1 or AOP2.
Since amyloid β (Aβ-42) recognizes STPS, isolated AOP1 and AOP2,
their complexes with albumin or the complete triplet, but not
purified albumin, can bind to this peptide (3).
Factors that prevent platelet-activating blood
factors such as ADP and fibrinogen from causing aberrant platelet
activation remain poorly understood. Notably, glycoprotein
GPIIb/IIIa, which is the most abundantly expressed protein on the
surface of platelets, serves as a fibrinogen receptor and acts as a
link in the ADP-mediated platelet activation cascade and
aggregation (4). GPIIb/IIIa is
heavily O-glycosylated and therefore rich in STPS, especially on
its IIb subunit (4). Furthermore,
the levels of immunoglobulins and albumin carried by platelets at
any given time are reported to vary in parallel (5). Platelets are also major carriers of
Aβ-42(6), which had been reported
to bind triplet O-glycoproteins in plasma (3). The aim of the present study was to
investigate the presence of anti-Gal/ABG-AOP1/AOP2-albumin
triplets, anchored using the unutilized binding sites of their
antibodies, on the surface of normal human platelets using the STPS
of O-glycoprotein(s) on platelet surface membrane as ligands. In
addition, the consequence of denuding the platelets by removal of
their triplet complexes were examined. Although hyperglycemia is
the hallmark of diabetes, the molecular mechanism underlying the
pathophysiological changes induced by hyperglycemia, including
vascular diseases, platelet dysfunction, platelet-leukocyte
adhesion and increased susceptibility to Alzheimer's disease,
remain elusive. In this context, the role of high concentrations of
glucose, which is an ABG ligand, assumes clinical relevance, as
diabetes has been reported to increase platelet aggregation
(7). Therefore, the results of the
present study may help to determine whether a shield containing a
natural immune complex, which carries the platelet-bound Aβ-42,
prevents platelet aggregation and masks reactive surface proteins
on the platelet. This may be a defence system used by the body
against diabetes-driven platelet malfunctions, such as premature
aggregation and platelet-leukocyte adhesion, which can lead to
vascular diseases.
Materials and methods
Reagents, proteins and conjugates
Fluorescein isothiocyanate (FITC), methyl
α-D-mannoside (MαM), methyl α-D-galactoside (MαG), cellobiose
(4-O-β-D-Glucopyranosyl-D-glucose), orthophenylene diamine (OPD),
horseradish peroxidase (HRP; cat. no. P-8375), neuraminidase from
Clostridium perfringens, soybean trypsin inhibitor,
galactose, Tween-20, Coomassie brilliant blue (G-250 and R-250),
soluble guar galactomannan, fibrinogen, ADP and rabbit antibodies
to human albumin (cat. no. A3293) were purchased from Sigma
Aldrich; Merck KGaA. HiLyte™ Fluor-488-labelled amyloid
β peptide Aβ-42 monomer (cat. no. AS-60479) was purchased from
AnaSpec, Inc. Polystyrene 96-well microplates (Maxisorb BREAKAPART
and Polysorb BREAKAPART) were purchased from Nunc; Thermo Fisher
Scientific, Inc. and antibodies to human IgA (cat. no. Q0332), IgG
(cat. no. Q0331) and IgM (cat. no. Q0333) raised in rabbit, were
from Dako, Agilent Technologies, Inc.
Jacalin was prepared from jackfruit (Artocarpus
integrifolia) seeds according to the method described by Suresh
Kumar et al (8). Using
affinity chromatography, lectins concanavalin A (ConA) was prepared
from the seeds of Canavalia ensiformis and peanut agglutinin
(PNA) was prepared from peanuts (1). Trypsin inhibitor-cellobiose (TIC) and
trypsin inhibitor-melibiose (TIM) were prepared by coupling
cellobiose or melibiose (Sigma Aldrich; Merck KGaA) to soybean
trypsin inhibitor by reductive amination (9). Yeast glycoprotein was isolated from
Saccharomyces cerevisiae (yeast type-II; Sigma Aldrich;
Merck KGaA). Briefly, yeast (5 g) was extracted to PBS by three
successive -20˚C to +2˚C freeze-thaw cycles. After homogenization
and sonication at 25˚C (six 30-sec bouts at 6 micron amplitude in
SONIPREP probe sonicator; MSE (UK) Ltd.), the yeast protein samples
were resolved by electrophoresis in 7% polyacrylamide gel and the
two slowest-moving proteins were electroeluted together (10). Previously reported procedures were
used to prepare affinity-purified samples of plasma anti-Gal
(APAG)- and ABG (APABG)-containing triplets using cross-linked guar
galactomannan gel (11) and
cellulose (12) in the affinity
matrices, respectively. APAG and APABG were triplet complexes
containing anti-Gal and ABG antibodies, respectively (1). The source of these samples were
outdated human plasma obtained between 1 March and 31 August, 2017
following approval from the Institutional Ethics Committee, Sree
Chitra Tirunal Institute for Medical Sciences and Technology,
Thiruvananthapuram (approval no. IEC/674). Plasma was prepared from
blood of 50 healthy voluntary donors (male, 40; female, 10) aged
22-45 years (32±6 years) selected without gender bias at the
Department of Transfusion Medicine of the Sree Chitra Tirunal
Institute for Medical Sciences and Technology. Donors with high
blood pressure, ongoing infections or dependence on any narcotics
were excluded. Plasma samples declared outdated by the Institute
after storage at -80˚C for ≥1 years were thawed at 4˚C and used
within 3 days thereafter. Purified samples of AOP1, AOP2, anti-Gal,
ABG and human serum albumin (HSA), without contamination by each
other due to non-covalent interactions, were isolated by the
alkaline electrophoretic separation of APAG or APABG from plasma or
platelets into their individual components (1). Electrophoretically purified samples of
albumin, anti-Gal, AOP1 and AOP2 were labelled with FITC by
treating with FITC (150 µg per mg protein) in 250 mM
carbonate-bicarbonate buffer, pH 9.0 overnight followed by dialysis
against PBS at 4˚C (13).
The number of platelets in PBS medium were counted
using the ABX Pentra 60 C+ haematology analyser (Horiba Medical),
which counts platelets using focused flow impedance measurement
through a double hydrodynamic sequential system. To prepare HRP
conjugates of proteins (antibodies against the following human
proteins: Albumin, IgG, IgM and IgA, and lectins ConA, PNA and
jacalin), HRP was oxidized using sodium periodate (1 h), dialyzed
overnight against sodium carbonate buffer pH 9.5 at 25˚C and
treated with the protein in a 2:3 ratio of HRP and protein for 2 h
at 4˚C before the product was changed to PBS medium by dialysis at
4˚C.
Preparation of native and denuded
(triplet-free) platelets
Fresh blood samples were collected from 50 (male,
30; female, 20) healthy volunteers aged 23-50 years (32±6 years),
using the procedure described by Jennings and Philips (14) after obtaining informed consent in
writing in accordance with the guidelines of the Institutional
Ethics Committee (permission no. IEC/1072). Blood samples collected
in anti-coagulant (heparin)-treated tubes (10 ml) were centrifuged
at 150 x g for 5 min at 25˚C. The supernatant was then mixed with
100 mM EDTA (1:10) to prevent platelet activation and centrifuged
at 25˚C, first at 230 x g for 10 min to remove erythrocytes and
again at 4,530 x g for 15 min. The final supernatant was removed
and the pellet was collected as platelets. This platelet pellet was
suspended in 300 µl PBS and one half diluted to 300 µl with PBS was
incubated for 2 h at 25˚C with a mixture of MαG and cellobiose
(both 15 mM), which are sugars specific for anti-Gal and ABG
respectively. The triplets extracted from the platelets were
collected as the supernatant after centrifugation at 4,530 x g for
15 min at 25˚C. The pellet was washed twice with PBS and collected
as denuded (triplet-free) platelets. The second half of platelets
that was treated similarly but with the non-specific sugar MαM (30
mM), yielded native non-denuded platelets.
Resolution of triplet components by
alkaline PAGE
Electrophoresis of triplets extracted from the
platelets after incubation with MαG and cellobiose was performed in
6% polyacrylamide gel tubes in Tris-glycine pH 8.3 buffer at 4˚C
with 50 µg triplet per tube. One gel was stained with Coomassie
Brilliant blue G-250 for 1 h at 25˚C. Bands of O-glycoproteins AOP1
and AOP2, albumin and antibodies (anti-Gal and ABG that move
together) were compared with the electrophoretic mobility of
components of plasma triplets (mixture of APAG and APABG; 25 µg
each) (1). Corresponding segments
of unstained gels were cut and each protein was electroeluted
separately. The two O-glycoprotein bands were also eluted together
where required to obtain AOP, which is a mixture of AOP1 and AOP2
in the ratio in which they occur on the platelet-bound
triplets.
ELISA for ligand binding and
inhibition assays General procedures
Macromolecules [thyroglobulin (Tg), TIC, TIM, guar
galactomannan or yeast glycoproteins] were coated on Nunc MAXISORB
microplate wells (maximum 1 µg in 200 µl PBS) by incubation at 37˚C
for 3 h or overnight at 4˚C. The wells were washed with PBS
containing 0.05% Tween-20 (PBST) and blocked by incubation for 30
min at 37˚C with PBS containing 0.5% Tween-20. After another wash
with PBST, the wells were incubated for 2 h at 4˚C with 200 µl PBST
containing the primary reactants (sugar-extracted platelet-bound
triplets or antibodies separated from them) pre-incubated with or
without specific sugars. Wells were washed with PBST again and
treated with HRP-labelled secondary reactants (anti-immunoglobulin
antibodies for antibody assay or anti-albumin antibody for triplet
assay). After washing the plates again three times with PBST, bound
HRP was measured by adding 200 µl OPD (0.5 mg/ml) dissolved in 0.1
M citrate-phosphate buffer (pH 5.0) containing 0.03%
H2O2 for 15 min at 25˚C, followed by the
addition of 50 µl 12.5% H2SO4 to stop the
reaction. Absorbance was measured at 490 nm using an
ELx800™ ELISA reader (BioTek Instruments, Inc.).
Anti-Gal and ABG assay
To measure anti-Gal or ABG in the platelet-derived
triplets, 100 ng immunoglobulin was pre-incubated with the
anti-Gal-specific sugar MαG, ABG-specific sugar cellobiose (each 25
mM) or no sugar in 200 µl PBS for 2 h at 4˚C. This was inoculated
into ELISA microplate wells coated with Tg or TIC (1 µg per well)
for 2 h at 4˚C and washed as described above. The levels of bound
antibody were determined by treatment with a mixture of equal
amounts of HRP-conjugated antibodies against human IgG, IgA and IgM
(0.3 µg total antibody in 200 µl PBST; 2 h at 4˚C), washing and
measurement of bound HRP as aforementioned.
Triplet assay
To assay sugar-extracted platelet-bound triplets,
300 µl platelet suspension (7.5x106 cells) in PBS was
incubated for 2 h at 25˚C with the non-specific sugar MαM, the
ABG-specific sugars cellobiose, glucose or the anti-Gal-specific
sugar MαG (15 mM each). Following centrifugation at 4,500 x g for
15 min at 25˚C, 250 µl supernatant containing the triplets
extracted from the platelets was dialyzed against PBS at 4˚C (6 h,
3 times) to remove the sugars. Afterwards, 1:100 dilution of this
dialysate (200 µl) was added into the microplate wells coated with
guar galactomannan (1 µg per well) to capture anti-Gal complexes or
with yeast glycoproteins to capture the ABG complexes. After
incubation for 2 h at 4˚C, wells were washed with PBST and bound
complexes were determined by incubation with rabbit anti-human
albumin-HRP (0.75 µg antibody in 200 µl PBST; 2 h at 4˚C). Bound
HRP was measured using ELISA as aforementioned.
Assay of triplet distribution in
platelets and plasma
From the platelet samples harvested as
aforementioned, 8x107 cells in 320 µl PBS were treated
with a mixture of MαG and cellobiose (15 mM each) at 25˚C for 2 h.
The mixture was centrifuged at 4,500 x g for 15 min at 25˚C and the
supernatant (250 µl) was dialyzed against PBS at 4˚C (two 6-h
dialysis cycles). This sample and cell-free plasma from the same
donor (supernatant after removing all cells by centrifugation at
5,000 x g for 15 min at 25˚C) were diluted 100X in PBST before 200
µl was added into microplate wells coated with TIM or TIC (1 µg per
well) to estimate the levels of triplets from anti-Gal and ABG,
respectively, using rabbit anti-human albumin-HRP. Bound HRP was
measured using ELISA as aforementioned (1).
Assay of triplet binding to
fibrinogen-treated denuded platelets
Platelets (8.75x107 cells in 350 µl PBS)
were incubated with 1 µM fibrinogen at 25˚C for 2 h. After
centrifugation at 4,500 x g for 15 min at 25˚C and removal of the
supernatant, the pellet was washed twice with PBS and incubated in
300 µl PBS containing 2 µg triplets released from normal platelets
(using MαG and cellobiose, both 25 mM) for 4 h at 25˚C. Unbound
triplets (200 µl supernatant collected after centrifugation at
4,500 x g for 15 min at 25˚C) was added to wells coated with a
mixture of TIM and TIC (0.5 µg each). After 2 h incubation at 4˚C
and washing, the level of triplets was assayed using incubation
with rabbit anti-human albumin-HRP (0.75 µg antibody in 200 µl
PBST). Bound HRP was measured using ELISA as aforementioned.
SDS electrophoresis, western blotting
and lectin staining of platelet surface proteins
Native platelets from normal blood (10 ml samples)
prepared as aforementioned were lysed in cold hypotonic buffer [2 h
in 7.5 mM phosphate buffer (pH 7.3) containing 10 mM EDTA; 600 µl].
The lysate was centrifuged at 100,000 x g for 1 h at 4˚C to
retrieve a fraction enriched with platelet surface proteins as the
pellet. Bradford protein assay was used to measure protein
concentration and SDS-PAGE (7.5%) of this sample was performed as
described by Laemmli (15), loading
~30 µg protein per lane. Separated proteins were transferred onto
PVDF membranes using protocols previously described by Towbin et
al (16). Strips 3-4 mm in
width were cut out from the transfer membrane, blocked with PBS
containing 0.2% Tween 20 at 25˚C for 2 h. The strips were then
treated with HRP-conjugated lectin [concanavalin A-HRP; 150 µg
lectin per ml: PNA-HRP: 75 µg lectin per ml before and after
neuraminidase treatment (100X dilution in PBS of 1.5 U per ml; 1 h
at 37˚C], and PNA-HRP as above in presence of lactose control (25
mM) after prior neuraminidase treatment of the strip or jacalin-HRP
(0.75 µg per ml) for 2 h at 4˚C. The strips were then washed twice
with 0.05% PBST and once with PBS alone, before being stained with
freshly prepared 4-chloronaphthol solution (1 mg in 0.4 ml methanol
mixed with 1.6 ml PBS containing 0.05% H2O2).
As the protein bands appeared, the strips were washed in PBS. To
assess relative O-glycan content in the protein bands, jacalin
binding and amido black (0.1% in 4% ethanol, 1% acetic acid)
binding responses to them were compared by digital scanning using
ImageJ 1.53e software (National Institutes of Health).
Platelet aggregation assay
In total, 6.3x107 denuded or non-denuded
platelets were treated with 20 µM ADP in 250 µl PBS at 25˚C and
absorbance was measured after 2 min at 405 nm using an
ELx800™ ELISA reader (BioTek Instruments, Inc.). A
reduction in absorbance was considered as a measure of
aggregation.
Fluorescence assay
FITC-labelled proteins and HiLyte-Fluor-488-labelled
amyloid β in free or platelet-bound form were assayed by measuring
the fluorescence of their solutions or suspensions of 300 µl PBS in
Nunc Polysorb BREAKAPART microplates using a FLx800™
fluorescence reader (BioTek Instruments, Inc.) with the excitation
wavelength of 485 nm and emission wavelength 528 nm (485/528 nm).
To confirm the identity of plasma triplets and platelet-bound
triplets, attachment of FITC-labelled anti-Gal and triplets to
denuded platelets was studied. To prepare de novo plasma
triplet samples 600 ng anti-Gal in 200 µl PBS was treated for 2 h
at 4˚C with 25 mM anti-Gal specific sugar MαG or the non-specific
sugar MαM and added to a pre-incubated (2 h at 4˚C) mixture of
AOP1, AOP2 and FITC-labeled albumin (200 ng each in 200 µl PBS) and
incubated again for 2 h at 4˚C. Denuded or native platelet
suspensions (7.5x107 cells in 300 µl PBS) were mixed
with either of the following: i) Glycoprotein-free anti-Gal-FITC
(600 ng in 400 µl PBS) pre-incubated for 2 h at 4˚C with 25 mM MαG
(specific) or MαM (non-specific) (1); or ii) de novo plasma triplets
as aforementioned (400 µl). The mixture was incubated for 2 h at
4˚C and centrifuged at 4,500 x g for 15 min at 4˚C to remove the
supernatant. Pellet was washed twice with PBS and re-suspended in
320 µl PBS. Cell-bound fluorescence was measured using a 300 µl
suspension at 485/528 nm.
To characterize the cell surface molecule recognized
by triplets for anchoring on platelets, denuded platelets
(6.3x107 cells in 250 µl PBS) were incubated with 50 ng
jacalin or heat-inactivated (95˚C for 2 min) jacalin for 1 h at 4˚C
before the supernatant was removed by centrifugation at 4,500 x g
for 15 min at 4˚C. Platelets in the pellet were washed twice with
PBS and incubated at 25˚C for 3 h in 400 µl PBS containing either
600 ng glycoprotein-free anti-Gal-FITC or 400 µl de novo
triplet reconstituted from components of platelet-derived samples,
with FITC-labelled albumin. After washing three times in PBS by
centrifugation at 4,500 x g at 25˚C for 15 min each, the pellet was
re-suspended in 320 µl PBS before cell-bound fluorescence was
measured at 485/528 nm.
To measure amyloid β binding to platelets, denuded
or native platelets in 320 µl PBS (8x106 cells) were
incubated for 3 h with 75 ng HiLyte™ Fluor-488-labelled
amyloid β at 25˚C. Following centrifugation at 4,500 x g for 15 min
at 25˚C fluorescence in the supernatant was measured at 485/528 nm
in a 300 µl aliquot. The pellet was washed twice with PBS and
incubated for 1 h at 25˚C with a mixture of anti-Gal- and
ABG-specific sugars MαG and cellobiose (15 mM each) in 350 µl PBS
before the triplet-bound amyloid β released from specific sugars
was measured using 300 µl of supernatant at 485/528 nm.
Density gradient ultracentrifugation
(DGUC)
Plasma made cell-free by centrifugation at 5,000 x g
for 30 min at 4˚C, AOP1-FITC or AOP2-FITC was incubated with or
without sugar molecules for 2 h at 4˚C before the density was
increased to 1.24 g/cm3 by adding solid KBr. The
resulting solution (1.1 ml in 1.3 ml tubes) was centrifuged in a
Himac CS 150 GXII microcentrifuge (Hitachi, Ltd.) at 535,000 x g
for 4 h at 4˚C. The middle (400 µl) and bottom (300 µl) layers were
separately saved and used for fluorescence assay.
Statistical analysis
Data are presented as the mean ± SD unless otherwise
specified. Statistical significance as judged from two-tailed
P-values, following analysis of the results using Microsoft Excel
2010 (Microsoft Corporation) and GraphPad Prism 5 (GraphPad
Software, Inc.), was determined by using unpaired student's t-test
except for Figs. 2, 4 and 7.
For Fig. 2, one-way ANOVA followed
by Dunnett's test and for Fig. 4,
one way ANOVA followed by Tukey's post hoc test were employed. For
Fig. 7, the data was analyzed using
two-way ANOVA followed by Sidak's post hoc tests, by using
native/denuded triplets as one factor and ADP/Jn as the second
factor. P<0.05 was considered to indicate a statistically
significant difference.
Results
Albumin and either of the two
O-glycosylated proteins are attached through anti-Gal or ABG to
platelets
Following alkaline polyacrylamide gel
electrophoresis, protein samples extracted from platelets using a
mixture of 15 mM each of MαG (specific for anti-Gal) and cellobiose
(specific for ABG), exhibited bands with mobilities identical to
those of a mixture of equal amounts of APAG and APABG (triplets of
anti-Gal and of ABG, respectively), in the plasma (Fig. 1) (1). The fastest and slowest moving bands
were identified to be HSA and immunoglobulins, respectively, by
electroelution, coating on polystyrene microplates and ELISA. Bands
with mobility equal to that of AOP1 or AOP2 were as heavily
O-glycosylated as the latter, judging by the observed capacity of
their microplate-coated forms to capture the HRP-conjugated
O-glycan-specific lectin jacalin (data not shown) (17). Platelet-bound immunoglobulins, which
were released by sugars and electrophoretically separated,
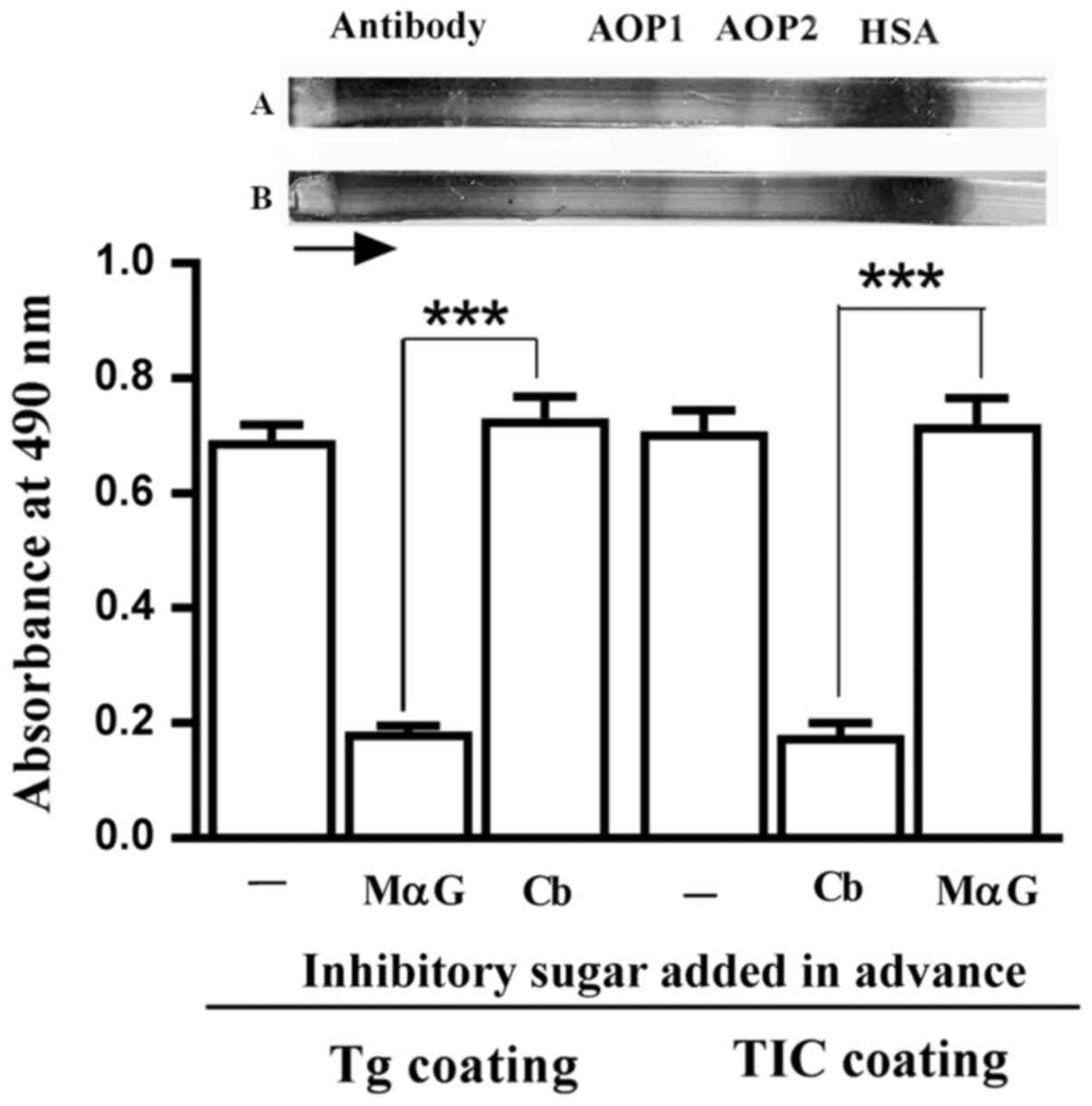
contained anti-Gal, as evidenced by the significant inhibition of
their binding to microplate wells coated with the
α-galactoside-bearing protein thyroglobulin, by MαG, but not by
cellobiose (Fig. 1). Electroeluted
immunoglobulins also contained ABG which bound to microplate wells
coated with TIC. This binding was significantly inhibited by
cellobiose, but not by MαG (Fig.
1). A mixture of guar galactomannan gel (11) and cellulose (12) that are respective affinity matrices
for anti-Gal and ABG (0.2 ml each) could capture all the
immunoglobulins out of 200 ng of the electroeluted immunoglobulin
aforementioned in 0.5 ml PBST, suggesting that anti-Gal and ABG
were the only two antibodies eluted from the platelet samples by
the mixture of MαG and cellobiose (data not shown). These results
suggested that attachment of the two O-glycoproteins and albumin to
platelets was mediated by anti-Gal or ABG. The procedure for
extraction of platelet-bound triplets ensured that any immune
complex bound non-specifically to platelets was not released from
the platelets (18).
Anti-Gal- or ABG-specific sugar
releases antibody-O-glycosylated protein-albumin triplets of both
antibodies from platelets
The triplet immune complexes in plasma, formed by
the simultaneous binding of either of the two antibodies anti-Gal
and ABG, and albumin to AOP1 or AOP2, were assayed by capturing
them using polystyrene plates coated with antibody ligands. This
exploits the free binding sites that remain on the antibodies,
whilst the albumin at the other end of the bound triplet was also
measured using an HRP-labelled anti-albumin antibody as a probe
(1). Proteins released from the
platelets by the anti-Gal- or ABG-specific sugars MαG or
cellobiose, respectively, were dialyzed to remove the sugar and
assayed by ELISA. The results demonstrated that the released
proteins contained significantly more albumin that was associated
directly or indirectly with anti-Gal or ABG compared with that in
the same dilution of proteins that was released by the non-specific
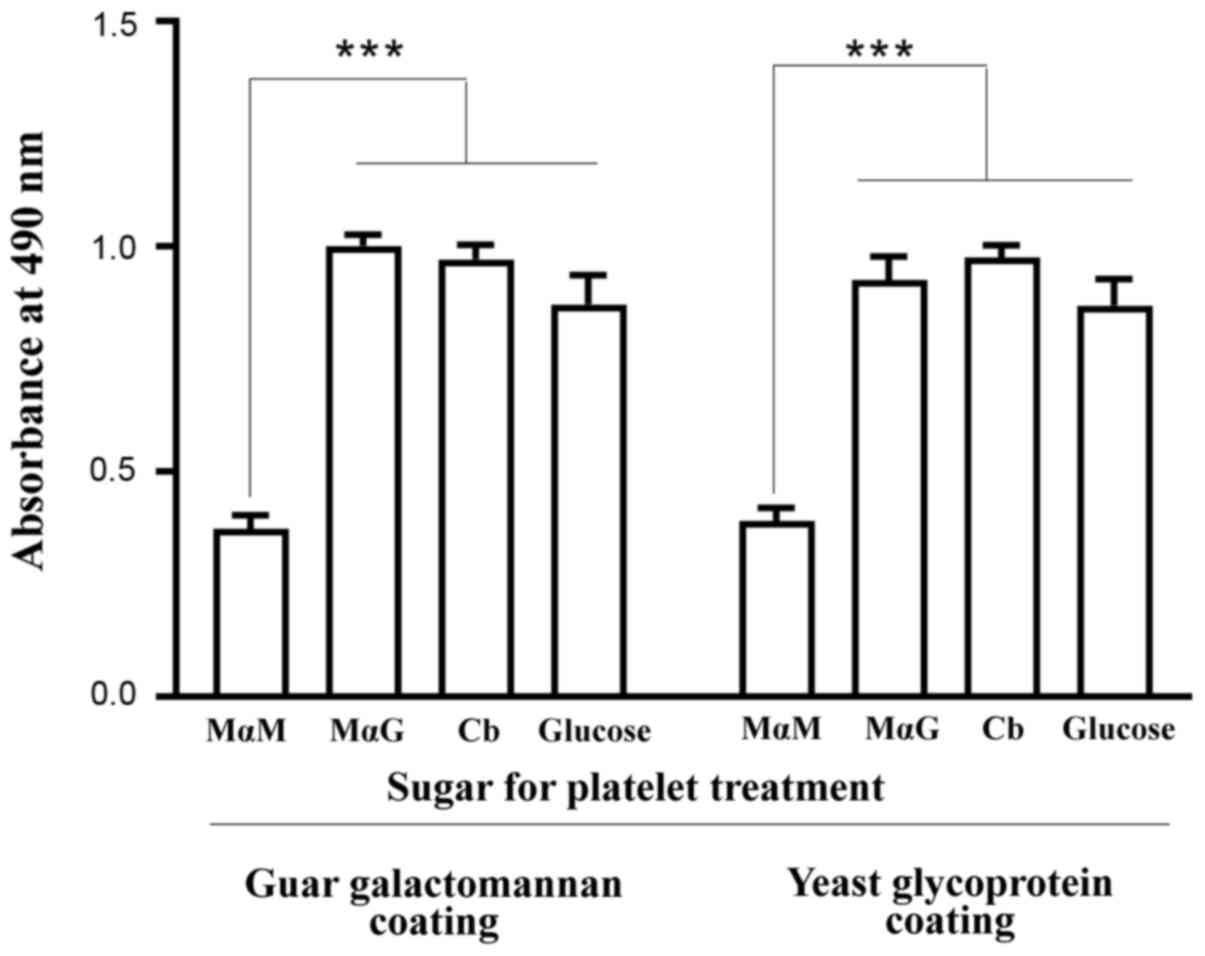
sugar MαM (Fig. 2). The presence of
O-glycoprotein-bound albumin in the proteins released by specific
sugars from the platelets was confirmed by capturing them on
microwells coated with jacalin and probing the bound proteins using
HRP-labelled anti-albumin as performed in the case of plasma
triplets previously (1). Albumin
has no direct association with anti-Gal or ABG, whilst
O-glycoproteins identical in mobility and O-glycan content with
those of AOP1 and AOP2 of plasma triplets were present in the
released proteins. These results suggest that the proteins released
by antibody-specific sugars from platelets contained O-glycoprotein
molecules that bridged between anti-Gal or ABG on one side and
albumin on the other to form triplets of the same structure as that
of plasma anti-Gal/ABG-AOP1/AOP2-albumin triplets (1). Since these triplets possessed binding
sites that remained free on their antibodies and were capable of
binding to ligand-bearing matrices and cells, the aforementioned
results suggested that platelet membranes carry receptor molecules
that possessed ligands for anti-Gal and ABG, which could capture
the triplets by utilizing their free binding sites. Notably,
glucose (15 mM) released nearly as many triplets from the platelets
as the same concentration of MαG or cellobiose did (Fig. 2). Since this level of serum glucose
in circulation is frequently observed under a diabetic setting this
finding suggests that the consequences of depriving platelets of
their triplets accompany diabetes.
Although anti-Gal and ABG share the affinity for
STPS (1), their specificities
towards small sugars are distinct, irrespective of if they were
isolated from plasma triplets or platelet-bound triplets (Fig. 1). Nevertheless, the same amounts of
anti-Gal triplets and ABG triplets were released by either the
ABG-specific sugar or the anti-Gal-specific sugar (Fig. 2).
To verify the observations involving the small
sugar-mediated release of free albumin and O-glycoproteins from
triplets, the differential distribution of AOP1-FITC or AOP2-FITC,
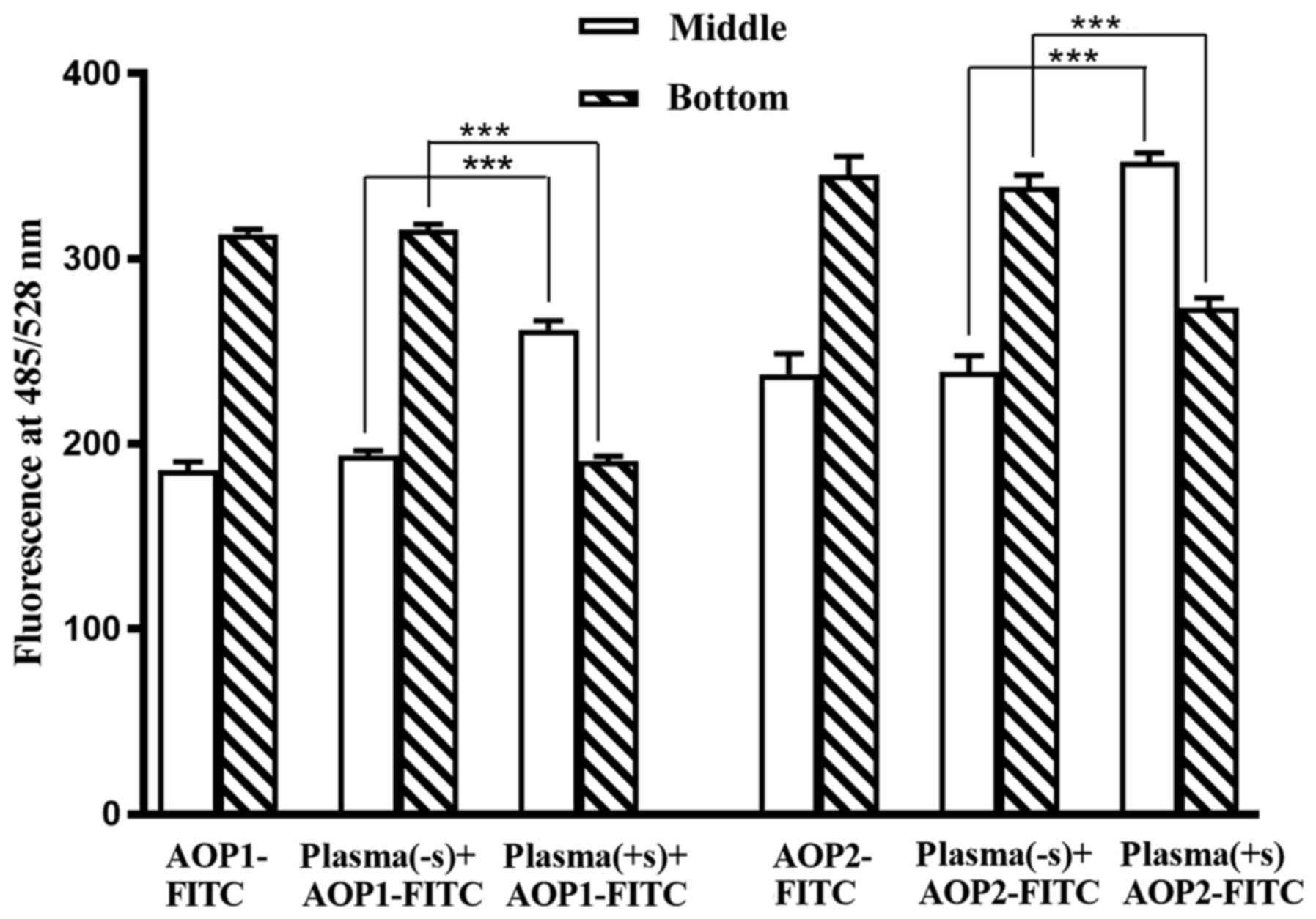
added to sugar-treated and untreated plasma, was examined.
Following DGUC of 1.1 ml KBr-treated plasma, undissociated triplets
were found predominantly in the bottom 300 µl, which was mostly due
to the presence of immunoglobulins (1). However, in the case of plasma treated
with antibody-specific sugar, the albumin-AOP1 and albumin-AOP2
complexes liberated from triplets migrated from the antibody-rich
bottom layer to the antibody-free and albumin-rich middle layer
(400 µl), apparently due to the high buoyancy of albumin, although
free AOP1/AOP2 mostly occupied the bottom layer under these
conditions (1). The results in
Fig. 3 show that the majority of
AOP1-FITC and AOP2-FITC added to PBS or untreated plasma remains in
the bottom 300 µl of the 1.1 ml sample subjected to DGUC. However,
AOP1-FITC and AOP2-FITC, added to plasma that was pre-treated with
anti-Gal- and ABG-specific sugars before DGUC, migrated mostly to
the middle layer, suggesting that free albumin that was ready to
interact with free AOP1/AOP2 or their FITC derivatives was released
in this case. Release of fresh free albumin is also accompanied by
the release of free AOP1 and AOP2, which were bound to the albumin.
Free AOP1 and AOP2 could react with either antibody in a similar
manner to the small sugar ligands (1), which possibly explains the phenomenon
of sugar specific to only one antibody ably releasing triplets of
both antibodies (Fig. 2).
Platelet-bound and plasma triplets are
identical
Platelets deprived of their attached triplets
following treatment with a mixture of anti-Gal- and ABG-specific
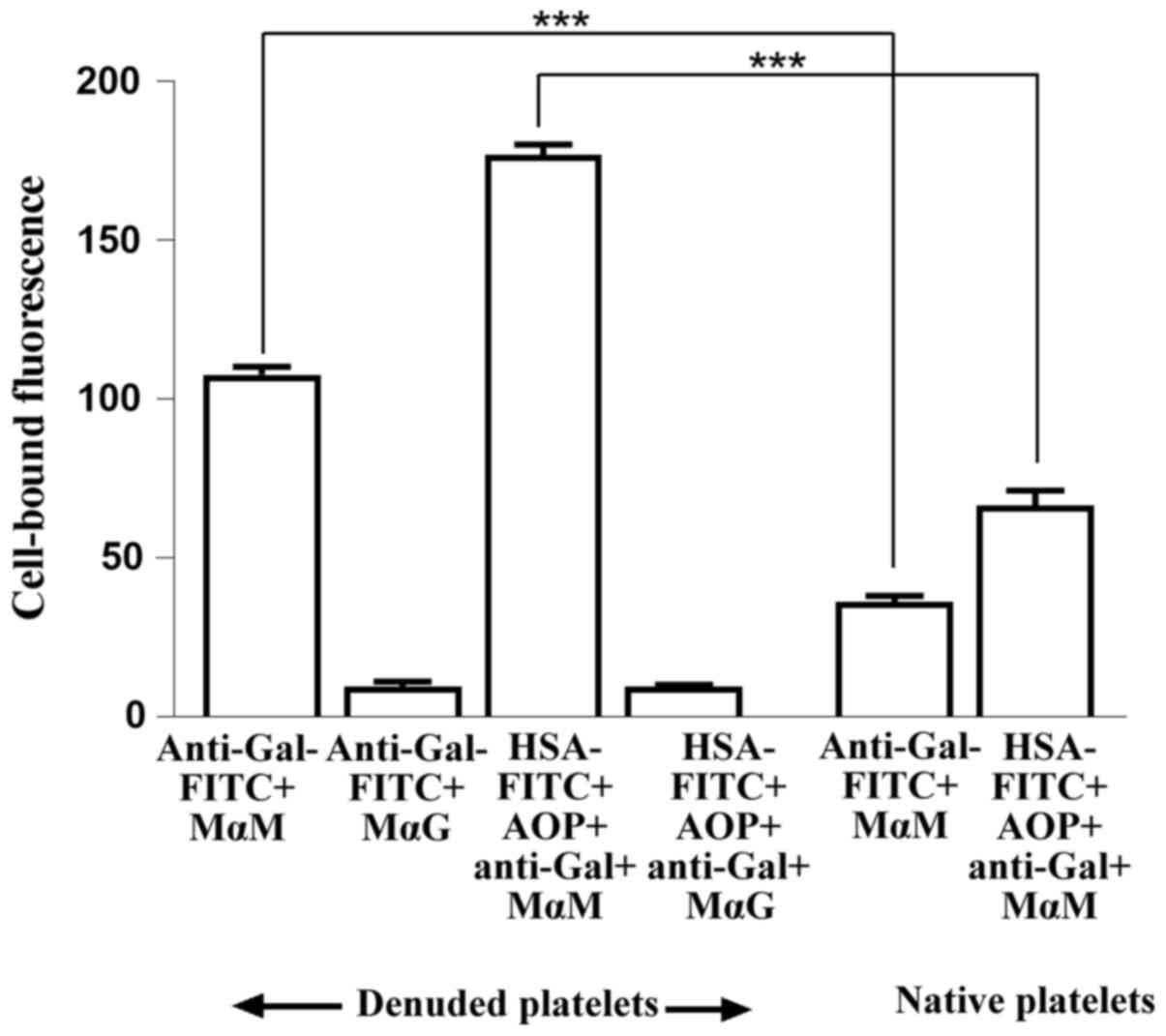
sugars (denuded platelets) were treated with FITC-labelled anti-Gal
or with plasma triplets reconstituted using FITC-labelled albumin,
anti-Gal and a mixture of AOP1 and AOP2. Plasma anti-Gal alone, in
addition to reconstituted plasma triplets, was captured by denuded
platelets but not in the presence of the specific sugars. By
contrast, native platelets captured significantly less of both free
anti-Gal and its corresponding reconstituted triplets (Fig. 4). Since reconstituted plasma
triplets added to the denuded platelets contained FITC-labelled
albumin and their attachment to these platelets was inhibited by
antibody-specific sugars, these results suggest that
antibody-O-glycoprotein-albumin triplets anchored onto the
platelets using the remaining free binding sites available on their
antibodies.
As triplets from cell-free plasma could substitute
for platelet-bound triplets and the O-glycoproteins contained
within the triplets from the plasma and platelets were equal, both
in terms of alkaline gel electrophoretic mobility and in O-glycan
content, it was hypothesized that the triplets of the same
composition and structure exist in free form in the plasma or bound
to platelets. These triplets have been previously found to be
consisted of albumin and either anti-Gal or ABG linked by AOP1 or
AOP2 as the bridging molecule (1).
The distribution of triplets between platelets and cell-free plasma
revealed that the triplets of anti-Gal and ABG are more likely to
be born by platelets than by free plasma in the same blood volume
(Table I).
 | Table ITriplet distribution in blood
(n=8). |
Table I
Triplet distribution in blood
(n=8).
| Triplet
antibody | Plasma (ng
albumin/mm3) | Platelet (ng
albumin/mm3) |
|---|
| Anti-Gal | 52.8±1.61 |
62.9±1.41a |
| ABG | 58.6±1.72 |
66.7±1.21a |
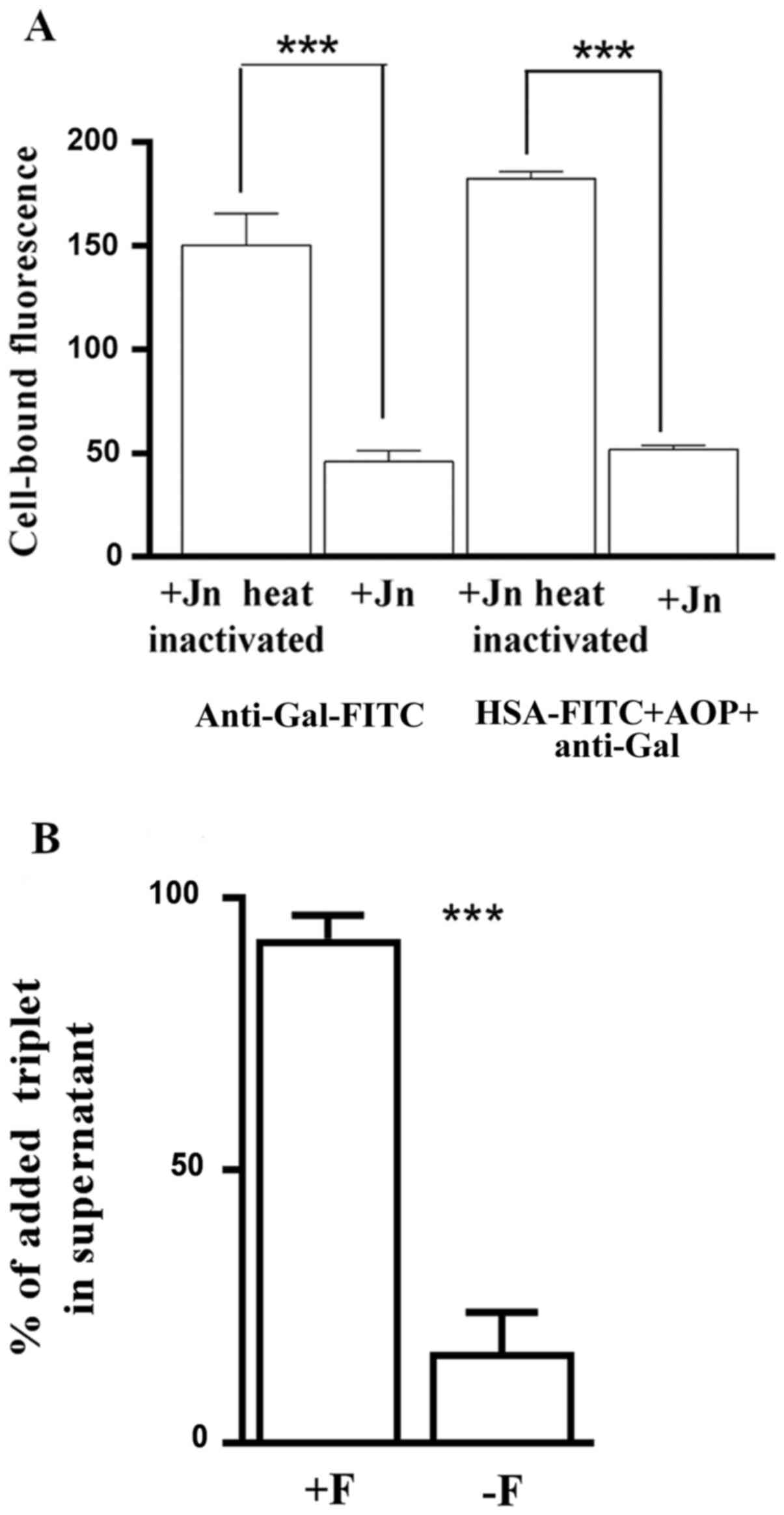
Anti-Gal and its triplets recognize
the O-glycosylated region of their platelet surface glycoprotein
receptor
Jacalin was used to block the O-glycosylated regions
of proteins on the surface of denuded platelets. FITC-labelled
anti-Gal and FITC-labelled triplets of anti-Gal (reconstituted
de novo using anti-Gal, AOP1, AOP2 and FITC-labelled
albumin) were prepared from components of sugar-extracted triplets
of platelets. Neither FITC-labelled anti-Gal nor FITC-labelled
triplets could bind to the denuded platelets that were pre-treated
with jacalin. Heat-inactivated jacalin, however, did not effect
this blocking of rebinding of anti-Gal or triplet (Fig. 5A). Since ABG shares the same STPS
specificity with anti-Gal, the aforementioned results suggest that
the platelet surface O-glycosylated protein(s) may serve as ligands
for the unoccupied binding sites of anti-Gal- and ABG-derived
triplets.
Subsequently, binding of triplets to the denuded
platelets was also assessed by determining the percentage of the
remaining unbound triplets in the supernatant of a limited quantity
of triplets added to the denuded platelets in suspension.
Pre-treatment of denuded platelets with fibrinogen (1 µM) resulted
in the complete blocking of triplet binding to denuded platelets
(Fig. 5B). Presence of 1 µM
fibrinogen did not affect the ELISA response of the triplet sample
used (data not shown).
O-glycans on the platelet surface are
carried predominantly by a 116-kDa protein subunit
Hypotonic lysis of platelets followed by
centrifugation yielded membranes completely devoid of adhering
triplets, since no triplet component was released following their
sugar treatment (result not shown). Western blotting of proteins
from these membranes revealed a number of concanavalin A-binding
N-glycosylated proteins, but only one O-glycosylated subunit
(arrow) was detected by jacalin, which recognizes the core-1 type
O-linked oligosaccharides regardless of the presence of terminal
sialic acid moieties on them (Fig.
6). The presence of the aforementioned oligosaccharides on this
subunit was confirmed by the sugar-dependent binding of peanut
agglutinin to its desialylated form. Furthermore, the ratio of
responses of individual protein subunit bands to jacalin-HRP and
amido black, represented by the areas under the peaks generated by
ImageJ analysis (Table II), were
0.8095 for the protein band recognized by jacalin and a mean of
0.0209 (SD: 2.38%) for four of the other prominent protein bands in
Fig. 6 (lane 1). The marked jacalin
reactivity of the strongly jacalin-reactive band, despite
lectin-carbohydrate interactions being much weaker [association
constant (Ka) ~1.67x105 M-1 for
jacalin binding to fetuin] (19)
than antigen-antibody interactions (Ka in the range
1x106-108 M-1) (20), suggesting its high O-glycan content.
The size of this dominant O-glycoprotein on the platelet surface
was determined to be 116 kDa (Fig.
6), a value close to the reported molecular weights of
120(21), 118(22) and 125±15 kDa (23) for the GPIIb subunit, implying the
possibility that GPIIb/IIIa acts as the dominant, if not the sole,
platelet ligand for anti-Gal and ABG in the triplets.
| Table IIJacalin binding relative to amido
black staining of platelet membrane proteins. |
Table II
Jacalin binding relative to amido
black staining of platelet membrane proteins.
| Platelet membrane
subunit | Jacalin binding
relative to protein contenta |
|---|
| a | 0.0204 |
| b | 0.0214 |
| c | 0.0212 |
| d | 0.0205 |
| j | 0.8095 |
Triplet-free platelets are more prone
to spontaneous aggregation whilst ADP-induced aggregation is
suppressed by prior jacalin treatment
Aggregation of platelets in suspension was monitored
in terms of reduction in the absorbance of the suspension at 405
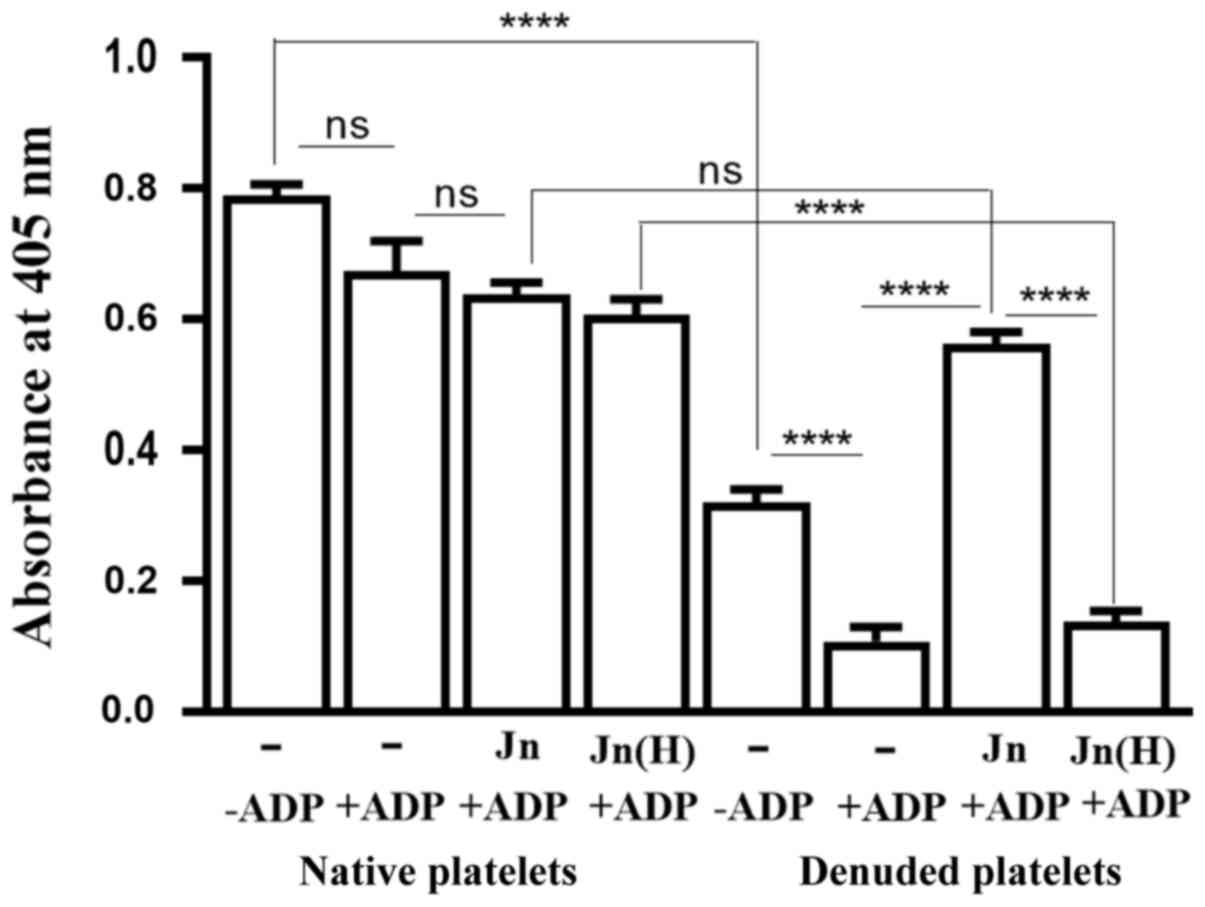
nm. Denuded platelets showed a significantly increased tendency for
spontaneous aggregation compared with native platelets (Fig. 7). However, ADP addition caused an
insignificant increase in the aggregation of native platelets,
though it caused a significant increase in the aggregation of
denuded platelets (Fig. 7).
Treatment with jacalin prior to ADP addition did not affect the
ADP-induced aggregation of native platelets (Fig. 7), but it significantly reduced the
aggregation of denuded platelets. This protection by jacalin of
denuded platelets from ADP-mediated aggregation involved the
binding by the lectin to O-glycan-bearing receptors on the latter,
since the heat-inactivated version of jacalin could not offer this
protection (Fig. 7). There was no
difference between the extent of aggregation of jacalin-treated
denuded platelets and their native counterparts (Fig. 7), whilst there was significantly
greater aggregation by denuded platelets compared with native
platelets when inactivated jacalin was used (Fig. 7). These results suggested that an
O-glycosylated membrane component that is crucial for ADP-mediated
platelet aggregation got exposed after the platelets were
denuded.
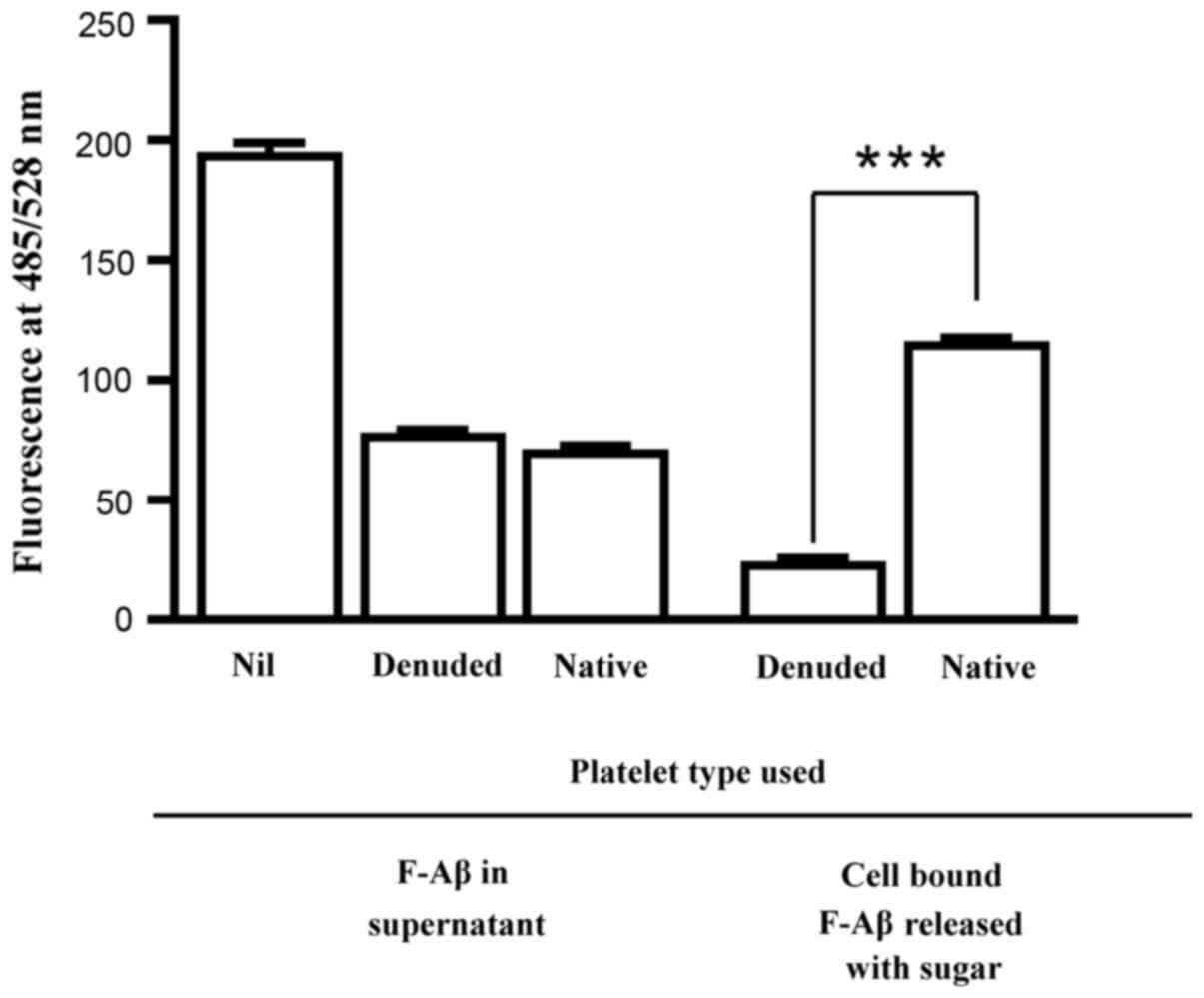
Amyloid β present on the circulating
platelets is bound to the O-glycoproteins of adhering triplets
rather than to GPIIb/IIIa
It was recently demonstrated that AOP1 and AOP2 in
free form, in complex with albumin or in the
anti-Gal/ABG-AOP1/AOP2-albumin triplet complex can capture amyloid
β (Aβ-42) by offering the STPS on these O-glycoproteins as ligands
for the peptide (3). The results in
Fig. 8 demonstrated that native and
denuded platelets captured comparable quantities of amyloid β
presented to them, suggesting that the newly exposed
O-glycoproteins on denuded platelets are capable of capturing
amyloid β. However, upon treatment of the resulting amyloid
β-bearing platelets with sugars that can release the triplets, the
amount of amyloid β released into the supernatant by the denuded
platelets was significantly lower compared with that by the native
platelets (Fig. 8). This suggests
that on native platelets, amyloid β are bound to the triplets using
the STPS on their AOP1 and AOP2 so that they could be released
along with the triplets by antibody-specific sugars. By contrast,
amyloid β bound to the denuded platelets could have utilized the
STPS on membrane O-glycoproteins that were newly exposed following
the release of triplets, such that the antibody-specific sugar
could not elute the peptide.
Discussion
The weaker binding of AOP1/AOP2 compared with
albumin or immunoglobulins to Coomassie blue G-250 in the present
study could be attributed to the abnormally high contents (~55% by
weight) of glycans in these O-glycoproteins (1). Being highly hydrophilic, glycans can
occupy the surfaces of glycoprotein molecules, but have little
affinity for hydrophobic dyes. By contrast, albumin is not
glycosylated whereas immunoglobulins are poorly glycosylated
(24), making them potent Coomassie
blue dye binders. This means that the minimal quantities of triplet
proteins loaded for SDS-PAGE for visualizing the AOP1/AOP2 bands
are sufficient to produce intense albumin and immunoglobulin bands.
Supporting this, Osset et al (25) previously demonstrated that
carbohydrate moieties can block Coomassie brilliant blue binding to
proteins and that dye binding to bovine pancreatic ribonuclease
increased with de-O-glycosylation. In addition, the presence of
sugars was shown to significantly reduce the absorbance of proteins
in a Bradford protein assay using Coomassie brilliant blue
G-250(26). This may explain why
the AOP1 and AOP2 bands were poorly stained even when triplet
protein quantity that produced intense albumin and immunoglobulin
bands was loaded in electrophoresis gels.
One possible reason for sugar specific for either
antibody being able to release triplets of both antibodies from the
platelets is that a small sugar specific for one antibody may
occupy all of its binding sites, resulting in the detachment of its
triplets from the platelets. This in turn releases albumin-bound
O-glycoproteins from the triplet antibody. If the latter event also
resulted in the temporary destabilization of the
O-glycoprotein-albumin complex, free O-glycoproteins would be
generated. Free O-glycoproteins, unlike those complexed with
albumin, resemble low molecular weight antibody-specific sugars in
that they occupy all available binding sites of either antibody
without steric hindrance (1). This
would result in the release of triplets of the other antibody. The
colligative effect of any sugar molecule per se as the
reason for the altered distribution upon DGUC of FITC-labelled
AOP1/AOP2 added to sugar-treated plasma had been previously ruled
out using sugars not specific to either antibody (1). O-Glycoprotein-free albumin that is
already present in the plasma before sugar treatment did not appear
to combine with the FITC-labelled AOP1 and AOP2, since their
distribution was not altered in the presence of sugar-free plasma.
A possible reason for the labelled AOP1/AOP2 to combine with
albumin liberated from the triplets is that the albumin molecules
not involved in triplet formation are likely to be engaged by ≥ one
of the other albumin-binding biomolecules in the plasma, unlike the
nascent albumin liberated from triplets by antibody-specific
sugars.
The results aforementioned demonstrated that STPS
that bear the O-glycans in the O-glycoproteins on the platelet
surface got blocked upon jacalin treatment, since this lectin
recognizes O-glycans. O-Glycan-bearing STPS also appear to be true
ligands for anti-Gal, since terminal α-linked galactose, the
monosaccharide ligand for this antibody, is absent in humans and
O-glycans per se are not ligands for anti-Gal (27), unlike for jacalin. Jacalin-mediated
blocking of anti-Gal access to STPS was demonstrated in the case of
O-glycoproteins in lipoprotein (a) [Lp(a)] (28) and in plasma AOP1 and AOP2(1). STPS underlying the O-glycans has been
confirmed to be anti-Gal and ABG ligands, since the
de-O-glycosylation of AOP1, AOP2 and Lp(a) without destruction of
peptide sequences only enhanced the binding of the antibodies
(1,28). Since ABG shares the same STPS
specificity as anti-Gal, the aforementioned results suggest that
platelet surface O-glycosylated protein(s) may serve as receptors
for the unoccupied binding sites of anti-Gal- and ABG-derived
triplets. Since fibrinogen recognizes the GPIIb/IIIa integrin
present on the platelet surface (4,21),
failure of fibrinogen-treated denuded platelets to capture triplets
pointed to GPIIb/IIIa as a possible receptor for anti-Gal and ABG
antibodies present in triplets. In support of this hypothesis,
GPIIb/IIIa is the dominant O-glycosylated protein expressed on the
platelet membrane (21-23).
The requirement for an O-glycosylated site for the binding of
anti-Gal or triplet to platelets found in the present study also
supports this hypothesis. Although plasma fibrinogen concentration
(8-9 µM) far exceeds the concentration used in the present study
for blocking triplet attachment, circulating platelets can remain
bound to triplets. This suggests that although fibrinogen and
triplets may bind to the same receptor(s) on the denuded platelets,
competitive displacement of bound triplets by fibrinogen is not
extensive.
The primary event in ADP-mediated platelet
aggregation is binding to the two G-protein-coupled receptors, P2Y1
and P2Y12, on the platelet surface (29). Although no data on the dependence of
this binding on O-glycosylated platelet surface molecules are
available, involvement of GPIIb/IIIa in subsequent events after
ADP-mediated platelet aggregation has been reported (21). In addition to explaining the
jacalin-mediated inhibition of the ADP-induced aggregation of
denuded platelets, the present study also suggested the possibility
that GPIIb/IIIa that is heavily O-glycosylated and becomes exposed
following denudation serves as a major platelet membrane receptor
for triplets. Compared with AOP1 and AOP2, albumin that was
electrophoretically separated from these O-glycoproteins was inert
towards amyloid β (3). In addition,
though it was previously reported that amyloid β could bind to an
albumin sample (30), this sample
also contained AOP1 and AOP2(3).
Amyloid β binding to AOP1 and AOP2 was consistent with the reported
binding of this peptide to the STPS-rich GPIIb/IIIa (31). Since GPIIb/IIIa is the most abundant
O-glycoprotein on the platelet surface and shares the amyloid
β-binding property of AOP1 and AOP2, the present study also
suggested a possible role for GPIIb/IIIa in triplet adhesion to
platelets. This is because GPIIb/IIIa-bound amyloid β, which cannot
be extracted from platelets using the anti-Gal- or ABG-specific
sugars, can be formed only on denuded platelets. This observation
suggests that GPIIb/IIIa is otherwise engaged and is inaccessible
to amyloid β on native platelets until the triplets have been
removed.
Beyond their role in clotting and homeostasis of
blood, platelets have also been found to adhere onto leukocytes,
resulting in the release of leukocyte and platelet constituents
(32). This damages vascular walls
and may be implicated in diseases, including myocardial infarction
(32) and ischemic stroke (33). A clinically important phenomenon,
largely unexplained on molecular level, is the increased
susceptibility of the platelets of patients with diabetes to
activation and aggregation (7,34).
This has been hypothesized to contribute to widespread vascular
damage (35). Furthermore, a role
for platelets as carriers of amyloid β, thereby acting as a
circulating sink for this peptide to reduce its concentration in
the brain, has been previously recognized (5), although the exact receptor for this
peptide on platelets remains unknown.
The availability of anti-Gal- and ABG-containing
triplets in the blood appears to be determined mostly by the
synthesis and/or plasma availability of these antibodies, since
~36% plasma albumin were found to be combined with AOP1 or AOP2 in
plasma (unpublished data), ensuring a large excess of
albumin-O-glycoprotein complexes over the antibodies.
Identification of a platelet surface molecule that can be
recognized by the antibodies in anti-Gal/ABG-AOP1/AOP2-albumin
triplets is important for understanding the contribution of the
latter towards platelet function. Complete blocking of triplet
attachment to denuded platelets by jacalin, which specifically
binds to O-glycoproteins and keeps them engaged, suggests the
O-glycoprotein abundance of the triplet receptor on platelet
surface. Furthermore, since the most jacalin-reactive
O-glycosylated molecule identified in the present study on the
platelet membrane was a subunit with an estimated molecular weight
of 116 kDa, which was comparable to the reported molecular weights
of the GPIIb subunit, GPIIb/IIIa appeared to be the most probable
ligand on the triplets. Another well-known O-glycosylated platelet
membrane protein, GP1bα, has a reported molecular weight of ≥135
kDa (36) and is far less abundant
than GPIIb/IIIa which is expressed at a density of ~80,000
molecules per platelet, considered the highest in any known cell
type (37). This could explain the
absence of any detectable jacalin-binding protein other than the
116-kDa subunit in the western blots of the platelet membrane.
Other evidence implicating GPIIb/IIIa to be the ligand for triplet
attachment includes the following: i) resistance of native
triplet-containing platelets to ADP-mediated aggregation, even when
all platelet components and plasma factors were available; ii)
blocking of the ADP-mediated aggregation of denuded platelets by
pre-bound jacalin, which for reasons aforementioned should be bound
predominantly to GPIIb/IIIa, an essential intermediate in
ADP-mediated platelet activation and aggregation (21,27,28);
and iii) complete blocking of triplet binding to denuded platelets
by pre-incubation with fibrinogen, which binds to GPIIb/IIIa.
Therefore, whilst direct evidence remains lacking, GPIIb/IIIa is
most likely to be a ligand for anti-Gal/ABG-AOP1/AOP2-albumin
triplets on platelets.
In addition to serving as the plasma sink for this
peptide to limit its availability in the brain (5), platelets have also been reported to
act as carriers of amyloid β to perivascular cells of the brain
following vascular damage (31).
Although GPIIb/IIIa has been reported to serve as a receptor for
amyloid β on platelets (31), the
present results indicate that this may be true only of denuded
platelets or of isolated platelet membranes. In addition, in native
circulating platelets anti-Gal/ABG-AOP1/AOP2-albumin triplets
mediate amyloid β binding. Supporting this conclusion are previous
reports that diabetes is the most common predisposing factor for
Alzheimer's disease (AD) (38,39).
Although glucose is nearly as efficient as cellobiose as a ligand
for ABG, it does not dissociate the ABG-triplet at normoglycemic
levels (~4.7 mM) (1). However,
concentrations of glucose in diabetes may reach up to 5-6 times
higher than normal, which is inhibitory to ABG binding and can
dissociate triplets containing ABG and anti-Gal (12), as shown in the present study. The
denuded platelets were capable of capturing amyloid β, presumably
through the newly exposed cell surface O-glycoprotein vacated by
triplet antibodies. However, they are more prone to aggregation and
are therefore less stable, resulting in substantial attenuation of
their amyloid β-arresting activity. A previous report that blood
samples from patients with AD exhibited a 39.57% increase in
platelet aggregates and a 53.3% increase in leukocyte-platelet
complexes (40) also supports this
conclusion. The myriad of platelet-mediated vascular injuries found
alongside diabetes also implicates the triplet-mediated protection
of platelets. Platelet-dependent thrombosis was found to be
proportional to blood glucose levels in coronary artery disease
(35). Majority of the tissue
toxicity associated with diabetes has been attributed to oxidative
and inflammatory stress due to the enhanced production of
mitochondrial superoxide dismutase, resulting in reduced
mitochondrial function and cell viability (41). However, short-term hyperglycaemia in
patients with diabetes has been reported to be sufficient in
causing vascular occlusion through platelet activation (34,42),
suggesting that events that occur earlier than oxidative stress
induction can account for vascular damage. In addition, the cascade
of inflammatory events leading to cerebral amyloid angiopathy
preceding AD has been reported to be triggered by amyloid β binding
to the exposed GPIIb/IIIa on platelets that adhere to vessel walls
(31).
Platelet-leukocyte adhesion facilitated by platelet
surface GpIIb/IIIa is a trigger for the synthesis and release of
inflammatory factors, such as leukotrienes and thromboxane A2, by
leukocytes and platelets (43).
These factors have been implicated in acute myocardial infarction
(44) and stroke (33). Since the protective cover for
GPIIb/IIIa presumably provided by triplets is diminished during
hyperglycaemia, the platelets become susceptible to leukocyte
adhesion. Results from the present study may explain in molecular
terms the contribution of diabetes towards the GPIIb/IIIa-mediated
pathophysiology of the aforementioned disorders.
In summary, the results of the present study
revealed the molecular basis for ABG/anti-Gal
antibody-AOP1/AOP2-albumin triplets anchoring onto platelets, for
the absence of platelet aggregation under normal conditions and for
platelets serving as an amyloid β sink in the circulation. These
data may offer an alternative direction for the investigation of
diabetes-mediated platelet vulnerability, platelet-leukocyte
adhesion and the contribution of diabetes towards AD. The main
limitation of the present study is that the data indicating
GPIIb/IIIa as the main O-glycoprotein anchor on platelets for
triplets remains indirect. Further investigations are therefore
required to confirm this using additional experiments, such as
blocking of triplet attachment to platelets using antibodies to
suspected receptors and variations in triplet binding to platelets
with GPIIb/IIIa O-glycosylation levels.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Sree Chitra
Tirunal Institute for Medical Sciences and Technology,
Thiruvananthapuram 695011, India (grant nos. Ph.D/2005/01/SS and
Ph.D/2013/05/SK).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SK and SSR conducted the experiments and
authenticate the raw data of this study. MG contributed to methods
development and adaptation, including optimizing glycoimmunology
protocols to the platelet context. PSA conceptualized the work and
analysed the data. All authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Ethics Committee of the Sree Chitra Tirunal Institute for Medical
Sciences And Technology, Thiruvanthapuram (approval nos. IEC-674
and IEC-1072). Written informed consent was obtained from voluntary
blood donors who participated.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sreedevi K, Subramanian SP, Mandagini G
and Appukuttan PS: Anti-α-galactoside and anti-β-glucoside
antibodies are partially occupied by either of two albumin-bound
O-glycoproteins and circulate as ligand-binding triplets. Immunol
Invest. 48:222–241. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sandrin MS, Vaughan HA, Xing PX and
McKenzie IF: Natural human anti-Gal alpha(1,3)Gal antibodies react
with human mucin peptides. Glycoconj J. 14:97–105. 1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Karthi S, Sumitha KC, Geetha M and
Appukuttan PS: Amyloid β binds to albumin-associated Lrp-like
plasma O-glycoproteins: Albumin prevents inhibition of binding by
LDL. Protein Pept Lett. 26:869–878. 2019.
|
|
4
|
Wang Y, Jobe SM, Ding X, Choo H, Archer
DR, Mi R, Ju T and Cummings RD: Platelet biogenesis and functions
require correct protein O-glycosylation. Proc Natl Acad Sci USA.
109:16143–16148. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
George JN: Platelet IgG: Measurement,
interpretation, and clinical significance. Prog Hemost Thromb.
10:97–126. 1991.PubMed/NCBI
|
|
6
|
Chen M, Inestrosa NC, Ross GS and
Fernandez HL: Platelets are the primary source of amyloid
beta-peptide in human blood. Biochem Biophys Res Commun.
213:96–103. 1995.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kwaan HC, Collwell JA, Cruz S, Sewanwela N
and Dobbie JG: Increased platelet aggregation in diabetes mellitus.
J Lab Clin Med. 80:236–246. 1972.PubMed/NCBI
|
|
8
|
Suresh Kumar G, Appukuttan PS and Basu D:
α-D-galactose-specific lectin from jack fruit (Artocarpus
integra) seed. J Biosci. 4:257–261. 1982.
|
|
9
|
Baues RJ and Grays GR: Lectin purification
on affinity columns containing reductively aminated disaccharides.
J Biol Chem. 252:57–60. 1977.PubMed/NCBI
|
|
10
|
Paul A, Antony M, Mathai J and Appukuttan
PS: High polymeric IgA content facilitates recognition of microbial
polysaccharide-natural serum antibody immune complexes by
immobilized human galectin-1. Immunol Lett. 136:55–60.
2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jaison PL and Appukuttan PS: Rapid
isolation of human plasma anti-alpha-galactoside antibody using
sugar-specific binding to guar galactomannan or agarose. Indian J
Biochem Biophys. 29:266–270. 1992.PubMed/NCBI
|
|
12
|
Geetha M, Annamma KI, Mathai J and
Appukuttan PS: Normal human plasma anti-beta-glucoside antibody has
markedly elevated IgA content and binds fungal and yeast
polysaccharides. Immunol Invest. 36:73–83. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hudson L and Hay FC: Practical immunology.
2nd edition. Blackwell Scientific Publications, Oxford, p222,
1980.
|
|
14
|
Jennings LK and Phillips DR: Purification
of glycoproteins IIb and III from human platelet plasma membranes
and characterization of a calcium-dependent glycoprotein IIb-III
complex. J Biol Chem. 257:10458–10466. 1982.PubMed/NCBI
|
|
15
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: Procedure and some applications. Proc Natl
Acad Sci USA. 76:4350–4354. 1979.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sastry MV, Banerjee P, Patanjali SR, Swamy
MJ, Swarnalatha GV and Surolia A: Analysis of saccharide binding to
Artocarpus integrifolia lectin reveals specific recognition
of T-antigen (beta-D-Gal(1→3)D-GalNAc). J Biol Chem.
261:11726–11733. 1986.PubMed/NCBI
|
|
18
|
Dolbeare FA: Platelet aggregation as a
quantitative immunologic technique. Immunol Commun. 2:65–76.
1973.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pedroso MM, Pesquero NC, Thomaz SM,
Roque-Barreira MC, Faria RC and Bueno PR: Jacalin interaction with
human immunoglobulin A1 and bovine immunoglobulin G1: Affinity
constant determined by piezoelectric biosensoring. Glycobiology.
22:326–331. 2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Pan Y, Sackmann EK, Wypisniak K, Hornsby
M, Datwani SS and Herr AE: Determination of equilibrium
dissociation constants for recombinant antibodies by
high-throughput affinity electrophoresis. Sci Rep.
6(39774)2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Coller BS and Shattil SJ: The GPIIb/IIIa
(integrin alphaIIbbeta3) odyssey: A technology-driven saga of a
receptor with twists, turns, and even a bend. Blood. 112:3011–3025.
2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jung SM, Yoshida N, Aoki N, Tanoue K,
Yamazaki H and Moroi M: Thrombasthenia with an abnormal platelet
membrane glycoprotein IIb of different molecular weight. Blood.
71:915–922. 1988.PubMed/NCBI
|
|
23
|
Rivas GA, Aznárez JA, Usobiaga P, Saiz JL
and González-Rodríguez J: Molecular characterization of the human
platelet integrin GPIIb/IIIa and its constituent glycoproteins. Eur
Biophys J. 19:335–345. 1991.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Arnold JN, Wormald MR, Sim RB, Rudd PM and
Dwek RA: The impact of glycosylation on the biological function and
structure of human immunoglobulins. Annu Rev Immunol. 25:21–50.
2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Osset M, Piñol M, Fallon MJ, de Llorence R
and Cuchillo CM: Interference of the carbohydrate moiety in
coomassie brilliant blue R-250 protein staining. Electrophoresis.
10:271–273. 1989.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Banik SP, Pal S, Gorai S, Chowdhury S and
Khowala S: Interference of sugars in the coomassie blue-G dye
binding assay of proteins. Anal Biochem. 386:113–115.
2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Galili U, Shohet SB, Kobrin E, Stults CL
and Macher BA: Man, apes, and old world monkeys differ from other
mammals in the expression of alpha-galactosyl epitopes on nucleated
cells. J Biol Chem. 263:17755–17762. 1988.PubMed/NCBI
|
|
28
|
Geetha M, Kalaivani V, Sabarinath PS and
Appukuttan PS: Plasma anti-α-galactoside antibody binds to serine-
and threonine-rich peptide sequence of apo(a) subunit in Lp(a).
Glycoconj J. 31:289–298. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Dorsam RT and Kunapuli SP: Central role of
the P2Y12 receptor in platelet activation. J Clin Invest.
113:340–345. 2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Biere AL, Ostaszewski B, Stimson ER, Hyman
BT, Maggio JE and Selkoe DJ: Amyloid beta-peptide is transported on
lipoproteins and albumin in human plasma. J Biol Chem.
271:32916–32922. 1996.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Donner L, Fälker K, Gremer L, Klinker S,
Pagani G, Ljungberg LU, Lothmann K, Rizzi F, Schaller M, Gohlke H,
et al: Platelets contribute to amyloid-β aggregation in cerebral
vessels through integrin αIIbβ3-induced outside-in signaling and
clusterin release. Sci Signal. 9(ra52)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Totani L and Evangelista V:
Platelet-leukocyte interactions in cardiovascular disease and
beyond. Arterioscler Thromb Vasc Biol. 30:2357–2361.
2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zeller JA, Lenz A, Eschenfelder CC, Zunker
P and Deuschl D: Platelet-leukocyte interaction and platelet
activation in acute stroke with and without preceding infection.
Arterioscler Thromb Vasc Biol. 25:1519–1523. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ferroni P, Basili S, Falco A and Davi G:
Platelet activation in type 2 diabetes mellitus. J Thromb Hemost.
2:1282–1291. 2004.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Shechter M, Merz CN, Paul-Labrador MJ and
Kaul S: Blood glucose and platelet-dependent thrombosis in patients
with coronary artery disease. J Am Coll Cardiol. 35:300–307.
2000.PubMed/NCBI View Article : Google Scholar
|
|
36
|
López JA, Ludwig EH and McCarthy BJ:
Polymorphism of human glycoprotein Ib alpha results from a variable
number of tandem repeats of a 13-amino acid sequence in the
mucin-like macroglycopeptide region. Structure/function
implications. J Biol Chem. 267:10055–10061. 1992.PubMed/NCBI
|
|
37
|
Wagner CL, Mascelli MA, Neblock DS,
Weisman HF, Coller BS and Jordan BE: Analysis of GPIIb/IIIa
receptor number by quantification of 7E3 binding to human
platelets. Blood. 88:907–914. 1996.PubMed/NCBI
|
|
38
|
Akter K, Lanza EA, Martin SA, Myronyuk N,
Rua M and Raffa RB: Diabetes mellitus and Alzheimer's disease:
Shared pathology and treatment? Brit J Clin Pharm. 71:365–376.
2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zheng F, Yan L, Yang Z, Zhong B and Xie W:
HbA1c, diabetes and cognitive decline: The English
Longitudinal study of ageing. Diabetologia. 61:839–848.
2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sevush S, JY W, Horstman LL, Mao WW,
Kolodny L and Abn YS: Platelet activation in Alzheimer disease.
Arch Neurol. 55:530–536. 1998.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820.
2001.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gresele P, Guglielmini G, De Angelis M,
Cifferi S, Ciofetta M, Falcinelli E, Lalli C, Ciabattoni G, Davi G
and Bolli BG: Acute, short-term hyperglycemia enhances shear
stress-induced platelet activation in patients with type II
diabetes mellitus. J Am Coll Cardiol. 41:1013–1020. 2003.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Totani L and Evangelista V:
Platelet-leukocyte interactions in cardiovascular diseases and
beyond. Arterioscler Thromb Vasc Biol. 30:2357–2361.
2010.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Neumann FJ, Zohlnhöfer D, Fakhoury L, Ott
I, Gawaz M and Schömig A: Effect of glycoprotein IIb/IIIa receptor
blockade on platelet-leukocyte interaction and surface expression
of the leukocyte integrin Mac-1 in acute myocardial infarction. J
Am Coll Cardiol. 34:1420–1426. 1999.PubMed/NCBI View Article : Google Scholar
|